
Glioblastoma Cells Counteract PARP Inhibition through Pro-Survival Induction of Lipid Droplets Synthesis and Utilization

 , , , , ,
, , , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Western Blot Analysis
2.3. Transfection of Small Interfering (si) RNA (siRNA)
2.4. Transfection of mCherry-GFP-LC3
2.5. Autophagy Flux Assay
2.6. RT-qPCR
2.7. GICs Immunofluorescence
2.8. LAMP1-Bodipy Immunofluorescence
2.9. Sudan-Red Staining
2.10. ATP Determination
2.11. Short-Term Proliferation Assay: MTT
2.12. SubG1 Analysis: Propidium Iodide
2.13. Seahorse Respirometry
2.14. Transmission Electron Microscopy
2.15. Statistical Analysis
3. Results
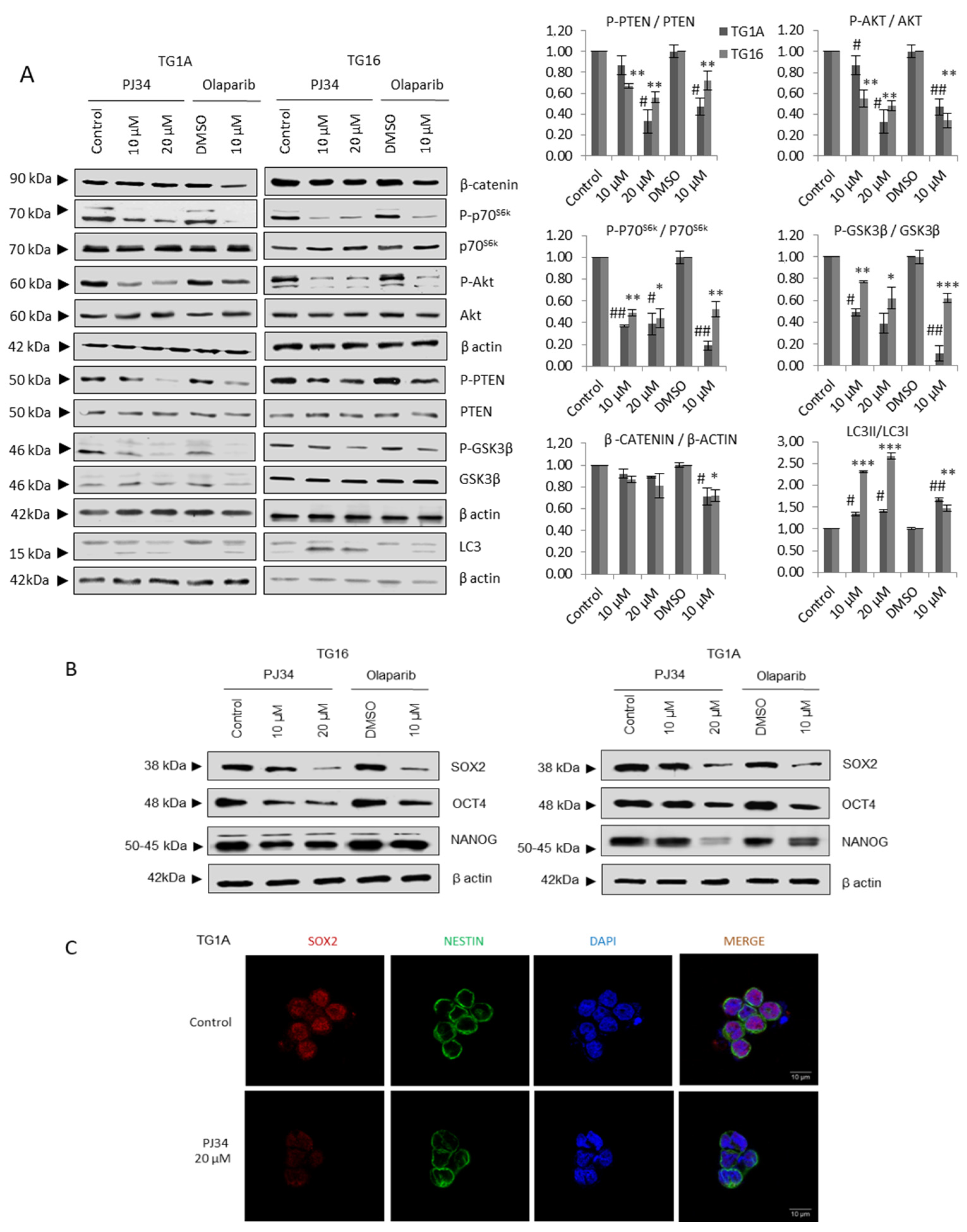
3.1. PARP Inhibition Down-Regulates Pro-Survival Pathways in Glioblastoma
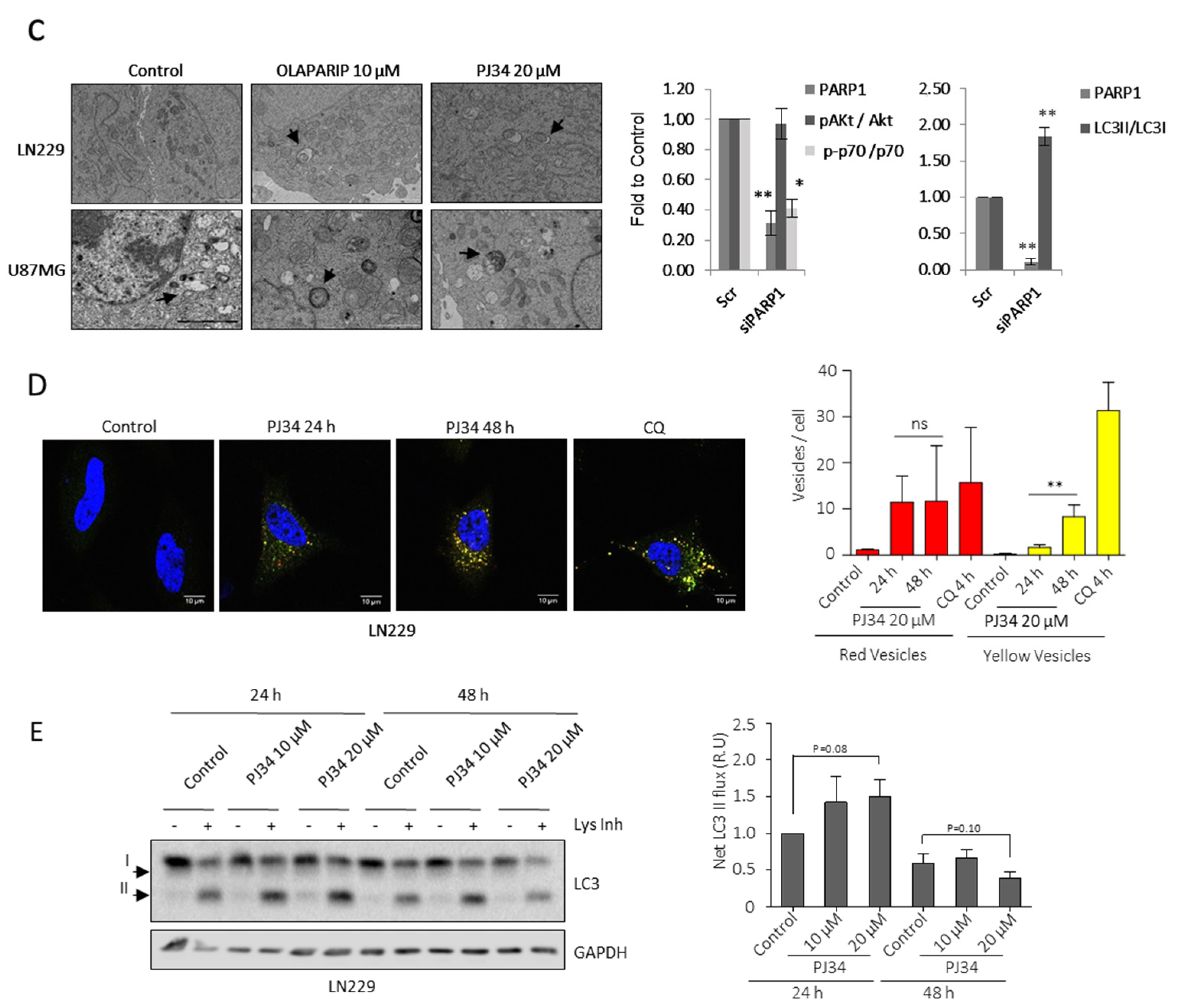
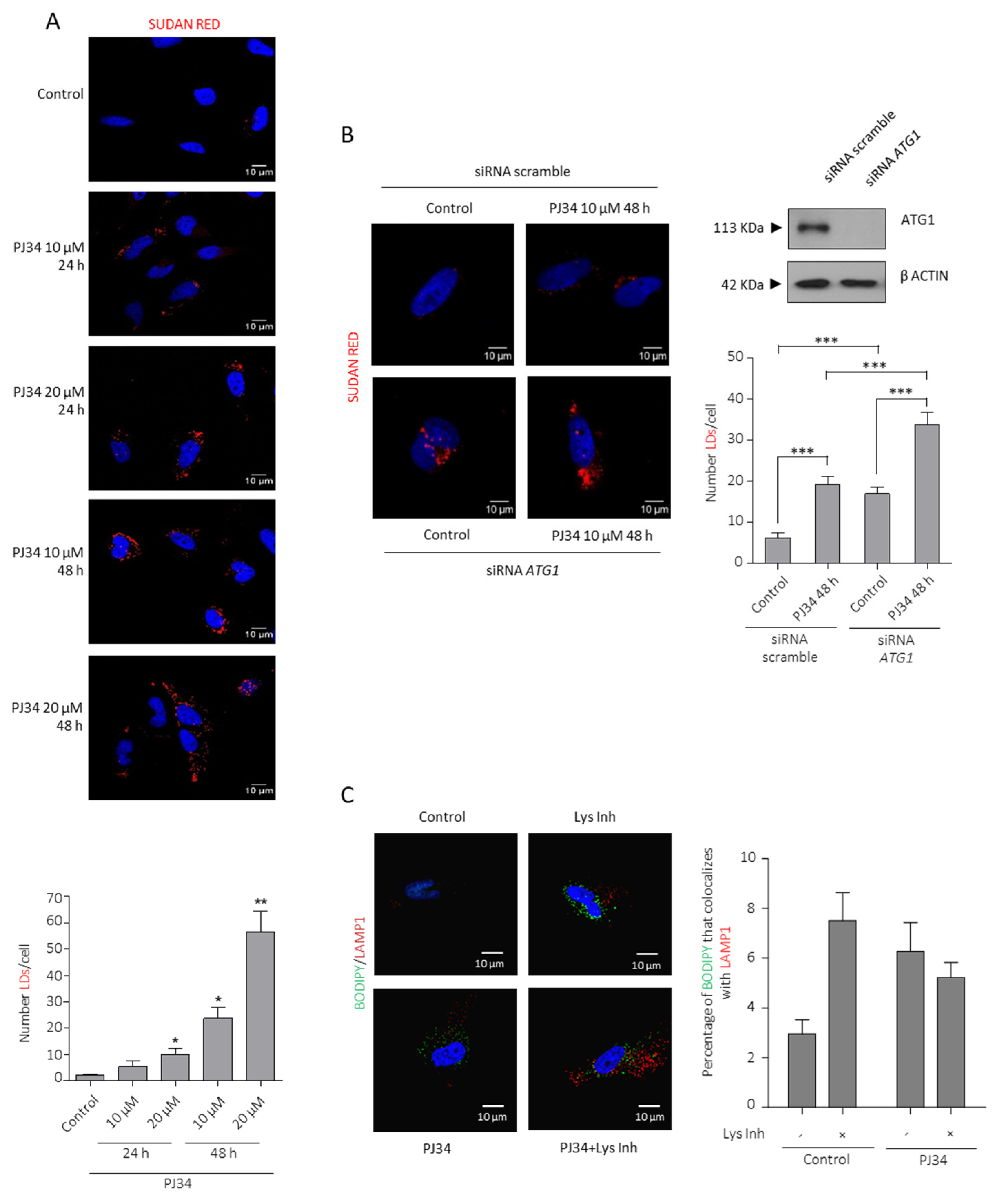
3.2. PARP Inhibition Induces Lipid Droplets (LDs) Formation and Utilization
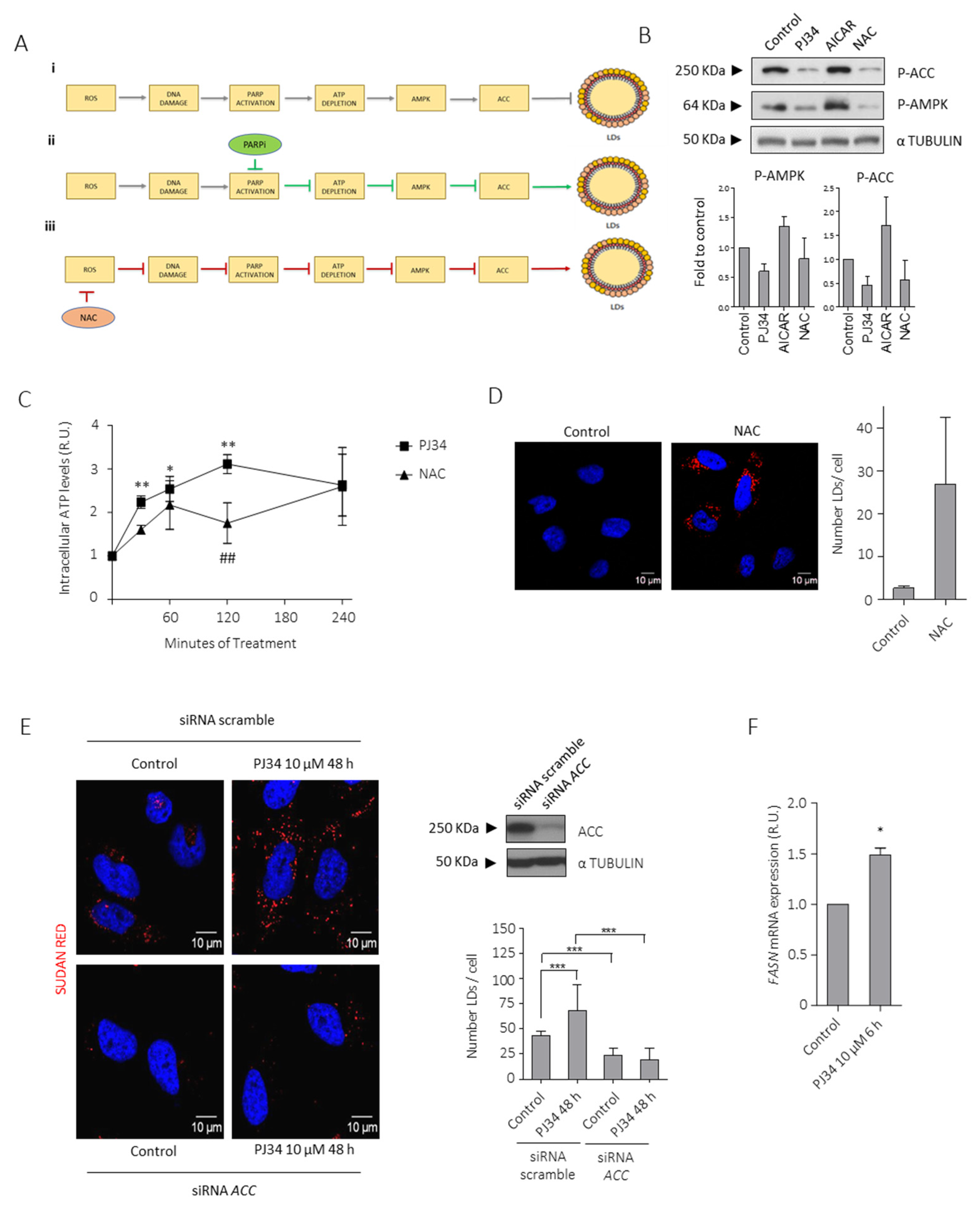
3.3. PARPi-Mediated Suppression of AMPK Modulates LDs Formation
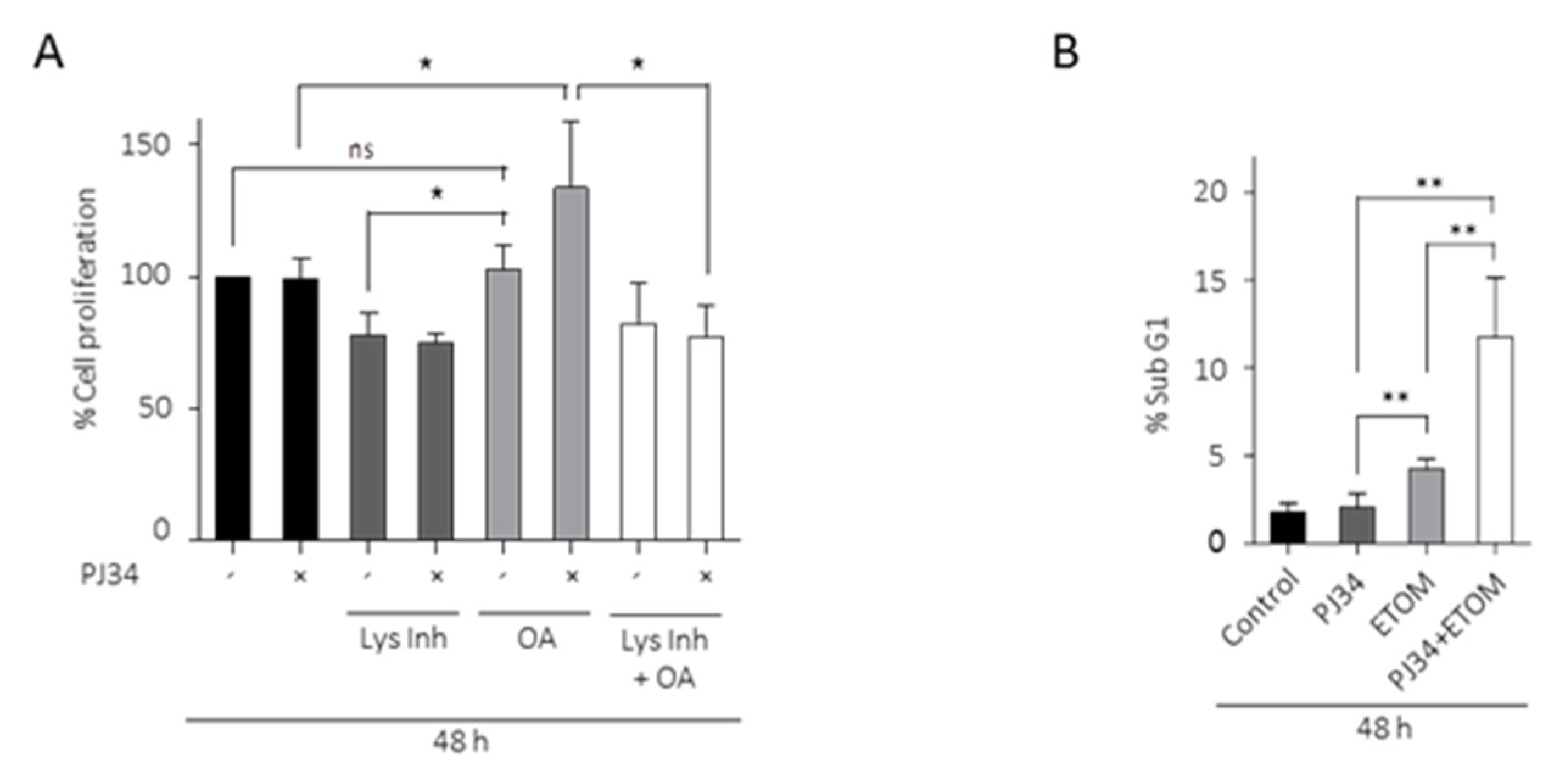
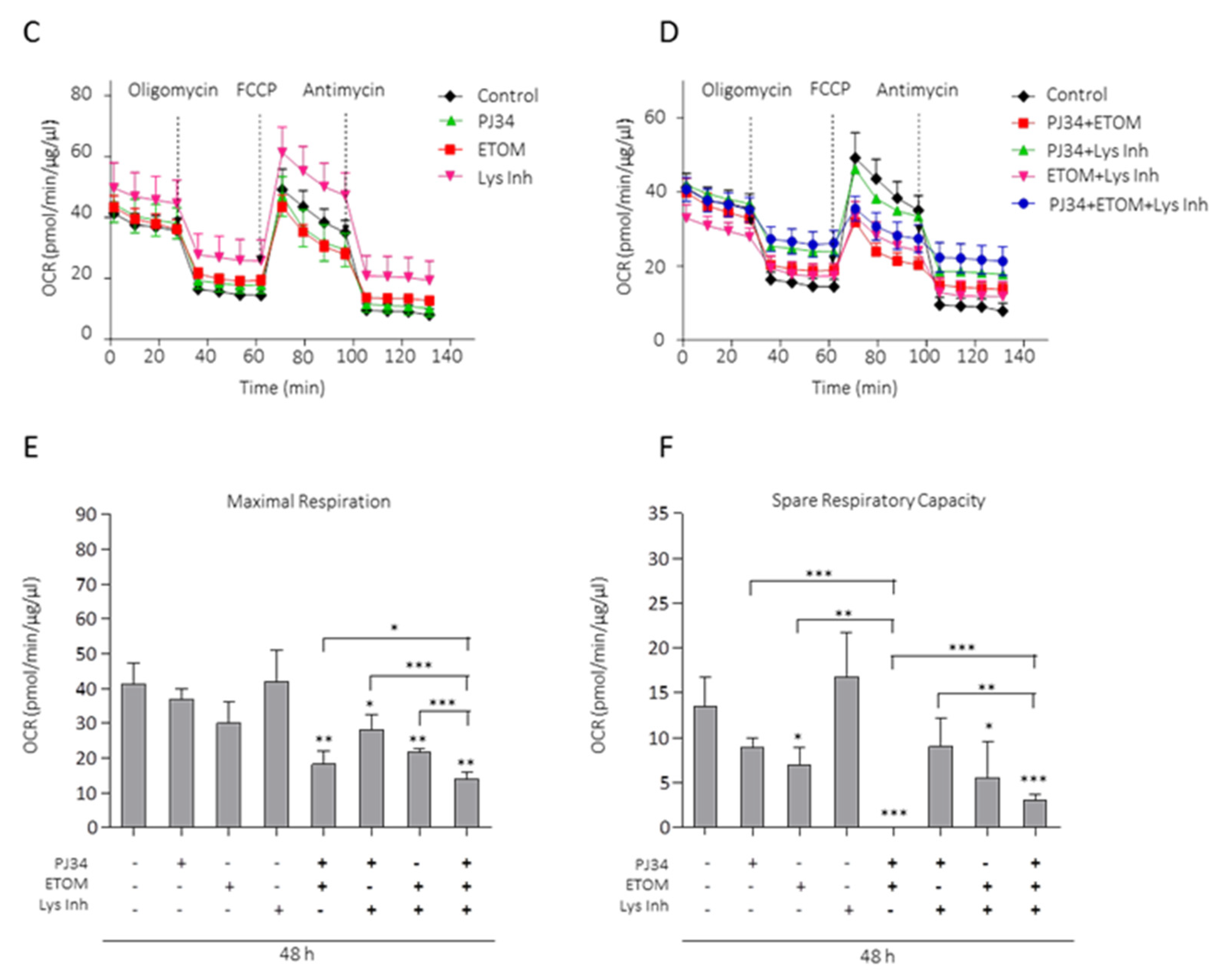
3.4. Lipids from LDs as Metabolic Fuel for Survival of Glioma Cells
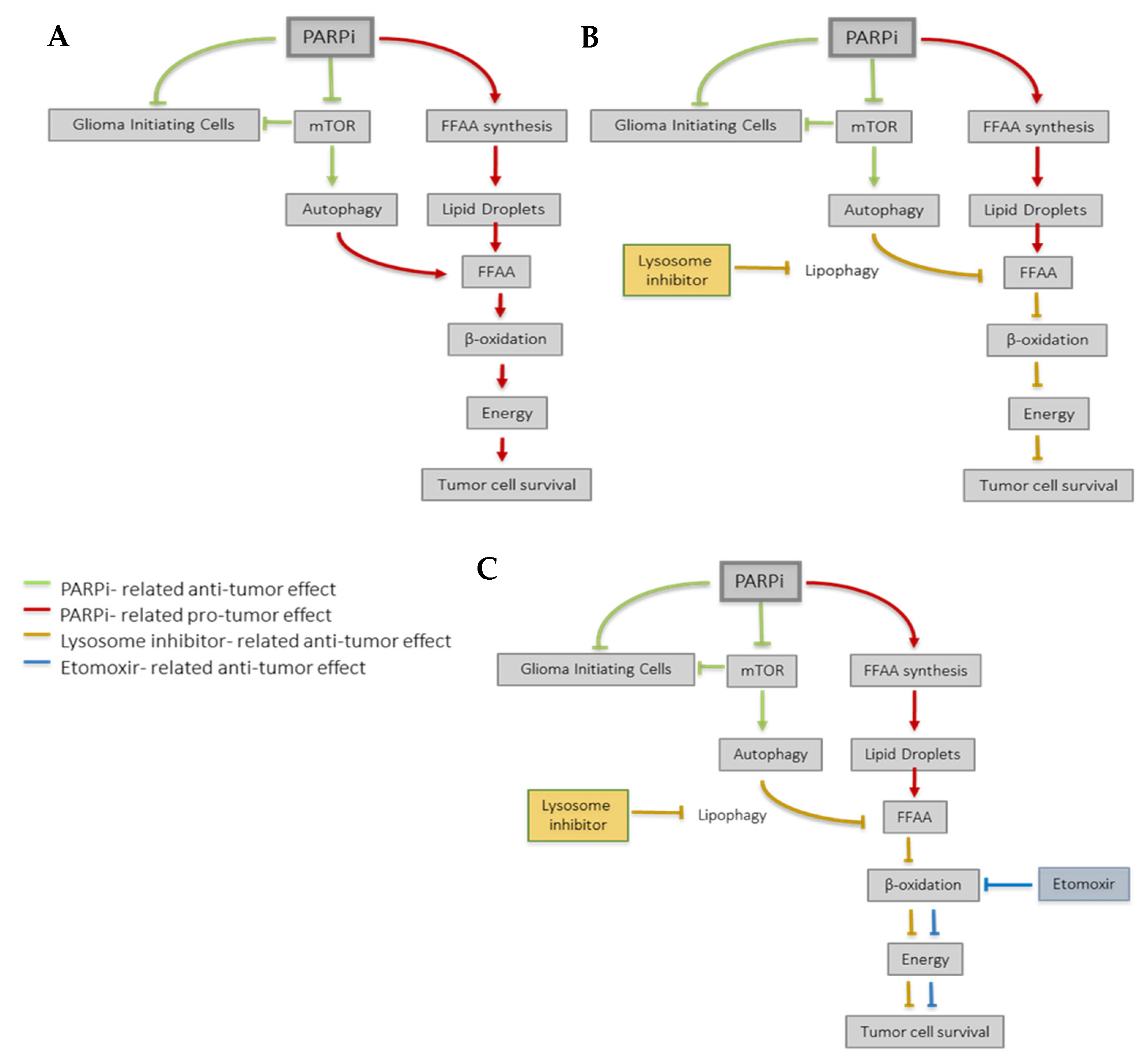
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Michels, J.; Obrist, F.; Castedo, M.; Vitale, I.; Kroemer, G. PARP and other prospective targets for poisoning cancer cell metabolism. Biochem. Pharmacol. 2014, 92, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Luna, A.; Aladjem, M.I.; Kohn, K.W. SIRT1/PARP1 crosstalk: Connecting DNA damage and metabolism. Genome Integr. 2013, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Białkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Zhang, Y.; Morris, J.; Ji, J.; Takeda, S.; Doroshow, J.H.; Pommier, Y. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharmacol. Exp. Ther. 2014, 349, 408–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venere, M.; Hamerlik, P.; Wu, Q.; Rasmussen, R.D.; Song, L.A.; Vasanji, A.; Tenley, N.; Flavahan, W.A.; Hjelmeland, A.B.; Bartek, J.; et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 2014, 21, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Curtin, N.J.; Wang, L.Z.; Yiakouvaki, A.; Kyle, S.; Arris, C.A.; Canan-Koch, S.; Webber, S.E.; Durkacz, B.W.; Calvert, H.A.; Hostomsky, Z.; et al. Novel poly(ADP-ribose) polymerase-1 inhibitor, AG14361, restores sensitivity to temozolomide in mismatch repair-deficient cells. Clin. Cancer Res. 2004, 10, 881–889. [Google Scholar] [CrossRef] [Green Version]
- Tentori, L.; Leonetti, C.; Scarsella, M.; d’Amati, G.; Vergati, M.; Portarena, I.; Xu, W.; Kalish, V.; Zupi, G.; Zhang, J.; et al. Systemic administration of GPI 15427, a novel poly(ADP-ribose) polymerase-1 inhibitor, increases the antitumor activity of temozolomide against intracranial melanoma, glioma, lymphoma. Clin. Cancer Res. 2003, 9, 5370–5379. [Google Scholar]
- Calabrese, C.R.; Almassy, R.; Barton, S.; Batey, M.A.; Calvert, A.H.; Canan-Koch, S.; Durkacz, B.W.; Hostomsky, Z.; Kumpf, R.A.; Kyle, S.; et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J. Natl. Cancer Inst. 2004, 96, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Majuelos-Melguizo, J.; Rodríguez, M.I.; López-Jiménez, L.; Rodríguez-Vargas, J.M.; Martín-Consuegra, J.M.M.; Serrano-Sáenz, S.; Gavard, J.; de Almodóvar, J.M.R.; Oliver, F.J. PARP targeting counteracts gliomagenesis through induction of mitotic catastrophe and aggravation of deficiency in homologous recombination in PTEN-mutant glioma. Oncotarget 2015, 6, 4790–4803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro. Oncol. 2017, 19, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patru, C.; Romao, L.; Varlet, P.; Coulombel, L.; Raponi, E.; Cadusseau, J.; Renault-Mihara, F.; Thirant, C.; Leonard, N.; Berhneim, A.; et al. CD133, CD15/SSEA-1, CD34 or side populations do not resume tumor-initiating properties of long-term cultured cancer stem cells from human malignant glio-neuronal tumors. BMC Cancer 2010, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Bleau, A.M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Galan-Moya, E.M.; Le Guelte, A.; Lima-Fernandes, E.; Thirant, C.; Dwyer, J.; Bidere, N.; Couraud, P.O.; Scott, M.G.; Junier, M.P.; Chneiweiss, H.; et al. Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep. 2011, 12, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Gupta, K.; Kalra, I.; Salunke, P.; Vasishta, R.K. Lipidized glioblastoma: A rare differentiation pattern. Neuropathology 2011, 31, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.; Migita, T.; Tang, D.; Loda, M. Fatty acid synthase: A metabolic oncogene in prostate cancer? J. Cell Biochem. 2004, 91, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Bell, E.H.; Chakravarti, A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol. 2013, 2, 289–299. [Google Scholar] [CrossRef]
- Srivastava, N.K.; Pradhan, S.; Gowda, G.A.; Kumar, R. In vitro, high-resolution 1H and 31P NMR based analysis of the lipid components in the tissue, serum, and CSF of the patients with primary brain tumors: One possible diagnostic view. NMR Biomed. 2010, 23, 113–122. [Google Scholar]
- Rodríguez-Vargas, J.M.; Ruiz-Magaña, M.J.; Ruiz-Ruiz, C.; Majuelos-Melguizo, J.; Peralta-Leal, A.; Rodríguez, M.I.; Muñoz-Gámez, J.A.; De Almodóvar, M.R.; Siles, E.; Rivas, A.L.; et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 2012, 22, 1181–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Vargas, J.M.; Rodríguez, M.I.; Majuelos-Melguizo, J.; García-Diaz, Á.; González-Flores, A.; López-Rivas, A.; Virág, L.; Illuzzi, G.; Schreiber, V.; Dantzer, F.; et al. Autophagy requires poly(adp-ribosyl)ation-dependent AMPK nuclear export. Cell Death Differ. 2016, 23, 2007–2018. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Ramiro, A.; Ramírez-Ortega, D.; Pérez de la Cruz, V.; Hérnandez-Pedro, N.Y.; González-Esquivel, D.F.; Sotelo, J.; Pineda, B. Role of Redox Status in Development of Glioblastoma. Front. Immunol. 2016, 7, 156. [Google Scholar] [CrossRef] [Green Version]
- Boonstra, J.; Post, J.A. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 2004, 337, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Lu, Y.; Yang, J.; Chen, G.; Kim, S.; Feng, L.; Ogasawara, M.; Hammoudi, N.; Lu, W.; Zhang, H.; et al. Metabolic activation of mitochondria in glioma stem cells promotes cancer development through a reactive oxygen species-mediated mechanism. Stem Cell Res. Ther. 2015, 6, 198. [Google Scholar] [CrossRef] [Green Version]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef] [Green Version]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13 C] glucose in human brain tumors in vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Zechner, R.; Madeo, F.; Kratky, D. Cytosolic lipolysis and lipophagy: Two sides of the same coin. Nat. Rev. Mol. Cell Biol 2017, 18, 671–684. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majuelos-Melguizo, J.; Rodríguez-Vargas, J.M.; Martínez-López, N.; Delgado-Bellido, D.; García-Díaz, Á.; Yuste, V.J.; García-Macía, M.; López, L.M.; Singh, R.; Oliver, F.J. Glioblastoma Cells Counteract PARP Inhibition through Pro-Survival Induction of Lipid Droplets Synthesis and Utilization. Cancers 2022, 14, 726. https://doi.org/10.3390/cancers14030726
Majuelos-Melguizo J, Rodríguez-Vargas JM, Martínez-López N, Delgado-Bellido D, García-Díaz Á, Yuste VJ, García-Macía M, López LM, Singh R, Oliver FJ. Glioblastoma Cells Counteract PARP Inhibition through Pro-Survival Induction of Lipid Droplets Synthesis and Utilization. Cancers. 2022; 14(3):726. https://doi.org/10.3390/cancers14030726
Chicago/Turabian StyleMajuelos-Melguizo, Jara, José Manuel Rodríguez-Vargas, Nuria Martínez-López, Daniel Delgado-Bellido, Ángel García-Díaz, Víctor J. Yuste, Marina García-Macía, Laura M. López, Rajat Singh, and F. J. Oliver. 2022. "Glioblastoma Cells Counteract PARP Inhibition through Pro-Survival Induction of Lipid Droplets Synthesis and Utilization" Cancers 14, no. 3: 726. https://doi.org/10.3390/cancers14030726
APA StyleMajuelos-Melguizo, J., Rodríguez-Vargas, J. M., Martínez-López, N., Delgado-Bellido, D., García-Díaz, Á., Yuste, V. J., García-Macía, M., López, L. M., Singh, R., & Oliver, F. J. (2022). Glioblastoma Cells Counteract PARP Inhibition through Pro-Survival Induction of Lipid Droplets Synthesis and Utilization. Cancers, 14(3), 726. https://doi.org/10.3390/cancers14030726