
Nonmalignant Features Associated with Inherited Colorectal Cancer Syndromes-Clues for Diagnosis

 ,
,  , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Inherited Colorectal Cancer Predisposition Syndromes
2.1. APC-Associated Familial Polyposis (FAP)
2.2. AXIN2-Associated Oligodontia–Colorectal Cancer Syndrome
2.3. BMPR1A- and SMAD4-Associated Juvenile Polyposis
2.4. MCM8- and MCM9-Associated CRC
2.5. MMR-Associated Polyposis
2.6. MUTYH-Associated Polyposis
2.7. NTHL1-Associated Polyposis
2.8. POLE- and POLD1-Associated Polyposis
2.9. PTEN Hamartoma Tumor Syndrome
2.10. STK11-Associated Peutz–Jeghers Syndrome (PJS)
3. The Clues
3.1. Congenital Hypertrophy of the Retinal Pigmented Epithelium
3.2. Osteomas
3.3. Dental Abnormalities
3.4. Desmoid Tumors
3.5. Skin Lesions
3.6. Café au Lait Macules
4. Pathways
4.1. Wnt Signaling Pathway
4.2. mTOR Signaling Pathway
4.3. TGF-β Signaling Pathway
4.4. BER Signaling Pathway
4.5. Homologous Recombination Signaling Pathway
4.6. Mismatch Repair System
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Czene, K.; Lichtenstein, P.; Hemminki, K. Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish Family-Cancer Database. Int. J. Cancer 2002, 99, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.L.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer susceptibility gene mutations in individuals with colorectal cancer. J. Clin. Oncol. 2017, 35, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Mork, M.E.; You, Y.N.; Ying, J.; Bannon, S.A.; Lynch, P.M.; Rodriguez-Bigas, M.A.; Vilar, E. High prevalence of hereditary cancer syndromes in adolescents and young adults with colorectal cancer. J. Clin. Oncol. 2015, 33, 3544–3549. [Google Scholar] [CrossRef] [Green Version]
- De Voer, R.M.; Diets, I.J.; van der Post, R.S.; Weren, R.D.A.; Kamping, E.J.; de Bitter, T.J.J.; Elze, L.; Verhoeven, R.H.A.; Vink-Börger, E.; Eijkelenboom, A.; et al. Clinical, pathology, genetic, and molecular features of colorectal tumors in adolescents and adults 25 years or younger. Clin. Gastroenterol. Hepatol. 2021, 19, 1642–1651.e8. [Google Scholar] [CrossRef]
- Hes, F.J.; Nielsen, M.; Bik, E.C.; Konvalinka, D.; Wijnen, J.T.; Bakker, E.; Vasen, H.F.; Breuning, M.H.; Tops, C.M. Somatic APC mosaicism: An underestimated cause of polyposis coli. Gut 2008, 57, 71–76. [Google Scholar] [CrossRef]
- Elsayed, F.A.; Tops, C.M.J.; Nielsen, M.; Morreau, H.; Hes, F.J.; van Wezel, T. Use of sanger and next-generation sequencing to screen for mosaic and intronic APC variants in unexplained colorectal polyposis patients. Fam. Cancer 2021, 1–5. [Google Scholar] [CrossRef]
- Lucia Jansen, A.M.; Goel, A. Mosaicism in patients with colorectal cancer or polyposis syndromes: A systematic review. Clin. Gastroenterol. Hepatol. 2020, 18, 1949–1960. [Google Scholar] [CrossRef]
- Gammon, A.; Jasperson, K.; Pilarski, R.; Prior, T.; Kuwada, S. PTEN mosaicism with features of Cowden syndrome. Clin. Genet. 2013, 84, 593–595. [Google Scholar] [CrossRef]
- Stormorken, A.T.; Berg, T.; Norum, O.J.; Hølmebakk, T.; Aaberg, K.; Steigen, S.E.; Grindedal, E.M. APC mosaicism in a young woman with desmoid type fibromatosis and familial adenomatous polyposis. Fam. Cancer 2018, 17, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, K.; Etzler, J. Constitutional mismatch repair-deficiency syndrome: Have we so far seen only the tip of an iceberg? Hum. Genet. 2008, 124, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, L. Recent discoveries in the genetics of familial colorectal cancer and polyposis. Clin. Gastroenterol. Hepatol. 2017, 15, 809–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Weren, R.D.; Ligtenberg, M.J.; Kets, C.M.; de Voer, R.M.; Verwiel, E.T.; Spruijt, L.; van Zelst-Stams, W.A.; Jongmans, M.C.; Gilissen, C.; Hehir-Kwa, J.Y.; et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 2015, 47, 668–671. [Google Scholar] [CrossRef]
- Nieminen, T.T.; O’Donohue, M.F.; Wu, Y.; Lohi, H.; Scherer, S.W.; Paterson, A.D.; Ellonen, P.; Abdel-Rahman, W.M.; Valo, S.; Mecklin, J.P.; et al. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology 2014, 147, 595–598. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.H.N.; Lai, J.C.W.; Ho, S.L.; Leung, W.K.; Law, W.L.; Lee, J.F.Y.; Chan, A.K.W.; Tsui, W.Y.; Chan, A.S.Y.; Lee, B.C.H.; et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut 2017, 66, 1645–1656. [Google Scholar] [CrossRef] [Green Version]
- Golubicki, M.; Bonjoch, L.; Acuña-Ochoa, J.G.; Díaz-Gay, M.; Muñoz, J.; Cuatrecasas, M.; Ocaña, T.; Iseas, S.; Mendez, G.; Cisterna, D.; et al. Germline biallelic Mcm8 variants are associated with early-onset Lynch-like syndrome. JCI Insight 2020, 5, e140698. [Google Scholar] [CrossRef]
- Nielsen, M.; Aretz, S. Familial adenomatous polyposis. In Hereditary Colorectal Cancer; Valle, L., Gruber, S., Capellá, G., Eds.; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Nieuwenhuis, M.H.; Vasen, H.F. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): A review of the literature. Crit. Rev. Oncol. Hematol. 2007, 61, 153–161. [Google Scholar] [CrossRef]
- Burt, R.W.; Leppert, M.F.; Slattery, M.L.; Samowitz, W.S.; Spirio, L.N.; Kerber, R.A.; Kuwada, S.K.; Neklason, D.W.; Disario, J.A.; Lyon, E.; et al. Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology 2004, 127, 444–451. [Google Scholar] [CrossRef]
- Gardner, E.J. A genetic and clinical study of intestinal polyposis, a predisposing factor for carcinoma of the colon and rectum. Am. J. Hum. Genet. 1951, 3, 167–176. [Google Scholar] [PubMed]
- Koskenvuo, L.; Ristimäki, A.; Lepistö, A. Comparison of sporadic and FAP-associated desmoid-type fibromatoses. J. Surg. Oncol. 2017, 116, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Kattentidt Mouravieva, A.A.; Geurts-Giele, I.R.; de Krijger, R.R.; van Noesel, M.M.; van de Ven, C.P.; van den Ouweland, A.M.; Kromosoeto, J.N.; Dinjens, W.N.; Dubbink, H.J.; Smits, R.; et al. Identification of familial adenomatous polyposis carriers among children with desmoid tumours. Eur. J. Cancer 2012, 48, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.L.; Nero, C.; Pappo, A.; Lev, D.; Lazar, A.J.; López-Terrada, D. CTNNB1 genotyping and APC screening in pediatric desmoid tumors: A proposed algorithm. Pediatr. Dev. Pathol. 2012, 15, 361–367. [Google Scholar] [CrossRef]
- Cojocaru, E.; Gennatas, S.; Thway, K.; Fisher, C.; Smrke, A.; Strauss, D.; Hayes, A.; Smith, M.; Jones, R.L.; Benson, C.; et al. Approach to screening for familial adenomatous polyposis (FAP) in a cohort of 226 patients with desmoid-type fibromatosis (DF): Experience of a specialist center in the UK. Fam. Cancer 2021, 1–6. [Google Scholar] [CrossRef]
- Dahl, N.A.; Sheil, A.; Knapke, S.; Geller, J.I. Gardner fibroma: Clinical and histopathologic implications of germline APC mutation association. J. Pediatr. Hematol. Oncol. 2016, 38, e154–e157. [Google Scholar] [CrossRef]
- Santoro, C.; Giugliano, T.; Bifano, D.; D’Anna, C.; D’Onofrio, V.; Perrotta, S. Gardner fibroma: Clinical and histopathologic implications of germline APC mutation association. Clin. Case. Rep. 2017, 5, 1557–1560. [Google Scholar] [CrossRef]
- Friedl, W.; Aretz, S. Familial adenomatous polyposis: Experience from a study of 1164 unrelated german polyposis patients. Hered. Cancer Clin. Pract. 2005, 3, 95–114. [Google Scholar] [CrossRef] [Green Version]
- Signoroni, S.; Piozzi, G.N.; Collini, P.; Cocco, I.M.F.; Biasoni, D.; Chiaravalli, S.; Ricci, M.T.; Vitellaro, M. Gardner-associated fibroma of the neck: Role of a multidisciplinary evaluation for familial adenomatous polyposis diagnosis. Tumori 2021, 107, NP73–NP76. [Google Scholar] [CrossRef]
- Aretz, S.; Koch, A.; Uhlhaas, S.; Friedl, W.; Propping, P.; von Schweinitz, D.; Pietsch, T. Should children at risk for familial adenomatous polyposis be screened for hepatoblastoma and children with apparently sporadic hepatoblastoma be screened for APC germline mutations? Pediatr. Blood Cancer 2006, 47, 811–818. [Google Scholar] [CrossRef]
- Yang, A.; Sisson, R.; Gupta, A.; Tiao, G.; Geller, J.I. Germline APC mutations in hepatoblastoma. Pediatr. Blood Cancer 2018, 65, e26892. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.; Clark, S.; Hyer, W.; Hadzic, N.; Tomlinson, I.; Hinds, R. Germline APC mutations are not commonly seen in children with sporadic hepatoblastoma. J. Pediatr. Gastroenterol. Nutr. 2008, 47, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Coleman, P.; Barnard, N.A. Congenital hypertrophy of the retinal pigment epithelium: Prevalence and ocular features in the optometric population. Ophthalmic. Physiol. Opt. 2007, 27, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Nusliha, A.; Dalpatadu, U.; Amarasinghe, B.; Chandrasinghe, P.C.; Deen, K.I. Congenital hypertrophy of retinal pigment epithelium (CHRPE) in patients with familial adenomatous polyposis (FAP); a polyposis registry experience. BMC Res. Notes 2014, 7, 734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonoda, H.; Shimizu, T.; Miyake, T.; Ohta, H.; Akabori, H.; Tani, M. Severe mental retardation patient with a de novo chromosomal deletion 5q14-22 can be a carrier of a rectal and duodenal cancer associated with over 200 colorectal polyps: A case report. Int. Surg. 2021, 105, 322–326. [Google Scholar] [CrossRef] [Green Version]
- Kadiyska, T.; Tourtourikov, I.; Petrov, A.; Chavoushian, A.; Antalavicheva, M.; König, E.M.; Klopocki, E.; Vessela, N.; Stanislavov, R. Interstitial deletion of 5q22.2q23.1 including APC and TSSK1B in a patient with adenomatous polyposis and asthenoteratozoospermia. Mol. Syndromol. 2019, 9, 235–240. [Google Scholar] [CrossRef]
- Lindgren, V.; Bryke, C.R.; Ozcelik, T.; Yang-Feng, T.L.; Francke, U. Phenotypic, cytogenetic, and molecular studies of three patients with constitutional deletions of chromosome 5 in the region of the gene for familial adenomatous polyposis. Am. J. Hum. Genet. 1992, 50, 988–997. [Google Scholar]
- Ofner, L.; Raedle, J.; Windpassinger, C.; Schwarzbraun, T.; Kroisel, P.M.; Wagner, K.; Petek, E. Phenotypic and molecular characterisation of a de novo 5q deletion that includes the APC gene. J. Hum. Genet. 2006, 51, 141–146. [Google Scholar] [CrossRef]
- Rosenberg, M.M.; Yang, F.; Giovanni, M.; Mohn, J.L.; Temburni, M.K.; Jacob, M.H. Adenomatous polyposis coli plays a key role, in vivo, in coordinating assembly of the neuronal nicotinic postsynaptic complex. Mol. Cell. Neurosci. 2008, 38, 138–152. [Google Scholar] [CrossRef] [Green Version]
- Mohn, J.L.; Alexander, J.; Pirone, A.; Palka, C.D.; Lee, S.Y.; Mebane, L.; Haydon, P.G.; Jacob, M.H. Adenomatous polyposis coli protein deletion leads to cognitive and autism-like disabilities. Mol. Psychiatry 2014, 19, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Correa, M.R.; Sala, A.C.; Cintrón, B.; Hernández, J.; Olivera, M.; Cora, A.; Moore, C.M.; Luciano, C.A.; Soto-Salgado, M.; Giardiello, F.M.; et al. Ubiquitous neurocognitive dysfunction in familial adenomatous polyposis: Proof-of-concept of the role of APC protein in neurocognitive function. Hered. Cancer Clin. Pract. 2020, 18, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azofra, A.S.; Kidambi, T.D.; Jeremy, R.J.; Conrad, P.; Blanco, A.; Myers, M.; Barkovich, J.; Terdiman, J.P. Differences in neuropsychological and behavioral parameters and brain structure in patients with familial adenomatous polyposis: A sibling-paired study. Hered. Cancer Clin. Pract. 2016, 14, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammi, L.; Arte, S.; Somer, M.; Jarvinen, H.; Lahermo, P.; Thesleff, I.; Pirinen, S.; Nieminen, P. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am. J. Hum. Genet. 2004, 74, 1043–1050. [Google Scholar] [CrossRef] [Green Version]
- Jensen, J.M.; Skakkebæk, A.; Gaustadness, M.; Sommerlund, M.; Gjørup, H.; Ljungmann, K.; Lautrup, C.K.; Sunde, L. Familial colorectal cancer and tooth agenesis caused by an AXIN2 variant: How do we detect families with rare cancer predisposition syndromes? Fam. Cancer 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rivera, B.; Perea, J.; Sánchez, E.; Villapún, M.; Sánchez-Tomé, E.; Mercadillo, F.; Robledo, M.; Benítez, J.; Urioste, M. A novel AXIN2 germline variant associated with attenuated FAP without signs of oligon- dontia or ectodermal dysplasia. Eur. J. Hum. Genet. 2014, 22, 423–426. [Google Scholar] [CrossRef]
- Marvin, M.L.; Mazzoni, S.M.; Herron, C.M.; Edwards, S.; Gruber, S.B.; Petty, E.M. AXIN2-associated autosomal dominant ecto-dermal dysplasia and neoplastic syndrome. Am. J. Med. Genet. A 2011, 155A, 898–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.; Liu, H.; Bai, B.; Chang, H.; Zhao, H.; Wang, Y.; Han, D.; Feng, H. Novel missense mutations in the AXIN2 gene associated with non-syndromic oligodontia. Arch. Oral Biol. 2014, 59, 349–353. [Google Scholar] [CrossRef]
- Hlouskova, A.; Bielik, P.; Bonczek, O.; Balcar, V.J.; Šerý, O. Mutations in AXIN2 gene as a risk factor for tooth agenesis and cancer: A review. Neuro Endocrinol. Lett. 2017, 38, 131–137. [Google Scholar]
- Aytac, E.; Sulu, B.; Heald, B.; O’Malley, M.; LaGuardia, L.; Remzi, F.H.; Kalady, M.F.; Burke, C.A.; Church, J.M. Genotype-defined cancer risk in juvenile polyposis syndrome. Br. J. Surg. 2015, 102, 114–118. [Google Scholar] [CrossRef]
- Wain, K.E.; Ellingson, M.S.; McDonald, J.; Gammon, A.; Roberts, M.; Pichurin, P.; Winship, I.; Riegert-Johnson, D.L.; Weitzel, J.N.; Lindor, N.M. Appreciating the broad clinical features of SMAD4 mutation carriers: A multicenter chart review. Genet. Med. 2014, 16, 588–593. [Google Scholar] [CrossRef] [Green Version]
- McDonald, J.; Bayrak-Toydemir, P.; Pyeritz, R.E. Hereditary hemorrhagic telangiectasia: An overview of diagnosis, management, and pathogenesis. Genet. Med. 2011, 13, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heald, B.; Rigelsky, C.; Moran, R.; LaGuardia, L.; O’Malley, M.; Burke, C.A.; Zahka, K. Prevalence of thoracic aortopathy in patients with juvenile Polyposis Syndrome-Hereditary Hemorrhagic Telangiectasia due to SMAD4. Am. J. Med. Genet. A 2015, 167A, 1758–1762. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Y.; Halpern, N.; Hubert, A.; Adler, S.N.; Cohen, S.; Plesser-Duvdevani, M.; Pappo, O.; Shaag, A.; Meiner, V. Mutated MCM9 is associated with predisposition to hereditary mixed polyposis and colorectal cancer in addition to primary ovarian failure. Cancer Genet. 2015, 208, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Y.; Aleme, O.; Peled-Perets, L.; Castellvi-Bel, S.; Nielsen, M.; Shalev, S.A. MCM9 is associated with germline predisposition to early-onset cancer-clinical evidence. NPJ Genom. Med. 2021, 23, 78. [Google Scholar] [CrossRef] [PubMed]
- Win, A.K.; Jenkins, M.A.; Dowty, J.G.; Antoniou, A.C.; Lee, A.; Giles, G.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Ahnen, D.J.; et al. Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiol. Biomarkers Prev. 2017, 26, 404–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessup, C.J.; Redston, M.; Tilton, E.; Reimann, J.D. Importance of universal mismatch repair protein immunohistochemistry in patients with sebaceous neoplasia as an initial screening tool for Muir-Torre syndrome. Hum. Pathol. 2016, 4, 1–9. [Google Scholar] [CrossRef]
- Akhtar, S.; Oza, K.K.; Khan, S.A.; Wright, J. MuirTorre syndrome: Case report of a patient with concurrent jejunal and ureteral cancer and a review of the literature. J. Am. Acad. Dermatol. 1999, 41 Pt 1, 681–686. [Google Scholar] [CrossRef]
- Roberts, M.E.; Riegert-Johnson, D.L.; Thomas, B.C.; Rumilla, K.M.; Thomas, C.S.; Heckman, M.G.; Purcell, J.U.; Hanson, N.B.; Leppig, K.A.; Lim, J.; et al. A clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir-Torre variant of Lynch syndrome. Genet. Med. 2014, 16, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Levi, Z.; Kariv, R.; Barnes-Kedar, I.; Goldberg, Y.; Half, E.; Morgentern, S.; Eli, B.; Baris, H.N.; Vilkin, A.; Belfer, R.G.; et al. The gastrointestinal manifestation of constitutional mismatch repair deficiency syndrome: From a single adenoma to polyposis-like phenotype and early onset cancer. Clin. Genet. 2015, 88, 474–478. [Google Scholar] [CrossRef]
- Suerink, M.; Ripperger, T.; Messiaen, L.; Menko, F.H.; Bourdeaut, F.; Colas, C.; Jongmans, M.; Goldberg, Y.; Nielsen, M.; Muleris, M.; et al. Constitutional mismatch repair deficiency as a differential diagnosis of neurofibromatosis type 1: Consensus guidelines for testing a child without malignancy. J. Med. Genet. 2019, 56, 53–62. [Google Scholar] [CrossRef]
- Toledano, H.; Orenstein, N.; Sofrin, E.; Ruhrman-Shahar, N.; Amarilyo, G.; Basel-Salmon, L.; Shuldiner, A.R.; Smirin-Yosef, P.; Aronson, M.; Al-Tarrah, H.; et al. Paediatric systemic lupus erythematosus as a manifestation of constitutional mismatch repair deficiency. J. Med. Genet. 2020, 57, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Al-Tassan, N.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.K.; Davies, D.R.; David, S.S.; Sampson, J.R.; et al. Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat. Genet. 2002, 30, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Mao de Ferro, S.; Suspiro, A.; Fildago, P. Aggressive phenotype of MYH associated polyposis with jejunal cancer and intra-abdominal desmoid tumor: Report of a case. Dis. Colon. Rectum. 2009, 52, 742–745. [Google Scholar] [CrossRef] [PubMed]
- Vogt, S.; Jones, N.; Christian, D.; Engel, C.; Nielsen, M.; Kaufmann, A.; Steinke, V.; Vasen, H.F.; Propping, P.; Sampson, J.R.; et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 2009, 137, 1976–1985.e10. [Google Scholar]
- Sieber, O.M.; Lipton, L.; Crabtree, M.; Heinimann, K.; Fidalgo, P.; Phillips, R.K.; Bisgaard, M.L.; Orntoft, T.F.; Aaltonen, L.A.; Hodgson, S.V.; et al. Multiple colorectal adenomas, classical adenomatous polyposis and germ-line mutations in MYH. N. Engl. J. Med. 2003, 348, 791–799. [Google Scholar] [CrossRef]
- Grolleman, J.E.; de Voer, R.M.; Elsayed, F.A.; Nielsen, M.; Weren, R.D.A.; Palles, C.; Ligtenberg, M.J.L.; Vos, J.R.; Ten Broeke, S.W.; de Miranda, N.F.C.C.; et al. Mutational signature analysis reveals NTHL1 deficiency to cause a multi-tumor phenotype. Cancer Cell 2019, 35, 256–266.e5. [Google Scholar] [CrossRef] [Green Version]
- Spier, I.; Holzapfel, S.; Altmüller, J.; Zhao, B.; Horpaopan, S.; Vogt, S.; Chen, S.; Morak, M.; Raeder, S.; Kayser, K.; et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int. J. Cancer 2015, 137, 320–331. [Google Scholar] [CrossRef]
- Yehia, L.; Eng, C. PTEN hamartoma tumor syndrome. 2001 Nov 29 [Updated 2021 Feb 11]. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2021. [Google Scholar]
- Pilarski, R. PTEN hamartoma tumor syndrome: A clinical overview. Cancers 2019, 11, 844. [Google Scholar] [CrossRef] [Green Version]
- Mester, J.; Eng, C. Estimate of de novo mutation frequency in probands with PTEN hamartoma tumor syndrome. Genet. Med. 2012, 14, 819–822. [Google Scholar] [CrossRef] [Green Version]
- Pilarski, R.; Stephens, J.A.; Noss, R.; Fisher, J.L.; Prior, T.W. Predicting PTEN mutations: An evaluation of Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome clinical features. J. Med. Genet. 2011, 48, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.H.; Mester, J.; Peterson, C.; Yang, Y.; Chen, J.L.; Rybicki, L.A.; Milas, K.; Pederson, H.; Remzi, B.; Orloff, M.S.; et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am. J. Hum. Genet. 2011, 88, 42–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drissen, M.M.C.M.; Schieving, J.H.; Schuurs-Hoeijmakers, J.H.M.; Vos, J.R.; Hoogerbrugge, N. Red flags for early recognition of adult patients with PTEN hamartoma tumour syndrome. Eur. J. Med. Genet. 2021, 64, 104364. [Google Scholar] [CrossRef] [PubMed]
- Innella, G.; Miccoli, S.; Colussi, D.; Pradella, L.M.; Amato, L.B.; Zuntini, R.; Salfi, N.C.M.; Collina, G.; Ferrara, F.; Ricciardiello, L.; et al. Colorectal polyposis as a clue to the diagnosis of Cowden syndrome: Report of two cases and literature review. Pathol. Res. Pract. 2021, 218, 153339. [Google Scholar] [CrossRef] [PubMed]
- Innella, G.; Bonora, E.; Neri, I.; Virdi, A.; Guglielmo, A.; Pradella, L.M.; Ceccarelli, C.; Amato, L.B.; Lanzoni, A.; Miccoli, S.; et al. PTEN hamartoma tumor syndrome: Skin manifestations and insights into their molecular pathogenesis. Front. Med. 2021, 8, 688105. [Google Scholar] [CrossRef]
- Robinson, S.; Cohen, A.R. Cowden disease and Lhermitte-Duclos disease: An update. Case report and review of the literature. Neurosurg. Focus. 2006, 20, E6. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, S.; Cheng, J.; Liu, W.; Hui, X. Lhermitte-Duclos disease: Clinical study with long-term follow-up in a single institution. Clin. Neurol. Neurosurg. 2017, 162, 53–58. [Google Scholar] [CrossRef]
- Heald, B.; Mester, J.; Rybicki, L.; Orloff, M.S.; Burke, C.A.; Eng, C. Frequent gastrointestinal polyps and colorectal adenocarcinomas in a prospective series of PTEN mutation carriers. Gastroenterology 2010, 139, 1927–1933. [Google Scholar] [CrossRef] [Green Version]
- Sweet, K.; Willis, J.; Zhou, X.P.; Gallione, C.; Sawada, T.; Alhopuro, P.; Khoo, S.K.; Patocs, A.; Martin, C.; Bridgeman, S.; et al. Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis. JAMA 2005, 294, 2465–2473. [Google Scholar] [CrossRef] [Green Version]
- Hearle, N.; Schumacher, V.; Menko, F.; Olschwang, S.; Boardman, L.A.; Gille, J.J.; Keller, J.J.; Westerman, A.M.; Scott, R.J.; Lim, W.; et al. Frequency and spectrum of cancers in the Peutz–Jeghers syndrome. Clin. Cancer Res. 2006, 12, 3209–3215. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, I.; Houlston, R. Peutz Jeghers syndrome. J. Med. Genet. 1997, 34, 1007–1011. [Google Scholar] [CrossRef] [Green Version]
- Van Lier, M.G.; Wagner, A.; Mathus-Vliegen, E.M.; Kuipers, E.J.; Steyerberg, E.W.; van Leerdam, M.E. High cancer risk in Peutz-Jeghers syndrome: A systematic review and surveillance recommendations. Am. J. Gastroenterol. 2010, 105, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Schriebman, I.; Baker, M.; Amos, C.; Mcgarrity, T. The hamartomatous polyposis syndromes: A clinical and molecular review. Am. J. Gastroenterol. 2005, 100, 476–490. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Phillips, K.D.; Grist, S.; Bennet, G.; Craig, J.E.; Muecke, J.S.; Suthers, G.K. Congenital hypertrophy of the retinal pigment epithelium (CHRPE) in familial colorectal cancer. Fam. Cancer 2006, 5, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Jung, S.H.; Yoon, T.M.; Lee, J.K.; Joo, Y.E.; Lim, S.C. Characteristics of paranasal sinus osteoma and treatment outcomes. Acta. Otolaryngol. 2015, 135, 602–607. [Google Scholar] [CrossRef]
- Butler, J.; Healy, C.; Toner, M.; Flint, S. Gardner’s syndrome—Review and report of a case. Oral. Oncol. Extr. 2005, 41, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Fernández Montenegro, P.; Valmaseda Castellón, E.; Berini Aytés, L.; Gay Escoda, C. Retrospective study of 145 supernumerary teeth. Med. Oral. Patol. Oral. Cir. Bucal. 2006, 11, E339–E344. [Google Scholar]
- Ye, X.; Attaie, A.B. Genetic basis of nonsyndromic and syndromic tooth agenesis. J. Pediatr. Genet. 2016, 5, 198–208. [Google Scholar] [CrossRef] [Green Version]
- De Coster, P.J.; Marks, L.A.; Martens, L.C.; Huysseune, A. Dental agenesis: Genetic and clinical perspectives. J. Oral. Pathol. Med. 2009, 38, 1–17. [Google Scholar]
- Ritwik, P.; Patterson, K.K. Diagnosis of tooth agenesis in childhood and risk for neoplasms in adulthood. Ochsner. J. 2018, 18, 345–350. [Google Scholar] [CrossRef]
- Reitamo, J.J.; Häyry, P.; Nykyri, E.; Saxén, E. The desmoid tumor. I. Incidence, sex-, age- and anatomical distribution in the Finnish population. Am. J. Clin. Pathol. 1982, 77, 665–673. [Google Scholar] [CrossRef]
- Koskenvuo, L.; Peltomaki, P.; Renkonen-Sinisalo, L.; Gylling, A.; Nieminen, T.T.; Ristimaki, A.; Lepisto, A. Desmoid tumor patients carry an elevated risk of familial adenomatous polyposis. J. Surg. Oncol. 2016, 113, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Ophir, G.; Sivan, S.; Hana, S.; Guy, R.; Nathan, G.; Naomi, F.I.; Joseph, K.; Ido, W.; Ofer, M.; Yael, G.; et al. Abdominal desmoid: Course, severe outcomes, and unique genetic background in a large local series. Cancers 2021, 13, 3673. [Google Scholar] [CrossRef] [PubMed]
- McLean, D.I.; Gallagher, R.P. “Sunburn” freckles, cafe-au-lait macules, and other pigmented lesions of schoolchildren: The Vancouver mole study. J. Am. Acad. Dermatol. 1995, 32, 565–570. [Google Scholar] [CrossRef]
- Bauer, A.J.; Stratakis, C.A. The lentiginoses: Cutaneous markers of systemic disease and a window to new aspects of tumourigenesis. J. Med. Genet. 2005, 42, 801–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alper, J.C.; Holmes, L.B. The incidence and significance of birthmarks in a cohort of 4,641 newborns. Pediatr. Dermatol. 1983, 1, 58–68. [Google Scholar] [CrossRef]
- Whitehouse, D. Diagnostic value of the cafe-au-lait spot in children. Arch. Dis. Child. 1966, 41, 316–319. [Google Scholar] [CrossRef] [Green Version]
- Shah, K.N. The diagnostic and clinical significance of café-au-lait macules. Pediatr. Clin. N. Am. 2010, 57, 1131–1153. [Google Scholar] [CrossRef]
- Cohen, B.A. Pediatric Dermatology, 4th ed.; Elsevier: Maryland, MD, USA, 2013. [Google Scholar]
- Friedman, J.M. Neurofibromatosis 1. 1998 Oct 2 [updated 2019 Jun 6]. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Messiaen, L.; Yao, S.; Brems, H.; Callens, T.; Sathienkijkanchai, A.; Denayer, E.; Spencer, E.; Arn, P.; Babovic-Vuksanovic, D.; Bay, C.; et al. Clinical and mutational spectrum of neurofibromatosis type 1-like syndrome. JAMA 2009, 302, 2111–2118. [Google Scholar] [CrossRef]
- Santos, A.C.; Heck, B.; Camargo, B.D.; Vargas, F.R. Prevalence of Café-au-Lait Spots in children with solid tumors. Genet. Mol. Biol. 2016, 39, 232–238. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, K.; Rosenbaum, T.; Messiaen, L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type 1. Clin. Genet. 2017, 91, 507–519. [Google Scholar] [CrossRef]
- Wimmer, K.; Beilken, A.; Nustede, R.; Ripperger, T.; Lamottke, B.; Ure, B.; Steinmann, D.; Reineke-Plaass, T.; Lehmann, U.; Zschocke, J. A novel germline POLE mutation causes. Fam. Cancer 2017, 16, 67–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, T.R.; Scatena, L.S.; Hoffenberg, E.J.; Gralla, J.; Lee, L.A. Café au lait macules and juvénile polyps. Pediatr. Dermatol. 2007, 24, E17–E21. [Google Scholar] [CrossRef] [PubMed]
- Phesse, T.; Flanagan, D.; Vincan, E. Frizzled7: A promising Achilles’ heel for targeting the Wnt receptor complex to treat cancer. Cancers 2016, 8, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groden, J.; Thliveris, A.; Samowitz, W.; Carlson, M.; Gelbert, L.; Albertsen, H.; Joslyn, G.; Stevens, J.; Spirio, L.; Robertson, M.; et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991, 66, 589–600. [Google Scholar] [CrossRef]
- Seidensticker, M.J.; Behrens, J. Biochemical interactions in the wnt pathway. Biochim. Biophys. Acta 2000, 1495, 168–182. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.M. Epithelial phenotype and the RPE: Is the answer blowing in the Wnt? Prog. Retin. Eye Res. 2008, 27, 579–595. [Google Scholar] [CrossRef] [Green Version]
- Gómez García, E.B.; Knoers, N.V. Gardner’s syndrome (familial adenomatous polyposis): A cilia-related disorder. Lancet Oncol. 2009, 10, 727–735. [Google Scholar] [CrossRef]
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733. [Google Scholar] [CrossRef] [Green Version]
- Qian, C.N.; Knol, J.; Igarashi, P.; Lin, F.; Zylstra, U.; The, B.T.; Williams, B.O. Cystic renal neoplasia following conditional inactivation of apc in mouse renal tubular epithelium. J. Biol. Chem. 2005, 280, 3938–3945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, Y.; Hayashi, S.; Levine, A.; Wieschaus, E. Regulation of armadillo by a Drosophila APC inhibits neuronal apoptosis during retinal development. Cell 1998, 93, 1171–1182. [Google Scholar] [CrossRef] [Green Version]
- Jenne, D.E.; Reimann, H.; Nezu, J.; Friedel, W.; Loff, S.; Jeschke, R.; Müller, O.; Back, W.; Zimmer, M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat. Genet. 1998, 18, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Hezel, A.F.; Bardeesy, N. LKB1; linking cell structure and tumor suppression. Oncogene 2008, 27, 6908–6919. [Google Scholar] [CrossRef] [Green Version]
- Tee, A.R.; Blenis, J. mTOR, translational control and human disease. Semin. Cell Dev. Biol. 2005, 16, 29–37. [Google Scholar] [CrossRef]
- Howe, J.R.; Roth, S.; Ringold, J.C.; Summers, R.W.; Järvinen, H.J.; Sistonen, P.; Tomlinson, I.P.; Houlston, R.S.; Bevan, S.; Mitros, F.A.; et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998, 280, 1086–1088. [Google Scholar] [CrossRef]
- Howe, J.R.; Bair, J.L.; Sayed, M.G.; Anderson, M.E.; Mitros, F.A.; Petersen, G.M.; Velculescu, V.E.; Traverso, G.; Vogelstein, B. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat. Genet. 2001, 28, 184–187. [Google Scholar] [CrossRef]
- Jung, B.; Staudacher, J.J.; Beauchamp, D. Transforming growth factor β superfamily signaling in development of colorectal cancer. Gastroenterology 2017, 152, 36–52. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, B.; Markkanen, E.; Hübscher, U. Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair 2010, 9, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Viel, A.; Bruselles, A.; Meccia, E.; Fornasarig, M.; Quaia, M.; Canzonieri, V.; Policicchio, E.; Urso, E.D.; Agostini, M.; Genuardi, M.; et al. A specific mutational signature associated with DNA 8-oxoguanine persistence in MUTYH-defective colorectal cancer. EBioMedicine 2017, 20, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, M.T.; Chaung, W.; Marenstein, D.R.; Chan, M.K.; Altamirano, A.; Basu, A.K.; Boorstein, R.J.; Cunningham, R.P.; Teebor, G.W. Targeted deletion of mNth1 reveals a novel DNA repair enzyme activity. Mol. Cell. Biol. 2002, 22, 6111–6121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood-Trageser, M.A.; Gurbuz, F.; Yatsenko, S.A.; Jeffries, E.P.; Kotan, L.D.; Surti, U.; Ketterer, D.M.; Matic, J.; Chipkin, J.; Jiang, H.; et al. MCM9 mutations are associated with ovarian failure, short stature, and chromosomal instability. Am. J. Hum. Genet. 2014, 95, 754–762. [Google Scholar] [CrossRef] [Green Version]
- AlAsiri, S.; Basit, S.; Wood-Trageser, M.A.; Yatsenko, S.A.; Jeffries, E.P.; Surti, U.; Ketterer, D.M.; Afzal, S.; Ramzan, K.; Faiyaz-Ul Haque, M.; et al. Exome sequencing reveals MCM8 mutation underlies ovarian failure and chromosomal instability. J. Clin. Investig. 2015, 125, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, K.; Ishiai, M.; Horikawa, K.; Fukagawa, T.; Takata, M.; Takisawa, H.; Kanemaki, M.T. Mcm8 and Mcm9 form a complex that functions in homologous recombination repair induced by DNA interstrand crosslinks. Mol. Cell. 2012, 47, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Peltomäki, P. Update on Lynch syndrome genomics. Fam. Cancer 2016, 15, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Clinical diagnosis | Raises clinical suspicions Influences decision to refer patients for genetic testing Makes early intervention possible when necessary. Prompts surveillance of asymptomatic at-risk patients. |
| Test choice | Single gene or multi-gene panel CNV testing Search for mosaicism |
| Test interpretation | VUS or likely pathogenic Mosaicism Family segregation |
| Variant class interpretation | POLE-Missense (PPAP) SMAD4–LOF (JPS-HHT) |
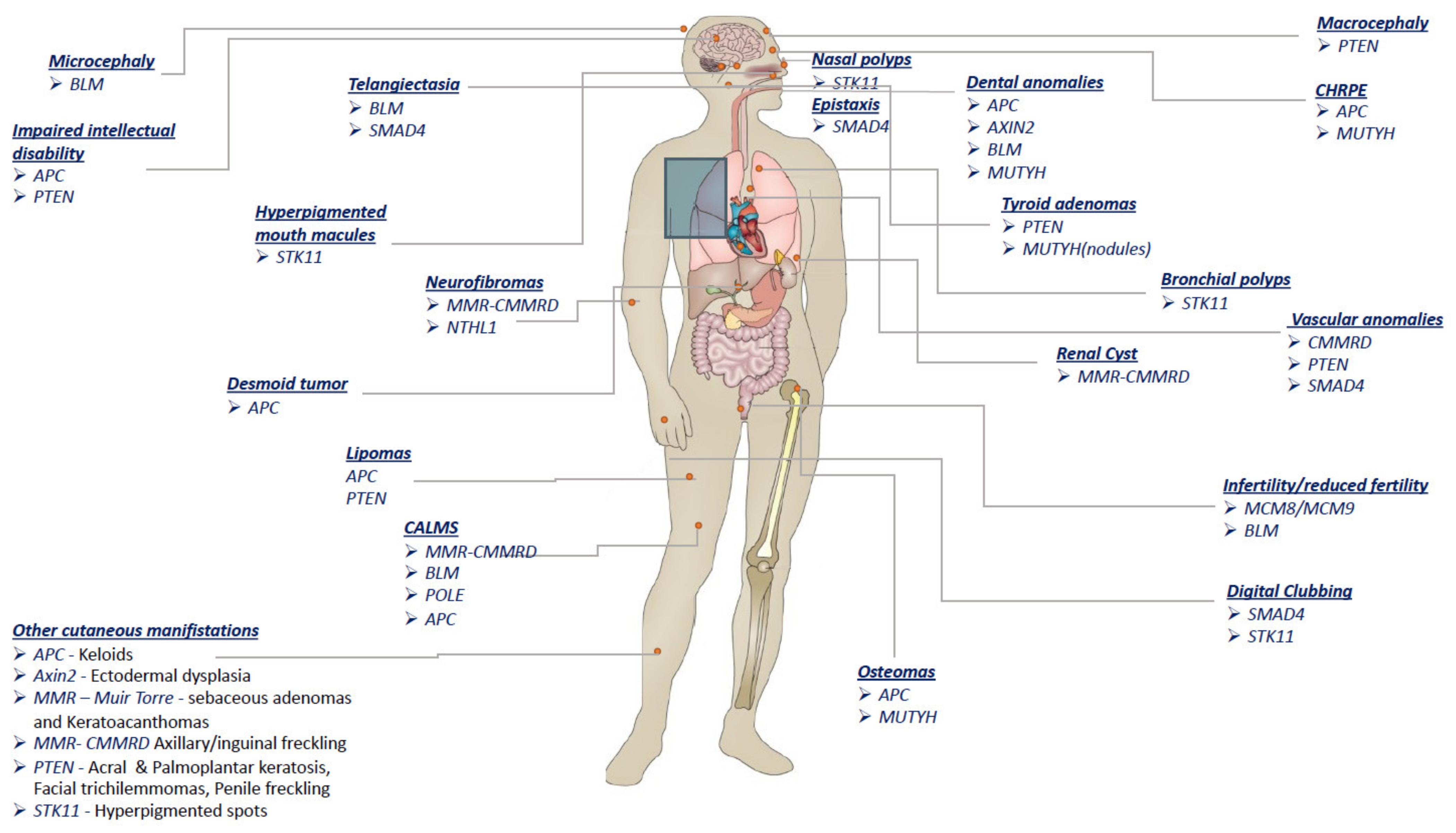
| Gene | Syndrome | Inheritance | Non-Malignant Features |
|---|---|---|---|
| APC | FAP | AD Inherited (75–80%) De novo (15–20%) | Eyes–CHRPE (>90%) often multiple bilateral Teeth—Dental anomalies: supernumerary teeth, unerupted teeth, dental caries, or odontomas (17%) Skull—osteomas, especially involving the mandibular angle Limbs—endosteal and exosteal osteomas (20%) Skin—Epidermoid inclusion cysts; fibromas; lipomas; lipofibromas, keloids; desmoid tumors (10–25%) Endocrine system—adrenal masses (7–13%) Central nervous system—mildly impaired intellectual abilities. |
| AFAP | AD | Eyes—CHRPE (rare) Skin—desmoid tumors (rare) | |
| 5q- | AD Mostly De novo | Central nervous system—mental retardation Face—dysmorphic features Reproductive system—asthenozoospermia (absence of TSSK1B) | |
| AXIN2 | ODCRCS | AD | Teeth—tooth agenesis (oligodontia) Skin—ectodermal dysplasia; sparse hair and eyebrows |
| BMPR1A | JPS | AD Inherited (33%) De novo (67%) | Not reported |
| BLM | BLOOM | AR | Growth retardation and growth failure Head—microcephaly Face—malar hypoplasia; prominent ears; prominent nose Teeth—absent upper lateral incisors Reproductive system—azoospermia; reduced fertility in females Skin—facial butterfly telangiectasia; spotty hypopigmentation; CALMs; photosensitivity Immune system—immune abnormalities |
| GREM1 | HMPS | AD | Not reported |
| MCM8 MCM9 | AR | Reproductive system—primary ovarian insufficiency/azoospermia | |
| MMR genes MLH1, MSH2, MSH6, PMS2 | LYNCH | AD | Skin—sebaceous adenomas and keratoacanthomas (Muir–Torre) |
| MMR genes | CMMRD | AR | Skin—CALMs (62–97%); neurofibromas; axillary/inguinal freckling (10%) Central nervous system—agenesis of the corpus callosum; gray matter heterotopia; intracerebral cyst; interhemispheric cyst Vascular system—developmental vascular abnormalities Kidneys—renal cysts Autoimmune system—pediatric SLE |
| MSH3 | FAP4 | AR | Not reported |
| MUTYH | MAP | AR | Endocrine system—thyroid nodules; benign adrenal lesions (18%) Eyes—CHRPE (5.5%) Limbs—osteomas Teeth—dental anomalies; jaw bone cysts |
| NTHL1 | NAP | AR | Skin—hemangiomas; neurofibromas Genitourinary system—ovarian cysts Liver—liver cysts |
| POLE | PPAP | AD | Skin—CALMs; |
| POLD1 | PPAP | AD | Not reported |
| PTEN | PHTS | AD De novo (44%) | Head—macrocephaly Mouth—oral papillomas Vascular system—vascular anomalies (50% of patients); intracranial developmental venous and arteriovenous malformations Genitourinary system—genitourinary malformation; uterine fibroids Skin—multiple facial papules; acral keratosis; palmoplantar keratosis; facial trichilemmomas; lipomas; fibromas; penile freckling Central nervous system—Cerebellar gangliocytoma manifesting as seizure and tremor (Lhermitte-Duclos disease; 6–15%); mental retardation (12%); autism Endocrine system—goiter; thyroid adenomas |
| RNF43 | SSPCS | AD | Not reported |
| SMAD4 | HHT | AD | Vascular system—AVMs (76%) Hands—digital clubbing (50%) Skin—telangiectasia (57%) Nose—epistaxis (61–71%) |
| STK11 | PJS | AD | Nose—nasal polyps Mouth—hyperpigmented macules of lips and buccal mucosa (65%) Respiratory system—bronchial polyps Genitourinary system—ovarian cysts; gynecomastia with sertoli cell tumors Hands—clubbing of fingers Skin—dark blue to dark brown melanocytic macules (which fade with age); hyperpigmented spots on hands/digits (especially palms; 73%), arms, feet (especially plantar areas), legs, or lips |
| TP53 | LI-FRAUMENI | AD | Not reported |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haimov, D.; Lieberman, S.; Castellvi-Bel, S.; Nielsen, M.; Goldberg, Y. Nonmalignant Features Associated with Inherited Colorectal Cancer Syndromes-Clues for Diagnosis. Cancers 2022, 14, 628. https://doi.org/10.3390/cancers14030628
Haimov D, Lieberman S, Castellvi-Bel S, Nielsen M, Goldberg Y. Nonmalignant Features Associated with Inherited Colorectal Cancer Syndromes-Clues for Diagnosis. Cancers. 2022; 14(3):628. https://doi.org/10.3390/cancers14030628
Chicago/Turabian StyleHaimov, Diana, Sari Lieberman, Sergi Castellvi-Bel, Maartje Nielsen, and Yael Goldberg. 2022. "Nonmalignant Features Associated with Inherited Colorectal Cancer Syndromes-Clues for Diagnosis" Cancers 14, no. 3: 628. https://doi.org/10.3390/cancers14030628
APA StyleHaimov, D., Lieberman, S., Castellvi-Bel, S., Nielsen, M., & Goldberg, Y. (2022). Nonmalignant Features Associated with Inherited Colorectal Cancer Syndromes-Clues for Diagnosis. Cancers, 14(3), 628. https://doi.org/10.3390/cancers14030628