
Advancements, Challenges, and Future Directions in Tackling Glioblastoma Resistance to Small Kinase Inhibitors



Abstract
:Simple Summary
Abstract
1. Introduction
2. Heterogeneity of GBM
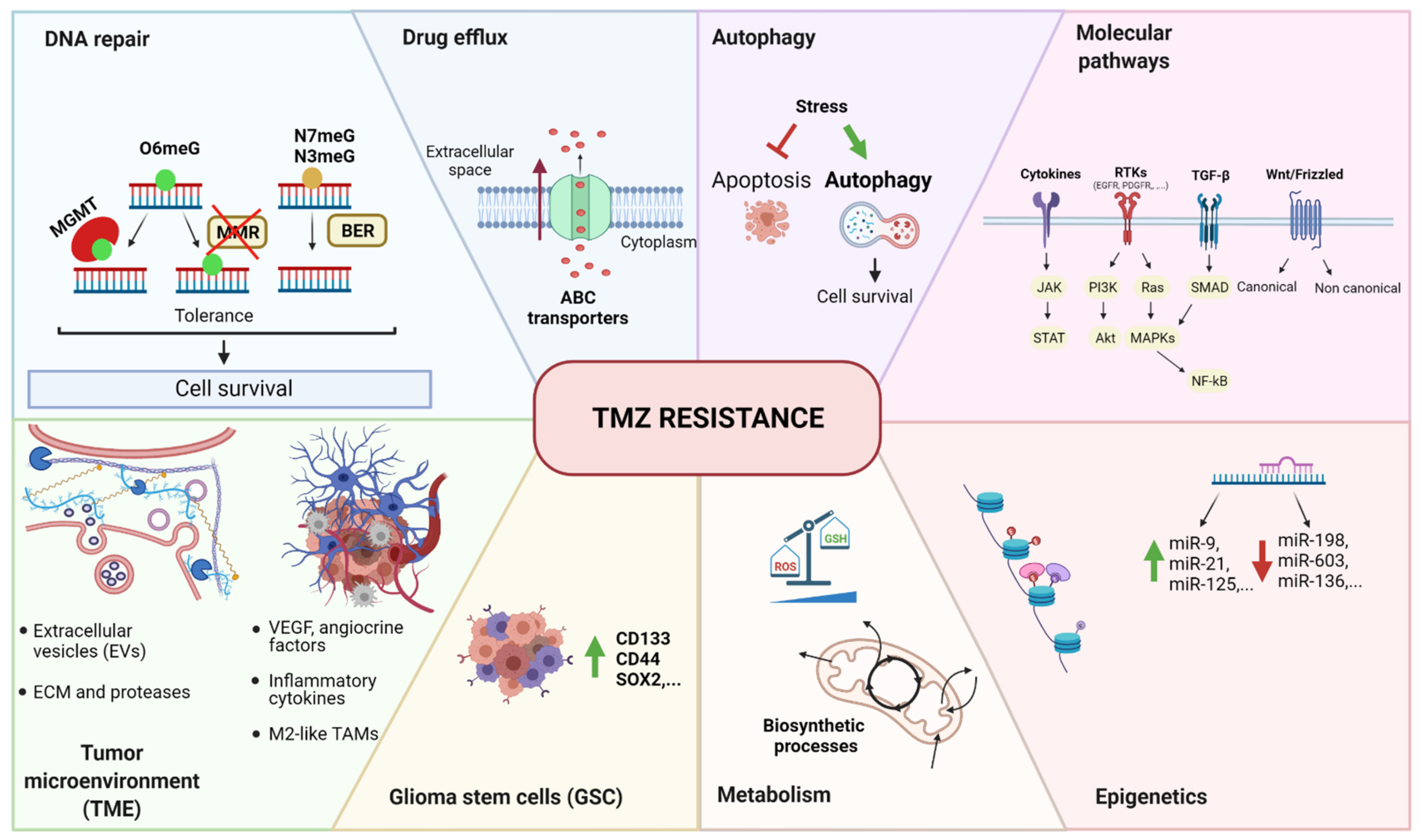
3. Temozolomide Resistance
4. Kinases in GBM
4.1. RTKs
4.1.1. EGFR
4.1.2. PDGFR
4.1.3. MET
4.1.4. FGFR
4.1.5. VEGF
4.1.6. IGF-1R
4.2. Downstream Intracellular Signaling
4.2.1. RAS–RAF–MEK–ERK Pathway
4.2.2. PI3K–AKT–mTOR Pathway
4.2.3. PKC Pathway
4.2.4. JAK/STAT Pathway
4.2.5. P53 Pathway
4.2.6. Rb Pathway
5. Small Kinase Inhibitors: Mechanisms of Resistance
5.1. Mutations
5.2. Coactivation or Transactivation of Other RTKs
5.3. Adaptation
5.4. Alternative Routes (Bypass)
5.5. Glioblastoma Stem Cells (GSC)
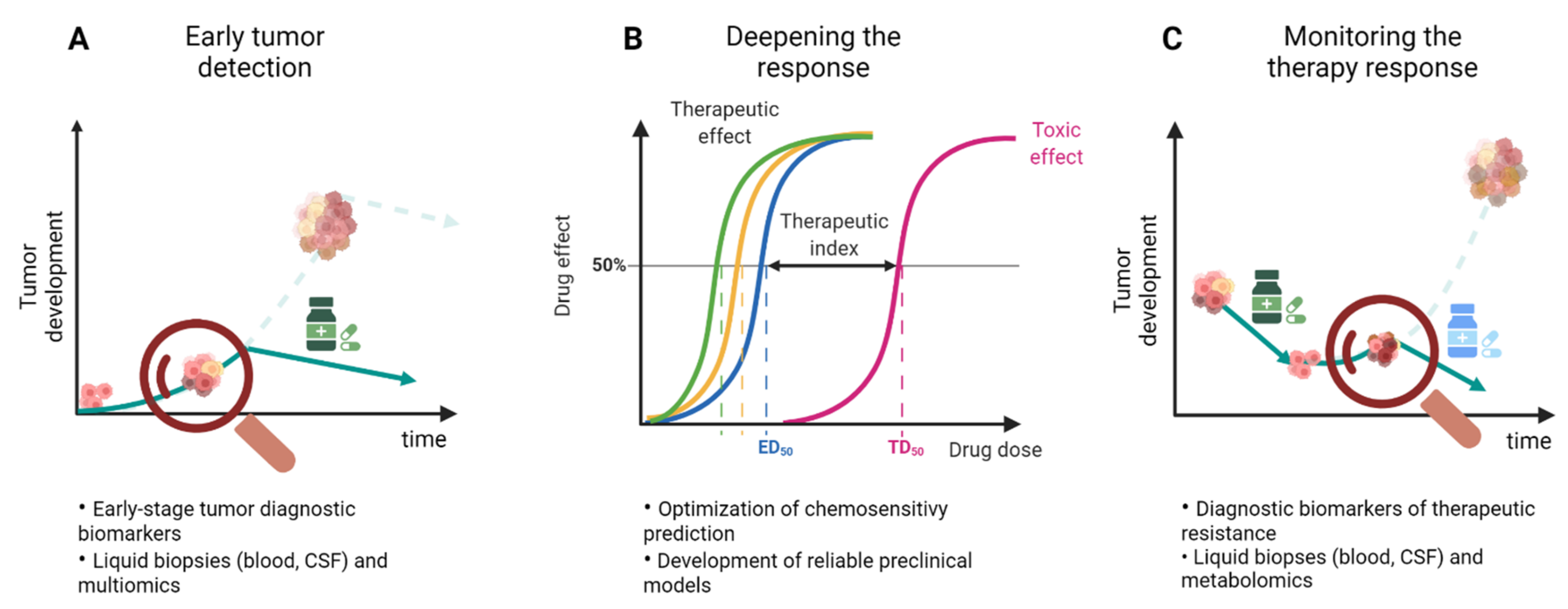
6. Overcoming Drug Resistance
6.1. In Vitro Models for Drug Resistance
6.2. Clinical Strategies
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-Dense Temozolomide for Newly Diagnosed Glioblastoma: A Randomized Phase III Clinical Trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef]
- Kolibaba, K.S.; Druker, B.J. Protein tyrosine kinases and cancer. Biochim. Biophys. Acta (BBA) Bioenerg. 1997, 1333, F217–F248. [Google Scholar] [CrossRef]
- Kim, G.; Ko, Y.T. Small molecule tyrosine kinase inhibitors in glioblastoma. Arch. Pharmacal Res. 2020, 43, 385–394. [Google Scholar] [CrossRef]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, E.C. Glioblastoma multiforme: The terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242–6244. [Google Scholar] [CrossRef] [Green Version]
- Urbańska, K.; Sokołowska, J.; Szmidt, M.; Sysa, P. Glioblastoma multiforme—An overview. Contemp. Oncol. 2014, 18, 307–312. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; Decarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, I.-H.; Cho, H.J.; Park, C.-K.; Jung, Y.-S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.-S.; et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational Analysis Reveals the Origin and Therapy-Driven Evolution of Recurrent Glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, K.; Meylan, M.; Bougoüin, A.; Connor, K.; Salvucci, M.; Bielle, F.; Prehn, J.H.M.; Verreault, M.; Idbaih, A.; Sautès-Fridman, C.; et al. TAMI-51. Identifying new tumor microenvironment (tme) contexts of vulnerability in glioblastoma. Neuro-Oncology 2020, 22 (Suppl. 2), ii224. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [Green Version]
- De Vleeschouwer, S.; Bergers, G. Glioblastoma: To Target the Tumor Cell or the Microenvironment? Glioblastoma 2017, 315–340. [Google Scholar] [CrossRef]
- Guan, X.; Hasan, N.; Maniar, S.; Jia, W.; Sun, D. Reactive Astrocytes in Glioblastoma Multiforme. Mol. Neurobiol. 2018, 55, 6927–6938. [Google Scholar] [CrossRef]
- Heiland, D.H.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef] [Green Version]
- Kenig, S.; Alonso, M.B.D.; Mueller, M.M.; Lah, T.T. Glioblastoma and endothelial cells cross-talk, mediated by SDF-1, enhances tumour invasion and endothelial proliferation by increasing expression of cathepsins B, S, and MMP-9. Cancer Lett. 2010, 289, 53–61. [Google Scholar] [CrossRef]
- Hadi, L.A.; Anelli, V.; Guarnaccia, L.; Navone, S.; Beretta, M.; Moccia, F.; Tringali, C.; Urechie, V.; Campanella, R.; Marfia, G.; et al. A bidirectional crosstalk between glioblastoma and brain endothelial cells potentiates the angiogenic and proliferative signaling of sphingosine-1-phosphate in the glioblastoma microenvironment. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2018, 1863, 1179–1192. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem Cell–like Glioma Cells Promote Tumor Angiogenesis through Vascular Endothelial Growth Factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [Green Version]
- Folkins, C.; Shaked, Y.; Man, S.; Tang, T.; Lee, C.R.; Zhu, Z.; Hoffman, R.M.; Kerbel, R.S. Glioma Tumor Stem-Like Cells Promote Tumor Angiogenesis and Vasculogenesis via Vascular Endothelial Growth Factor and Stromal-Derived Factor 1. Cancer Res. 2009, 69, 7243–7251, Correction in Cancer Res. 2009, 69, 8216. [Google Scholar] [CrossRef] [Green Version]
- Tong, N.; He, Z.; Ma, Y.; Wang, Z.; Huang, Z.; Cao, H.; Xu, L.; Zou, Y.; Wang, W.; Yi, C.; et al. Tumor Associated Macrophages, as the Dominant Immune Cells, Are an Indispensable Target for Immunologically Cold Tumor—Glioma Therapy? Front. Cell Dev. Biol. 2021, 9, 706286. [Google Scholar] [CrossRef]
- Andersen, R.S.; Anand, A.; Harwood, D.S.L.; Kristensen, B.W. Tumor-Associated Microglia and Macrophages in the Glioblastoma Microenvironment and Their Implications for Therapy. Cancers 2021, 13, 4255. [Google Scholar] [CrossRef]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy; Exon Publications: Brisbane City, Australia, 2017; pp. 197–241. [Google Scholar]
- Danson, S.J.; Middleton, M.R. Temozolomide: A novel oral alkylating agent. Expert Rev. Anticancer. Ther. 2001, 1, 13–19. [Google Scholar] [CrossRef]
- Denny, B.J.; Wheelhouse, R.T.; Stevens, M.F.G.; Tsang, L.L.H.; Slack, J.A. NMR and Molecular Modeling Investigation of the Mechanism of Activation of the Antitumor Drug Temozolomide and Its Interaction with DNA. Biochemistry 1994, 33, 9045–9051. [Google Scholar] [CrossRef]
- Bobola, M.S.; Kolstoe, D.D.; Blank, A.; Chamberlain, M.C.; Silber, J.R. Repair of 3-methyladenine and abasic sites by base excision repair mediates glioblastoma resistance to temozolomide. Front. Oncol. 2012, 2, 176. [Google Scholar] [CrossRef] [Green Version]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef]
- Karran, P.; Bignami, M. DNA damage tolerance, mismatch repair and genome instability. BioEssays 1994, 16, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Ziouta, A.; Ochs, K.; Coquerelle, T. Chromosomal instability, reproductive cell death and apoptosis induced by O6-methylguanine in Mex−, Mex+ and methylation-tolerant mismatch repair compromised cells: Facts and models. Mutat. Res. Mol. Mech. Mutagen. 1997, 381, 227–241. [Google Scholar] [CrossRef]
- Ochs, K.; Kaina, B. Apoptosis induced by DNA Damage O-Methylguanine is Bcl-2 and Caspase-9/3 regulated and Fas/Caspase-8 independent. Cancer Res. 2000, 60, 5815–5824. [Google Scholar]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Spiegl-Kreinecker, S.; Pirker, C.; Filipits, M.; Lötsch, D.; Buchroithner, J.; Pichler, J.; Silye, R.; Weis, S.; Micksche, M.; Fischer, J.; et al. O6-Methylguanine DNA methyltransferase protein expression in tumor cells predicts outcome of temozolomide therapy in glioblastoma patients. Neuro-Oncology 2009, 12, 28–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro-Oncology 2009, 11, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergström, T.; Jarvius, M.; Xie, Y.; Westermark, A.; Sönmez, D.; Hermansson, A.; Kastemar, M.; et al. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016, 17, 2994–3009. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2020, 4, 17–43. [Google Scholar] [CrossRef]
- Poon, W.-S.; Woo, P.Y.M.; Li, Y.; Chan, A.H.Y.; Ng, S.C.P.; Loong, H.H.F.; Chan, D.T.M.; Wong, G.K.C. A multifaceted review of temozolomide resistance mechanisms in glioblastoma beyond O-6-methylguanine-DNA methyltransferase. Glioma 2019, 2, 68. [Google Scholar] [CrossRef]
- Akgül, S.; Patch, A.-M.; D’Souza, R.C.; Mukhopadhyay, P.; Nones, K.; Kempe, S.; Kazakoff, S.H.; Jeffree, R.L.; Stringer, B.W.; Pearson, J.V.; et al. Intratumoural Heterogeneity Underlies Distinct Therapy Responses and Treatment Resistance in Glioblastoma. Cancers 2019, 11, 190. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Van Vliet, M.; Zhao, J.; De Ståhl, T.D.; Lindström, M.S.; Cheng, H.; Heller, S.; Nistér, M.; Hägerstrand, D. Identification of functionally distinct and interacting cancer cell subpopulations from glioblastoma with intratumoral genetic heterogeneity. Neuro-Oncol. Adv. 2020, 2, vdaa061. [Google Scholar] [CrossRef]
- Lahiry, P.; Torkamani, A.; Schork, N.J.; Hegele, R.A. Kinase mutations in human disease: Interpreting genotype–phenotype relationships. Nat. Rev. Genet. 2010, 11, 60–74. [Google Scholar] [CrossRef]
- Gross, S.; Rahal, R.; Stransky, N.; Lengauer, C.; Hoeflich, K.P. Targeting cancer with kinase inhibitors. J. Clin. Investig. 2015, 125, 1780–1789. [Google Scholar] [CrossRef]
- Nørøxe, D.S.; Poulsen, H.S.; Lassen, U. Hallmarks of glioblastoma: A systematic review. ESMO Open 2016, 1, e000144. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Hamer, P.C.D.W. Small molecule kinase inhibitors in glioblastoma: A systematic review of clinical studies. Neuro-Oncology 2010, 12, 304–316. [Google Scholar] [CrossRef]
- Hermanson, M.; Funa, K.; Hartman, M.; Claesson-Welsh, L.; Heldin, C.H.; Westermark, B.; Nistér, M. Platelet-derived growth factor and its receptors in human glioma tissue: Expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992, 52, 3213–3219. [Google Scholar]
- Nishikawa, R.; Ji, X.D.; Harmon, R.C.; Lazar, C.S.; Gill, G.N.; Cavenee, W.K.; Huang, H.J. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. USA 1994, 91, 7727–7731. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.J.; Bigner, S.H.; Bigner, D.D.; Kinzler, K.W.; Hamilton, S.R.; Vogelstein, B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc. Natl. Acad. Sci. USA 1987, 84, 6899–6903. [Google Scholar] [CrossRef] [Green Version]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Boerner, J.L.; Demory, M.L.; Silva, C.; Parsons, S.J. Phosphorylation of Y845 on the Epidermal Growth Factor Receptor Mediates Binding to the Mitochondrial Protein Cytochrome c Oxidase Subunit II. Mol. Cell. Biol. 2004, 24, 7059–7071. [Google Scholar] [CrossRef] [Green Version]
- Liccardi, G.; Hartley, J.A.; Hochhauser, D. EGFR Nuclear Translocation Modulates DNA Repair following Cisplatin and Ionizing Radiation Treatment. Cancer Res. 2011, 71, 1103–1114. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-C.; Hung, M.-C. Nuclear Translocation of the Epidermal Growth Factor Receptor Family Membrane Tyrosine Kinase Receptors. Clin. Cancer Res. 2009, 15, 6484–6489. [Google Scholar] [CrossRef] [Green Version]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Ozawa, T.; Brennan, C.W.; Wang, L.; Squatrito, M.; Sasayama, T.; Nakada, M.; Huse, J.T.; Pedraza, A.; Utsuki, S.; Yasui, Y.; et al. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev. 2010, 24, 2205–2218. [Google Scholar] [CrossRef] [Green Version]
- Alentorn, A.; Marie, Y.; Carpentier, C.; Boisselier, B.; Giry, M.; Labussière, M.; Mokhtari, K.; Hoang-Xuan, K.; Sanson, M.; Delattre, J.-Y.; et al. Prevalence, clinico-pathological value, and co-occurrence of PDGFRA abnormalities in diffuse gliomas. Neuro-Oncology 2012, 14, 1393–1403. [Google Scholar] [CrossRef]
- Clarke, I.D.; Dirks, P.B. A human brain tumor-derived PDGFR-α deletion mutant is transforming. Oncogene 2003, 22, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Koochekpour, S.; Jeffers, M.; Rulong, S.; Taylor, G.; Klineberg, E.; Hudson, E.A.; Resau, J.H.; Woude, G.F.V. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res. 1997, 57, 5391–5398. [Google Scholar]
- Abounader, R.; Laterra, J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro-Oncology 2005, 7, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Guo, D. MET in glioma: Signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Navis, A.C.; Van Lith, S.; Van Duijnhoven, S.M.J.; De Pooter, M.; Yetkin-Arik, B.; Wesseling, P.; Hendriks, W.J.A.J.; Venselaar, H.; Timmer, M.; Van Cleef, P.; et al. Identification of a novel MET mutation in high-grade glioma resulting in an auto-active intracellular protein. Acta Neuropathol. 2015, 130, 131–144. [Google Scholar] [CrossRef] [Green Version]
- Bao, Z.-S.; Chen, H.-M.; Yang, M.-Y.; Zhang, C.-B.; Yu, K.; Ye, W.-L.; Hu, B.-Q.; Yan, W.; Zhang, W.; Akers, J.; et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res. 2014, 24, 1765–1773. [Google Scholar] [CrossRef] [Green Version]
- International Cancer Genome Consortium PedBrain Tumor Project. Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat. Med. 2016, 22, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Pascual, A.; Siebzehnrubl, F.A. Fibroblast Growth Factor Receptor Functions in Glioblastoma. Cells 2019, 8, 715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasorella, A.; Sanson, M.; Iavarone, A. FGFR-TACC gene fusions in human glioma. Neuro-Oncology 2016, 19, now240–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming Fusions of FGFR and TACC Genes in Human Glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Hamerlik, P.; Lathia, J.D.; Rasmussen, R.; Wu, Q.; Bartkova, J.; Lee, M.; Moudry, P.; Bartek, J., Jr.; Fischer, W.; Lukas, J.; et al. Autocrine VEGF–VEGFR2–Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J. Exp. Med. 2012, 209, 507–520. [Google Scholar] [CrossRef] [Green Version]
- Kessler, T.; Sahm, F.; Blaes, J.; Osswald, M.; Rübmann, P.; Milford, D.; Urban, S.; Jestaedt, L.; Heiland, S.; Bendszus, M.; et al. Glioma cell VEGFR-2 confers resistance to chemotherapeutic and antiangiogenic treatments in PTEN-deficient glioblastoma. Oncotarget 2015, 6, 31050–31068. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wu, X.; Zhu, J. VEGF Promotes Proliferation of Human Glioblastoma Multiforme Stem-Like Cells through VEGF Receptor 2. Sci. World J. 2013, 2013, 417413. [Google Scholar] [CrossRef] [Green Version]
- Maris, C.; D’Haene, N.; Trepant, A.-L.; Le Mercier, M.; Sauvage, S.; Allard, J.; Rorive, S.; Demetter, P.; Decaestecker, C.; Salmon, I. IGF-IR: A new prognostic biomarker for human glioblastoma. Br. J. Cancer 2015, 113, 729–737. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, J.; Li, M.; Zhang, S.; Lu, Y.; Liang, Y.; Zhao, K.; Li, Y. Insulin-like growth factor 1/insulin-like growth factor 1 receptor signaling protects against cell apoptosis through the PI3K/AKT pathway in glioblastoma cells. Exp. Ther. Med. 2018, 16, 1477–1482. [Google Scholar] [CrossRef]
- Kondo, Y.; Hollingsworth, E.F.; Kondo, S. Molecular targeting for malignant gliomas (Review). Int. J. Oncol. 2004, 24, 1101–1109. [Google Scholar] [CrossRef]
- Qiu, X.Y.; Hu, D.X.; Chen, W.-Q.; Chen, R.Q.; Qian, S.R.; Li, C.Y.; Li, Y.J.; Xiong, X.X.; Liu, D.; Pan, F.; et al. PD-L1 confers glioblastoma multiforme malignancy via Ras binding and Ras/Erk/EMT activation. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 1754–1769. [Google Scholar] [CrossRef]
- Bos, J.L. The ras gene family and human carcinogenesis. Mutat. Res. Genet. Toxicol. 1988, 195, 255–271. [Google Scholar] [CrossRef]
- Jeuken, J.; Broecke, C.V.D.; Gijsen, S.; Boots-Sprenger, S.; Wesseling, P. RAS/RAF pathway activation in gliomas: The result of copy number gains rather than activating mutations. Acta Neuropathol. 2007, 114, 121–133. [Google Scholar] [CrossRef]
- Knobbe, C.B.; Reifenberger, J.; Reifenberger, G. Mutation analysis of the Ras pathway genes NRAS, HRAS, KRAS and BRAF in glioblastomas. Acta Neuropathol. 2004, 108, 467–470. [Google Scholar] [CrossRef]
- Behling, F.; Schittenhelm, J. Oncogenic BRAF Alterations and Their Role in Brain Tumors. Cancers 2019, 11, 794. [Google Scholar] [CrossRef] [Green Version]
- Pelloski, C.E.; Lin, E.; Zhang, L.; Yung, W.A.; Colman, H.; Liu, J.-L.; Woo, S.Y.; Heimberger, A.B.; Suki, D.; Prados, M.; et al. Prognostic Associations of Activated Mitogen-Activated Protein Kinase and Akt Pathways in Glioblastoma. Clin. Cancer Res. 2006, 12, 3935–3941. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Luo, J.; Cantley, L. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Langhans, J.; Schneele, L.; Trenkler, N.; Von Bandemer, H.; Nonnenmacher, L.; Karpel-Massler, G.; Siegelin, M.D.; Zhou, S.; Halatsch, M.-E.; Debatin, K.-M.; et al. The effects of PI3K-mediated signalling on glioblastoma cell behaviour. Oncogenesis 2017, 6, 398. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, C.; Bartels, G.; Gehlhaar, C.; Holtkamp, N.; von Deimling, A. PIK3CA mutations in glioblastoma multiforme. Acta Neuropathol. 2005, 109, 639–642. [Google Scholar] [CrossRef]
- Mizoguchi, M.; Nutt, C.L.; Mohapatra, G.; Louis, D.N. Genetic Alterations of Phosphoinositide 3-kinase Subunit Genes in Human Glioblastomas. Brain Pathol. 2006, 14, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Batchelor, T.T.; Iafrate, A.J.; Dias-Santagata, D.; Borger, D.R.; Ellisen, L.W.; Yang, D.; Louis, D.N.; Cahill, D.P.; Chi, A.S. PIK3CA activating mutations are associated with more disseminated disease at presentation and earlier recurrence in glioblastoma. Acta Neuropathol. Commun. 2019, 7, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis. Markers 2018, 2018, 9230479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, A.J.; Procyk, K.J.; Leitges, M.; Parker, P.J. PKC alpha protein but not kinase activity is critical for glioma cell proliferation and survival. Int. J. Cancer 2008, 123, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; McNulty, A.M.; Hanna, K.R.; Konicek, B.W.; Lynch, R.L.; Bailey, S.N.; Banks, C.; Capen, A.; Goode, R.; Lewis, J.E.; et al. The Protein Kinase Cβ–Selective Inhibitor, Enzastaurin (LY317615.HCl), Suppresses Signaling through the AKT Pathway, Induces Apoptosis, and Suppresses Growth of Human Colon Cancer and Glioblastoma Xenografts. Cancer Res. 2005, 65, 7462–7469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmo, A.D.; Balça-Silva, J.; Matias, D.; Lopes, M.C. PKC signaling in glioblastoma. Cancer Biol. Ther. 2013, 14, 287–294. [Google Scholar] [CrossRef] [Green Version]
- Ou, A.; Ott, M.; Fang, D.; Heimberger, A. The Role and Therapeutic Targeting of JAK/STAT Signaling in Glioblastoma. Cancers 2021, 13, 437. [Google Scholar] [CrossRef] [PubMed]
- Brantley, E.C.; Benveniste, E.N. Signal Transducer and Activator of Transcription-3: A Molecular Hub for Signaling Pathways in Gliomas. Mol. Cancer Res. 2008, 6, 675–684. [Google Scholar] [CrossRef] [Green Version]
- De La Iglesia, N.; Puram, S.V.; Bonni, A. STAT3 Regulation of Glioblastoma Pathogenesis. Curr. Mol. Med. 2009, 9, 580–590. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Dube, C.; Gibert, J.M.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef] [Green Version]
- England, B.; Huang, T.; Karsy, M. Current understanding of the role and targeting of tumor suppressor p53 in glioblastoma multiforme. Tumor Biol. 2013, 34, 2063–2074. [Google Scholar] [CrossRef]
- Nevins, J.R. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 2001, 10, 699–703. [Google Scholar] [CrossRef]
- Biasoli, D.; Kahn, S.A.; Cornélio, T.A.; Furtado, M.; Campanati, L.; Chneiweiss, H.; Moura-Neto, V.; Borges, H.L. Retinoblastoma protein regulates the crosstalk between autophagy and apoptosis, and favors glioblastoma resistance to etoposide. Cell Death Dis. 2013, 4, e767. [Google Scholar] [CrossRef] [Green Version]
- Juric, V.; Murphy, B. Cyclin-dependent kinase inhibitors in brain cancer: Current state and future directions. Cancer Drug Resist. 2020, 3, 48–62. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten things you should know about protein kinases: IUPHAR Review 14. J. Cereb. Blood Flow Metab. 2015, 172, 2675–2700. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, S.; Lamfers, M.L.M.; Dirven, C.M.F.; Leenstra, S. Genetic biomarkers of drug response for small-molecule therapeutics targeting the RTK/Ras/PI3K, p53 or Rb pathway in glioblastoma. CNS Oncol. 2016, 5, 77–90. [Google Scholar] [CrossRef]
- Thorne, A.H.; Zanca, C.; Furnari, F. Epidermal growth factor receptor targeting and challenges in glioblastoma. Neuro-Oncology 2016, 18, 914–918. [Google Scholar] [CrossRef] [Green Version]
- Pearson, J.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, A.S.; Sorensen, A.G.; Jain, R.K.; Batchelor, T.T. Angiogenesis as a Therapeutic Target in Malignant Gliomas. Oncology 2009, 14, 621–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Turner, S.; Peters, K.B.; Desjardins, A.; Gururangan, S.; Sampson, J.; McLendon, R.E.; Herndon, J.E.; Jones, L.W.; Kirkpatrick, J.; et al. A Review of VEGF/VEGFR-Targeted Therapeutics for Recurrent Glioblastoma. J. Natl. Compr. Cancer Netw. 2011, 9, 414–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, H.-W. Targeting Ras-RAF-ERK and its Interactive Pathways as a Novel Therapy for Malignant Gliomas. Curr. Cancer Drug Targets 2010, 10, 840–848. [Google Scholar] [CrossRef]
- Schreck, K.C.; Patel, M.P.; Wemmer, J.; Grossman, S.A.; Peters, K.B. RAF and MEK inhibitor therapy in adult patients with brain tumors: A case-based overview and practical management of adverse events. Neuro-Oncol. Pr. 2020, 7, 369–375. [Google Scholar] [CrossRef]
- Zhao, H.F.; Wang, J.; Shao, W.; Wu, C.P.; Chen, Z.P.; To, S.T.; Li, W.P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [Green Version]
- Riggins, G.J.; Gallia, G.L. Targeting the AKT Pathway in Glioblastoma. Curr. Pharm. Des. 2011, 17, 2411–2420. [Google Scholar] [CrossRef]
- Gerber, D.E. EGFR inhibition in the treatment of non-small cell lung cancer. Drug Dev. Res. 2008, 69, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Marie, Y.; Carpentier, A.F.; Omuro, A.; Sanson, M.; Thillet, J.; Hoang-Xuan, K.; Delattre, J.-Y. EGFR tyrosine kinase domain mutations in human gliomas. Neurology 2005, 64, 1444–1445. [Google Scholar] [CrossRef]
- Vivanco, I.; Robins, H.I.; Rohle, D.; Campos, C.; Grommes, C.; Nghiemphu, P.L.; Kubek, S.; Oldrini, B.; Chheda, M.G.; Yannuzzi, N.; et al. Differential Sensitivity of Glioma- versus Lung Cancer–Specific EGFR Mutations to EGFR Kinase Inhibitors. Cancer Discov. 2012, 2, 458–471. [Google Scholar] [CrossRef] [Green Version]
- Burger, K.; Bakker, M.A.D.; Kros, J.M.; De Bruin, A.M.; Oosterhuis, W.; Ingh, H.F.V.D.; Van Der Harst, E.; De Schipper, H.P.; Wiemer, E.A.; Nooter, K.; et al. Activating mutations in c-KIT and PDGFRα are exclusively found in gastrointestinal stromal tumors and not in other tumors overexpressing these imatinib mesylate target genes. Cancer Biol. Ther. 2005, 4, 1270–1274. [Google Scholar] [CrossRef] [Green Version]
- Mocellin, S.; Pasquali, S.; Campana, L.; Yuan, Y.; Gronchi, A.; Griffiths, E.; Vohra, R. Tyrosine kinase inhibitor therapies for gastrointestinal stromal tumours. Cochrane Database Syst. Rev. 2018, 2018, CD012951. [Google Scholar] [CrossRef]
- De Luca, A.; Abate, R.E.; Rachiglio, A.M.; Maiello, M.R.; Esposito, C.; Schettino, C.; Izzo, F.; Nasti, G.; Normanno, N. FGFR Fusions in Cancer: From Diagnostic Approaches to Therapeutic Intervention. Int. J. Mol. Sci. 2020, 21, 6856. [Google Scholar] [CrossRef]
- Inda, M.-d.-M.; Bonavia, R.; Seoane, J. Glioblastoma Multiforme: A Look Inside Its Heterogeneous Nature. Cancers 2014, 6, 226–239. [Google Scholar] [CrossRef] [Green Version]
- Stommel, J.M.; Kimmelman, A.C.; Ying, H.; Nabioullin, R.; Ponugoti, A.H.; Wiedemeyer, R.; Stegh, A.H.; Bradner, J.E.; Ligon, K.L.; Brennan, C.; et al. Coactivation of Receptor Tyrosine Kinases Affects the Response of Tumor Cells to Targeted Therapies. Science 2007, 318, 287–290. [Google Scholar] [CrossRef]
- Szerlip, N.J.; Pedraza, A.; Chakravarty, D.; Azim, M.; McGuire, J.; Fang, Y.; Ozawa, T.; Holland, E.C.; Huse, J.T.; Jhanwar, S.; et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc. Natl. Acad. Sci. USA 2012, 109, 3041–3046. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Yuan, Y.; Lin, Y.; Wang, Y.-X.; Zhou, J.; Gai, Q.-J.; Zhang, L.; Mao, M.; Yao, X.-X.; Qin, Y.; et al. ERBB3, IGF1R, and TGFBR2 expression correlate with PDGFR expression in glioblastoma and participate in PDGFR inhibitor resistance of glioblastoma cells. Am. J. Cancer Res. 2018, 8, 792–809. [Google Scholar]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.Y.; Chute, D.J.; et al. Molecular Determinants of the Response of Glioblastomas to EGFR Kinase Inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef] [Green Version]
- Mellinghoff, I.K.; Cloughesy, T.F.; Mischel, P.S. PTEN-Mediated Resistance to Epidermal Growth Factor Receptor Kinase Inhibitors. Clin. Cancer Res. 2007, 13, 378–381. [Google Scholar] [CrossRef] [Green Version]
- Greenall, S.; Donoghue, J.; Van Sinderen, M.; Dubljevic, V.; Budiman, S.; Devlin, M.J.; Street, I.P.; Adams, T.; Johns, T. EGFRvIII-mediated transactivation of receptor tyrosine kinases in glioma: Mechanism and therapeutic implications. Oncogene 2015, 34, 5277–5287. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Narayan, R.N.; Horton, L.; Patel, T.R.; Habib, A.A. The Role of EGFR-Met Interactions in the Pathogenesis of Glioblastoma and Resistance to Treatment. Curr. Cancer Drug Targets 2017, 17, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Mukasa, A.; Bonavia, R.; Flynn, R.A.; Brewer, Z.E.; Cavenee, W.K.; Furnari, F.B.; White, F.M. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc. Natl. Acad. Sci. USA 2007, 104, 12867–12872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, H.J.; Acquaviva, J.; Chi, D.; Lessard, J.; Zhu, H.; Woolfenden, S.; Bronson, R.T.; Pfannl, R.; White, F.; Housman, D.E.; et al. Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme. Oncogene 2011, 31, 3039–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, B.; Goodwin, C.R.; Sang, Y.; Foss, C.A.; Cornet, K.; Muzamil, S.; Pomper, M.G.; Kim, J.; Laterra, J. EGFRvIII and c-Met pathway inhibitors synergize against PTEN-null/EGFRvIII+ glioblastoma xenografts. Mol. Cancer Ther. 2009, 8, 1751–1760. [Google Scholar] [CrossRef] [Green Version]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET Receptor Drives Resistance to Anti-EGFR Therapies in Colorectal Cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [Green Version]
- Cappuzzo, F.; Jänne, P.A.; Skokan, M.; Finocchiaro, G.; Rossi, E.; Ligorio, C.; Zucali, P.A.; Terracciano, L.; Toschi, L.; Roncalli, M.; et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann. Oncol. 2008, 20, 298–304. [Google Scholar] [CrossRef]
- Nathanson, D.A.; Gini, B.; Mottahedeh, J.; Visnyei, K.; Koga, T.; Gomez, G.; Eskin, A.; Hwang, K.; Wang, J.; Masui, K.; et al. Targeted Therapy Resistance Mediated by Dynamic Regulation of Extrachromosomal Mutant EGFR DNA. Science 2014, 343, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Akhavan, D.; Pourzia, A.L.; Nourian, A.A.; Williams, K.J.; Nathanson, D.; Babic, I.; Villa, G.R.; Tanaka, K.; Nael, A.; Yang, H.; et al. De-Repression of PDGFRβ Transcription Promotes Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in Glioblastoma Patients. Cancer Discov. 2013, 3, 534–547. [Google Scholar] [CrossRef] [Green Version]
- Rubin, B.P.; Duensing, A. Mechanisms of resistance to small molecule kinase inhibition in the treatment of solid tumors. Lab. Investig. 2006, 86, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Day, E.K.; Sosale, N.G.; Xiao, A.; Zhong, Q.; Purow, B.; Lazzara, M.J. Glioblastoma Cell Resistance to EGFR and MET Inhibition Can Be Overcome via Blockade of FGFR-SPRY2 Bypass Signaling. Cell Rep. 2020, 30, 3383–3396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnin, D.A.A.; Ran, C.; Havrda, M.C.; Liu, H.; Hitoshi, Y.; Zhang, Z.; Cheng, C.; Ung, M.; Israel, M.A. Insulin-Mediated Signaling Facilitates Resistance to PDGFR Inhibition in Proneural hPDGFB-Driven Gliomas. Mol. Cancer Ther. 2017, 16, 705–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastorino, S.; Langley, E.J.; Chao, Y.; Jiang, P.; Mukthavaram, R.; Pingle, S.C.; Kim, P.S.; Singh, S.; Kesari, S. Mechanisms of resistance to PDGFR inhibition in glioblastoma. J. Clin. Oncol. 2014, 32, e13030. [Google Scholar] [CrossRef]
- Chakravarti, A.; Loeffler, J.S.; Dyson, N.J. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res. 2002, 62, 200–207. [Google Scholar] [PubMed]
- Ma, Y.; Tang, N.; Thompson, R.C.; Mobley, B.C.; Clark, S.W.; Sarkaria, J.N.; Wang, J. InsR/IGF1R Pathway Mediates Resistance to EGFR Inhibitors in Glioblastoma. Clin. Cancer Res. 2015, 22, 1767–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruickshanks, N.; Zhang, Y.; Hine, S.; Gibert, M.; Yuan, F.; Oxford, M.; Grello, C.M.; Pahuski, M.; Dube, C.; Guessous, F.; et al. Discovery and Therapeutic Exploitation of Mechanisms of Resistance to MET Inhibitors in Glioblastoma. Clin. Cancer Res. 2018, 25, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Bielen, A.; Perryman, L.; Box, G.M.; Valenti, M.; Brandon, A.D.H.; Martins, V.; Jury, A.; Popov, S.; Gowan, S.; Jeay, S.; et al. Enhanced Efficacy of IGF1R Inhibition in Pediatric Glioblastoma by Combinatorial Targeting of PDGFRα/β. Mol. Cancer Ther. 2011, 10, 1407–1418. [Google Scholar] [CrossRef] [Green Version]
- Carapancea, M.; Cosaceanu, D.; Budiu, R.; Kwiecinska, A.; Tataranu, L.G.; Ciubotaru, V.; Alexandru, O.; Banita, M.; Pisoschi, C.; Bäcklund, M.; et al. Dual targeting of IGF-1R and PDGFR inhibits proliferation in high-grade gliomas cells and induces radiosensitivity in JNK-1 expressing cells. J. Neuro-Oncol. 2007, 85, 245–254. [Google Scholar] [CrossRef]
- Carrasco-Garcia, E.; Martinez-Lacaci, I.; Mayor-López, L.; Tristante, E.; Carballo-Santana, M.; García-Morales, P.; Ventero Martin, M.P.; Fuentes-Baile, M.; Rodriguez-Lescure, Á.; Saceda, M. PDGFR and IGF-1R Inhibitors Induce a G2/M Arrest and Subsequent Cell Death in Human Glioblastoma Cell Lines. Cells 2018, 7, 131. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.J.; Hsieh, J.K.; Manjila, S.; Lathia, J.D.; Sloan, A. The role of cancer stem cells in glioblastoma. Neurosurg. Focus 2014, 37, E6. [Google Scholar] [CrossRef] [Green Version]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Dong, J.; Haiech, J.; Kilhoffer, M.-C.; Zeniou, M. Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int. 2016, 2016, 1740936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Rothe, K.; Babaian, A.; Nakamichi, N.; Chen, M.; Chafe, S.C.; Watanabe, A.; Forrest, D.L.; Mager, D.L.; Eaves, C.J.; Dedhar, S.; et al. Integrin-Linked Kinase Mediates Therapeutic Resistance of Quiescent CML Stem Cells to Tyrosine Kinase Inhibitors. Cell Stem Cell 2020, 27, 110–124.e9. [Google Scholar] [CrossRef]
- Del Re, M.; Arrigoni, E.; Restante, G.; Passaro, A.; Rofi, E.; Crucitta, S.; De Marinis, F.; Di Paolo, A.; Danesi, R. Concise Review: Resistance to Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer: The Role of Cancer Stem Cells. Stem Cells 2018, 36, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Gianì, F.; Vella, V.; Tumino, D.; Malandrino, P.; Frasca, F. The Possible Role of Cancer Stem Cells in the Resistance to Kinase Inhibitors of Advanced Thyroid Cancer. Cancers 2020, 12, 2249. [Google Scholar] [CrossRef]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-Dimensional Cell Culture Systems and Their Applications in Drug Discovery and Cell-Based Biosensors. ASSAY Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Duval, K.; Grover, H.; Han, L.-H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Fontoura, J.C.; Viezzer, C.; dos Santos, F.G.; Ligabue, R.A.; Weinlich, R.; Puga, R.D.; Antonow, D.; Severino, P.; Bonorino, C. Comparison of 2D and 3D cell culture models for cell growth, gene expression and drug resistance. Mater. Sci. Eng. C 2019, 107, 110264. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, L.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Paolillo, M.; Comincini, S.; Schinelli, S. In Vitro Glioblastoma Models: A Journey into the Third Dimension. Cancers 2021, 13, 2449. [Google Scholar] [CrossRef]
- Lenin, S.; Ponthier, E.; Scheer, K.; Yeo, E.; Tea, M.; Ebert, L.; Mansilla, M.O.; Poonnoose, S.; Baumgartner, U.; Day, B.; et al. A Drug Screening Pipeline Using 2D and 3D Patient-Derived In Vitro Models for Pre-Clinical Analysis of Therapy Response in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 4322. [Google Scholar] [CrossRef]
- Fernandez-Fuente, G.; Mollinedo, P.; Grande, L.; Vazquez-Barquero, A.; Fernandez-Luna, J.L. Culture Dimensionality Influences the Resistance of Glioblastoma Stem-like Cells to Multikinase Inhibitors. Mol. Cancer Ther. 2014, 13, 1664–1672. [Google Scholar] [CrossRef] [Green Version]
- Kaur, G.; Dufour, J.M. Cell lines. Spermatogenesis 2012, 2, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Gillet, J.-P.; Varma, S.; Gottesman, M.M. The Clinical Relevance of Cancer Cell Lines. J. Natl. Cancer Inst. 2013, 105, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Stringer, B.W.; Day, B.W.; D’Souza, R.; Jamieson, P.R.; Ensbey, K.S.; Bruce, Z.C.; Lim, Y.C.; Goasdoué, K.; Offenhäuser, C.; Akgul, S.; et al. A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci. Rep. 2019, 9, 4902. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Xu, K.; Zhu, X.; Dunphy, P.S.; Gudenas, B.; Lin, W.; Twarog, N.; Hover, L.D.; Kwon, C.-H.; Kasper, L.H.; et al. Patient-derived models recapitulate heterogeneity of molecular signatures and drug response in pediatric high-grade glioma. Nat. Commun. 2021, 12, 4089. [Google Scholar] [CrossRef]
- Patrizii, M.; Bartucci, M.; Pine, S.R.; Sabaawy, H.E. Utility of Glioblastoma Patient-Derived Orthotopic Xenografts in Drug Discovery and Personalized Therapy. Front. Oncol. 2018, 8, 23. [Google Scholar] [CrossRef]
- Golebiewska, A.; Hau, A.-C.; Oudin, A.; Stieber, D.; Yabo, Y.A.; Baus, V.; Barthelemy, V.; Klein, E.; Bougnaud, S.; Keunen, O.; et al. Patient-derived organoids and orthotopic xenografts of primary and recurrent gliomas represent relevant patient avatars for precision oncology. Acta Neuropathol. 2020, 140, 919–949. [Google Scholar] [CrossRef] [PubMed]
- Stanković, T.; Ranđelović, T.; Dragoj, M.; Burić, S.S.; Fernández, L.; Ochoa, I.; Pérez-García, V.M.; Pešić, M. In vitro biomimetic models for glioblastoma-a promising tool for drug response studies. Drug Resist. Updat. 2021, 55, 100753. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Yan, T.; Zhu, H.; Liang, K.X.; Leiss, L.; Sakariassen, P.; Skaftnesmo, K.O.; Huang, B.; Costea, D.E.; Enger, P.; et al. A co-culture model with brain tumor-specific bioluminescence demonstrates astrocyte-induced drug resistance in glioblastoma. J. Transl. Med. 2014, 12, 278. [Google Scholar] [CrossRef] [PubMed]
- Civita, P.; Leite, D.M.; Pilkington, G.J. Pre-Clinical Drug Testing in 2D and 3D Human In Vitro Models of Glioblastoma Incorporating Non-Neoplastic Astrocytes: Tunneling Nano Tubules and Mitochondrial Transfer Modulates Cell Behavior and Therapeutic Response. Int. J. Mol. Sci. 2019, 20, 6017. [Google Scholar] [CrossRef] [Green Version]
- Azambuja, J.H.; da Silveira, E.F.; de Carvalho, T.R.; Oliveira, P.S.; Pacheco, S.; Couto, C.T.D.; Beira, F.T.; Stefanello, F.M.; Spanevello, R.M.; Braganhol, E. Glioma sensitive or chemoresistant to temozolomide differentially modulate macrophage protumor activities. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 2652–2662. [Google Scholar] [CrossRef]
- Mahesparan, R.; Read, T.-A.; Lund-Johansen, M.; Skaftnesmo, K.; Bjerkvig, R.; Engebraaten, O. Expression of extracellular matrix components in a highly infiltrative in vivo glioma model. Acta Neuropathol. 2003, 105, 49–57. [Google Scholar] [CrossRef]
- Xia, S.; Lal, B.; Tung, B.; Wang, S.; Goodwin, C.R.; Laterra, J. Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro Oncol. 2016, 18, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Wang, S.; Zhang, R.; Sohrabi, A.; Yu, Q.; Liu, S.; Ehsanipour, A.; Liang, J.; Bierman, R.D.; Nathanson, D.A.; et al. Bioengineered scaffolds for 3D culture demonstrate extracellular matrix-mediated mechanisms of chemotherapy resistance in glioblastoma. Matrix Biol. 2019, 85–86, 128–146. [Google Scholar] [CrossRef]
- Wang, K.; Kievit, F.M.; Erickson, A.E.; Silber, J.R.; Ellenbogen, R.G.; Zhang, M. Culture on 3D Chitosan-Hyaluronic Acid Scaffolds Enhances Stem Cell Marker Expression and Drug Resistance in Human Glioblastoma Cancer Stem Cells. Adv. Heal. Mater. 2016, 5, 3173–3181. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef]
- Patwardhan, G.A.; Marczyk, M.; Wali, V.B.; Stern, D.F.; Pusztai, L.; Hatzis, C. Treatment scheduling effects on the evolution of drug resistance in heterogeneous cancer cell populations. Npj Breast Cancer 2021, 7, 60. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Saenz-Antoñanzas, A.; Auzmendi-Iriarte, J.; Carrasco-Garcia, E.; Moreno-Cugnon, L.; Ruiz, I.; Villanua, J.; Egaña, L.; Otaegui, D.; Samprón, N.; Matheu, A. Liquid Biopsy in Glioblastoma: Opportunities, Applications and Challenges. Cancers 2019, 11, 950. [Google Scholar] [CrossRef] [Green Version]
- Linhares, P.; Carvalho, B.; Vaz, R.; Costa, B.M. Glioblastoma: Is There Any Blood Biomarker with True Clinical Relevance? Int. J. Mol. Sci. 2020, 21, 5809. [Google Scholar] [CrossRef]
- Verheul, C.; Kleijn, A.; Lamfers, M.L.M. Cerebrospinal Fluid Biomarkers of Malignancies Located in The Central Nervous System. In Handbook of Clinical Neurology; Chapter 10; Deisenhammer, F., Teunissen, C.E., Tumani, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 139–169. [Google Scholar]
- Hishii, M.; Matsumoto, T.; Arai, H. Diagnosis and treatment of early-stage glioblastoma. Asian J. Neurosurg. 2019, 14, 589. [Google Scholar] [CrossRef]
- Lippert, T.H.; Ruoff, H.-J.; Volm, M. Current Status of Methods to Assess Cancer Drug Resistance. Int. J. Med. Sci. 2011, 8, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Howard, C.; Valluri, J.; Alberico, A.; Julien, T.; Mazagri, R.; Marsh, R.; Alastair, H.; Cortese, A.; Griswold, M.; Wang, W.; et al. Analysis of Chemopredictive Assay for Targeting Cancer Stem Cells in Glioblastoma Patients. Transl. Oncol. 2017, 10, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Stockslager, M.A.; Malinowski, S.; Touat, M.; Yoon, J.C.; Geduldig, J.; Mirza, M.; Kim, A.S.; Wen, P.Y.; Chow, K.-H.; Ligon, K.L.; et al. Functional drug susceptibility testing using single-cell mass predicts treatment outcome in patient-derived cancer neurosphere models. Cell Rep. 2021, 37, 109788. [Google Scholar] [CrossRef]
- Piccioni, D.E.; Achrol, A.S.; Kiedrowski, L.A.; Banks, K.; Boucher, N.; Barkhoudarian, G.; Kelly, D.F.; Juarez, T.; Lanman, R.B.; Raymond, V.M.; et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol. 2019, 8, CNS34. [Google Scholar] [CrossRef]
- Jain, K.K. A Critical Overview of Targeted Therapies for Glioblastoma. Front. Oncol. 2018, 8, 419. [Google Scholar] [CrossRef]
- Ghosh, D.; Nandi, S.; Bhattacharjee, S. Combination therapy to checkmate Glioblastoma: Clinical challenges and advances. Clin. Transl. Med. 2018, 7, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Target Kinase | Small Kinase Inhibitors | Reference |
|---|---|---|
| EGFR | Gefitinib, erlotinib, lapatinib, afatinib, dacomitinib, neratinib | [111] |
| PDGFR | Imatinib, tandutinib, lenvatinib, nintedanib, thyrophostin | [112] |
| MET | Crizotinib, volitinib, cabozantinib, altiratinib, SGX523, INCB28060, PLB-1001 | [69] |
| FGFR | Dovitinib, nintedanib, lenvatinib, brivanib, orantinib, ponatinib, E3810, ENMD-2076, AZD4547, BGJ398, LY2874455 | [73] |
| VEGFR | Imatinib, cediranib, pazopanib, sorafenib, sunitinib, vandetanib, vatalanib, AEE788, CT-322, XL184 | [113,114] |
| BRAF | Sorafenib, vemurafenib, dabrafenib, encorafenib | [115,116] |
| MEK | Combimetinib, trametinib, binimetinib | [116] |
| PI3K | Pictilisib, buparlisib, pilaralisib, sonolisib, dactolisib, voxtalisib, PQR309 | [117] |
| AKT | Perofisine, MK2206 | [118,119] |
| mTOR | Sirolimus, everolimus, temsirolimus, ridaforolimus, onatasertib, dactolisib, voxtalisib, PQR309, gedatolisib, sapanisertib | [117,118] |
| PKCβ | Enzastaurin | [97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabro, F.; Lamfers, M.L.M.; Leenstra, S. Advancements, Challenges, and Future Directions in Tackling Glioblastoma Resistance to Small Kinase Inhibitors. Cancers 2022, 14, 600. https://doi.org/10.3390/cancers14030600
Fabro F, Lamfers MLM, Leenstra S. Advancements, Challenges, and Future Directions in Tackling Glioblastoma Resistance to Small Kinase Inhibitors. Cancers. 2022; 14(3):600. https://doi.org/10.3390/cancers14030600
Chicago/Turabian StyleFabro, Federica, Martine L. M. Lamfers, and Sieger Leenstra. 2022. "Advancements, Challenges, and Future Directions in Tackling Glioblastoma Resistance to Small Kinase Inhibitors" Cancers 14, no. 3: 600. https://doi.org/10.3390/cancers14030600
APA StyleFabro, F., Lamfers, M. L. M., & Leenstra, S. (2022). Advancements, Challenges, and Future Directions in Tackling Glioblastoma Resistance to Small Kinase Inhibitors. Cancers, 14(3), 600. https://doi.org/10.3390/cancers14030600