
Combined Inhibition of Polo-Like Kinase-1 and Wee1 as a New Therapeutic Strategy to Induce Apoptotic Cell Death in Neoplastic Mast Cells


 ,
,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
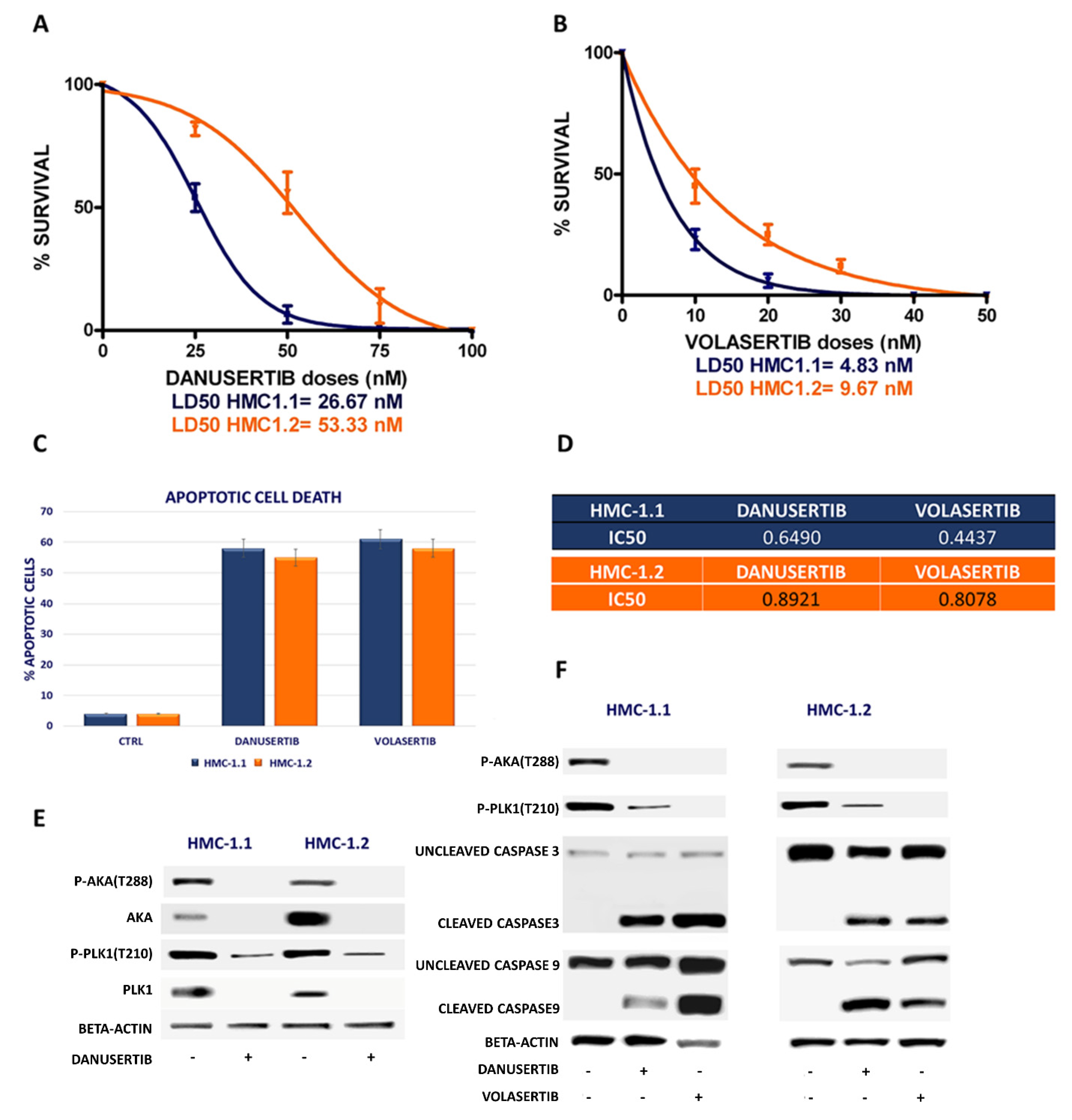
2.1. Danusertib (AKA Inhibitor) and Volasertib (Plk1 Inhibitor) Inhibited Growth in HMC-1.1 and -1.2 Cells
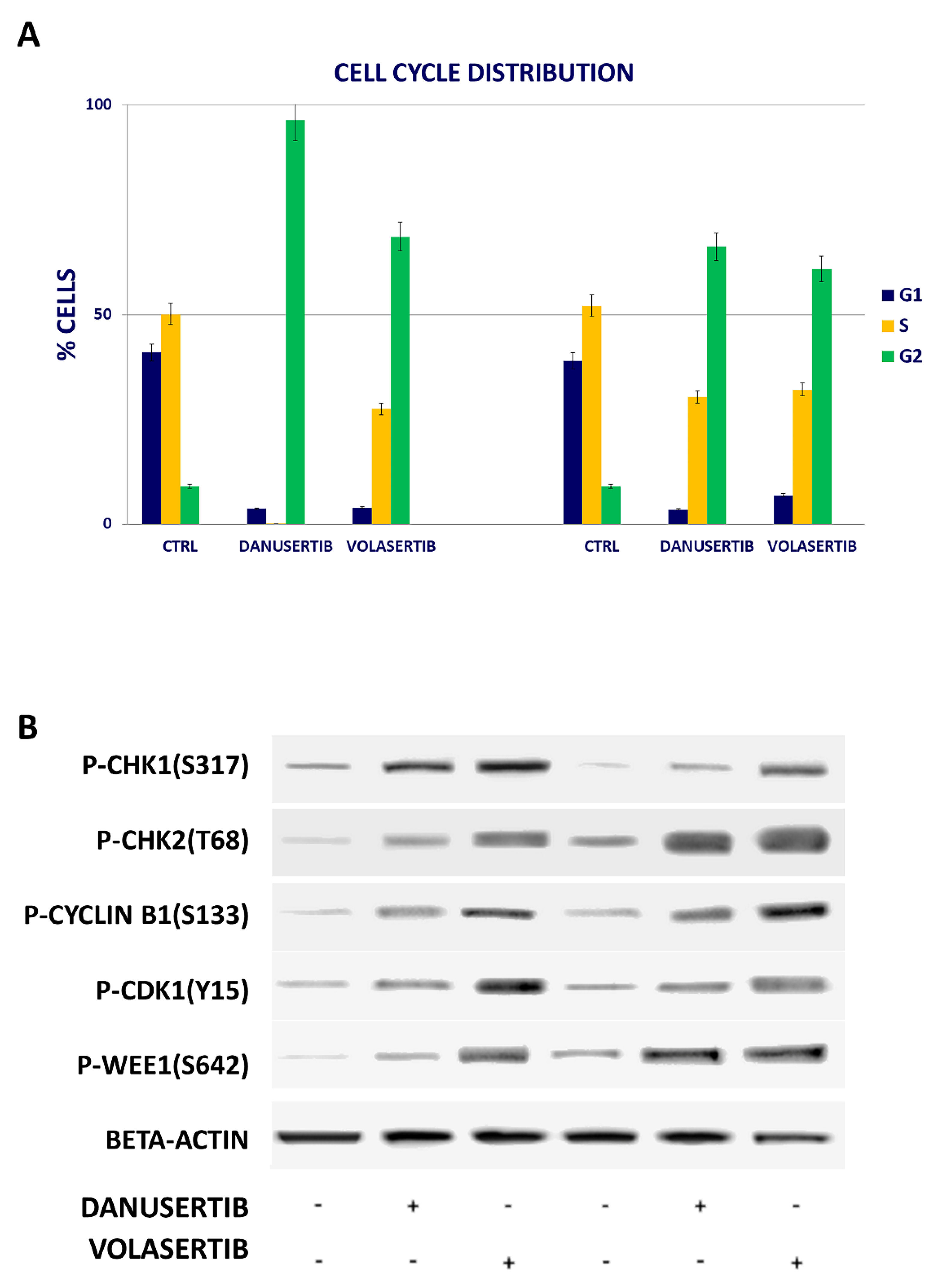
2.2. Inhibition of Either AKA or Plk1 Induces Cell Cycle Arrest and Affects G2/M Checkpoint Proteins
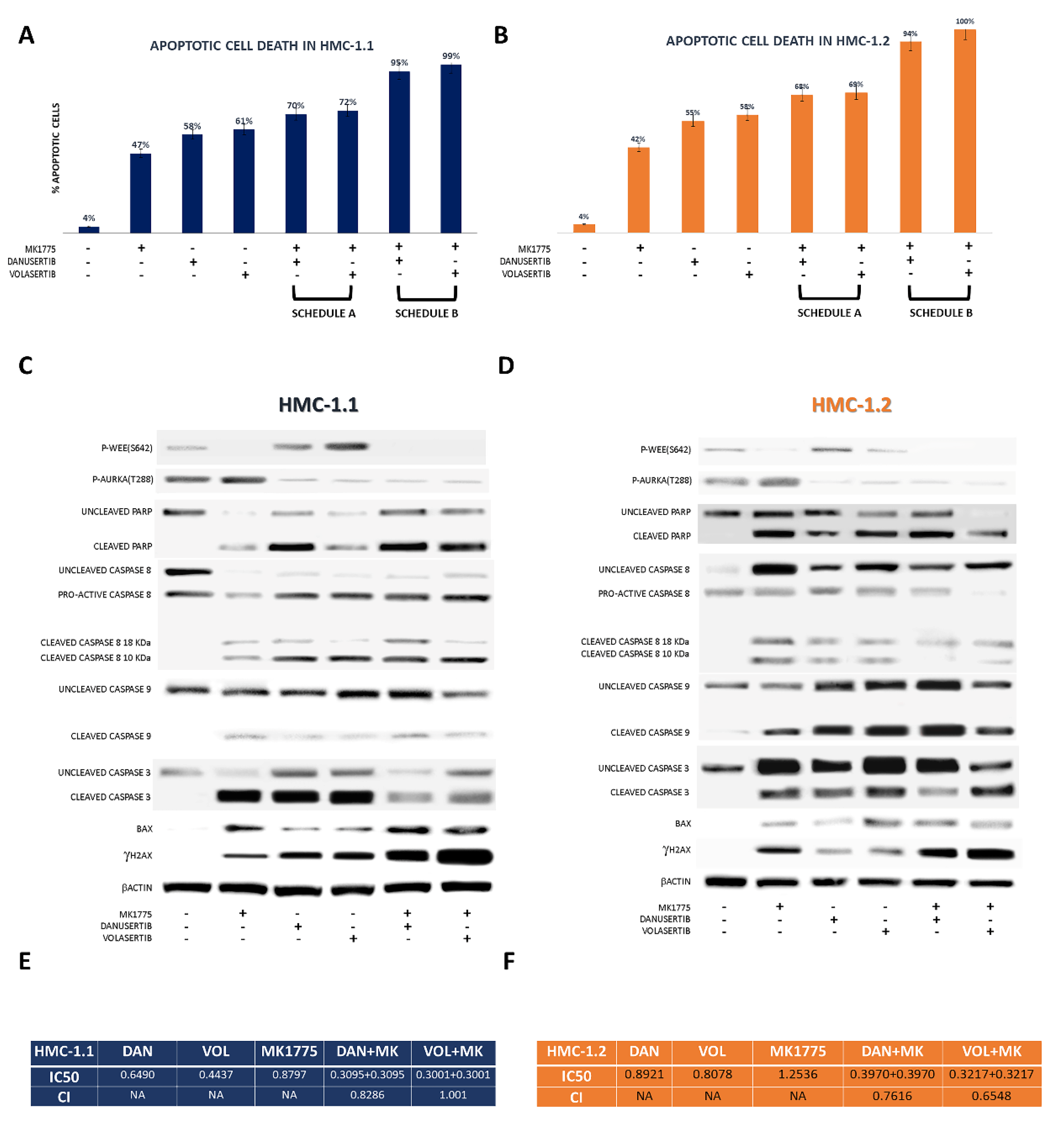
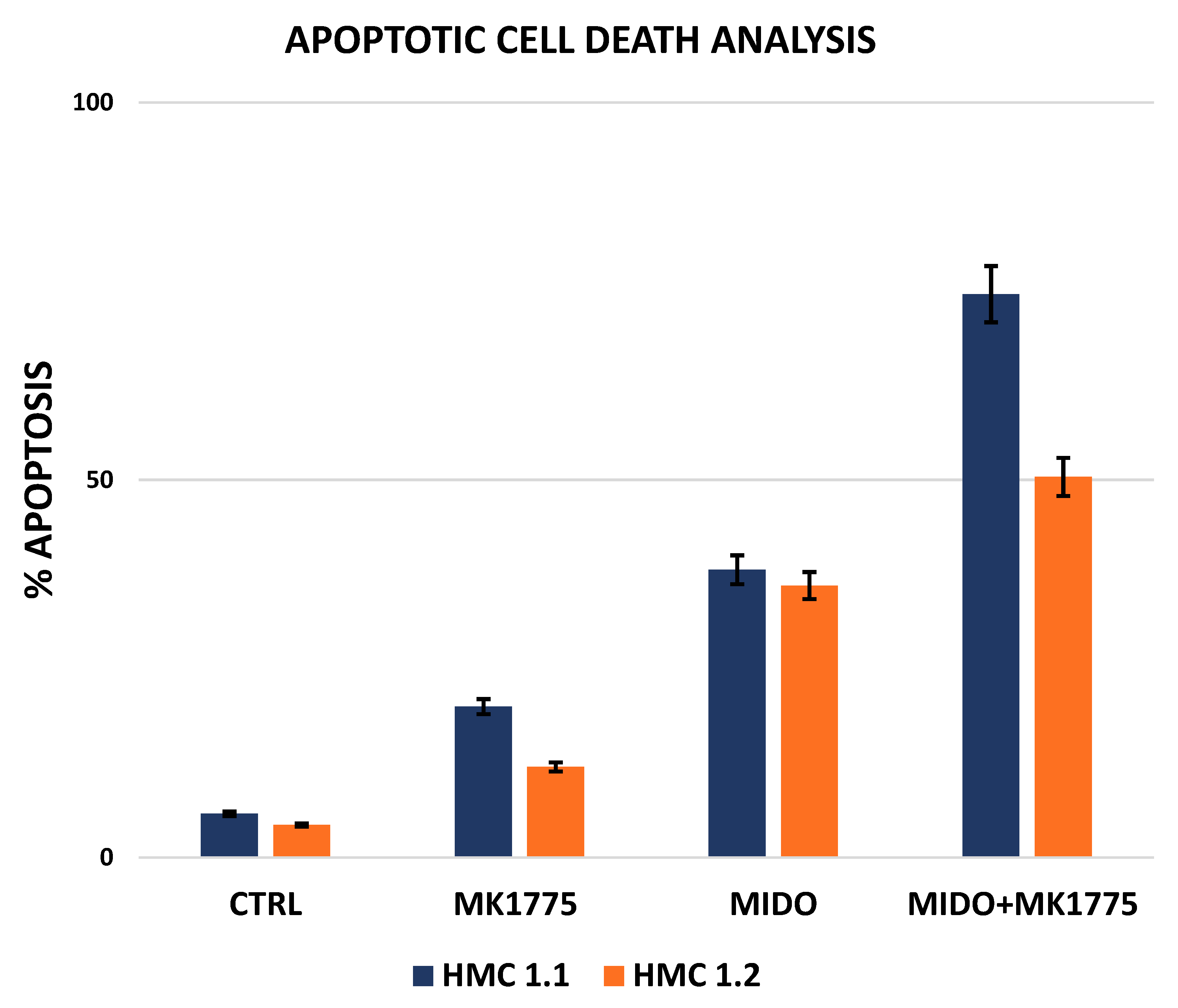
2.3. Combined WEE1 and AKA, or PLK1, Inhibition Has Synergistic Effects in HMC-1 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Drug Treatments
4.3. Protein Analysis
4.4. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Valent, P. Biology, classification and treatment of human mastocytosis. Wien. Klin. Wochenschr. 1996, 108, 385–397. [Google Scholar] [PubMed]
- Horny, H.P.; Valent, P. Diagnosis of mastocytosis: General histopathological aspects, morphological criteria, and immunohistochemical findings. Leuk Res. 2001, 25, 543–551. [Google Scholar] [CrossRef]
- Escribano, L.; Akin, C.; Castells, M.; Orfao, A.; Metcalfe, D.D. Mastocytosis: Current concepts in diagnosis and treatment. Ann. Hematol. 2002, 81, 677–690. [Google Scholar]
- Valent, P.; Akin, C.; Sperr, W.R.; Horny, H.P.; Arock, M.; Lechner, K.; Bennett, J.M.; Metcalfe, D.D. Diagnosis and treatment of systemic mastocytosis: State of the art. Br. J. Haematol. 2003, 122, 695–717. [Google Scholar] [CrossRef]
- Akin, C.; Metcalfe, D.D. Systemic mastocytosis. Annu. Rev. Med. 2004, 55, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Sumbly, V.; Landry, I.; Iqbal, S.; Bhatti, Z.; Alshamam, M.S.; Ashfaq, S.; Rizzo, V. The Role of Avapritinib for the Treatment of Systemic Mastocytosis. Cureus 2021, 13, e18385. [Google Scholar] [CrossRef]
- Sammarco, G.; Gallo, G.; Vescio, G.; Picciariello, A.; De Paola, G.; Trompetto, M.; Currò, G.; Ammendola, M. Mast Cells, microRNAs and Others: The Role of Translational Research on Colorectal Cancer in the Forth-coming Era of Precision Medicine. J. Clin. Med. 2020, 9, 2852. [Google Scholar] [CrossRef]
- Conti, P.; Castellani, M.L.; Kempuraj, D.; Salini, V.; Vecchiet, J.; Tetè, S.; Mastrangelo, F.; Perrella, A.; De Lutiis, M.A.; Tagen, M.; et al. Role of mast cells in tumor growth. Ann. Clin. Lab. Sci. 2007, 37, 315–322. [Google Scholar]
- Marech, I.; Ammendola, M.; Gadaleta, C.; Zizzo, N.; Oakley, C.; Gadaleta, C.D.; Ranieri, G. Possible bio-logical and translational significance of mast cells density in colorectal cancer. World J. Gastroenterol. 2014, 20, 8910–8920. [Google Scholar]
- Malfettone, A.; Silvestris, N.; Saponaro, C.; Ranieri, G.; Russo, A.; Caruso, S.; Popescu, O.; Simone, G.; Paradiso, A.; Mangia, A. High density of tryptase-positive mast cells in human colorectal cancer: A poor prognostic factor related to protease-activated receptor 2 expression. J. Cell. Mol. Med. 2013, 17, 1025–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degenhardt, Y.; Lampkin, T. Targeting polo-like kinase in cancer therapy. Clin. Cancer Res. 2010, 16, 384–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lens, S.M.A.; Voest, E.E.; Medema, R.H. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat. Rev. Cancer 2010, 10, 825–841. [Google Scholar] [CrossRef]
- Strebhardt, K.; Ullrich, A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 2006, 6, 321–330. [Google Scholar] [CrossRef]
- Schöffski, P. Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology. Oncologist 2009, 14, 559–570. [Google Scholar] [CrossRef] [Green Version]
- van de Weerdt, B.C.; Medema, R.H. Polo-like kinases: A team in control of the division. Cell Cycle 2006, 5, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Mundt, K.E.; Golsteyn, R.M.; Lane, H.A.; Nigg, E.A. On the regulation and function of human polo-like kinase 1 (PLK1): Effects of overexpression on cell cycle progression. Biochem. Biophys. Res. Commun. 1997, 239, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.R.; Carmena, M.; Earnshaw, W.C. Chromosomal passengers and the (aurora) ABCs of mitosis. Trends Cell Biol. 2001, 11, 49–54. [Google Scholar] [CrossRef]
- Carmena, M.; Earnshaw, W.C. The cellular geography of aurora kinases. Nat. Rev. Mol. Cell Biol. 2003, 4, 842–854. [Google Scholar] [CrossRef]
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2010, 2, 21–32. [Google Scholar] [CrossRef]
- Mancini, M.; Monaldi, C.; De Santis, S.; Papayannidis, C.; Rondoni, M.; Bavaro, L.; Martelli, M.; Chiara, A.M.; Curti, A.; Ficarra, E.; et al. MDM2 and Aurora Kinase a Contribute to SETD2 Loss of Function in Advanced Systemic Mastocytosis: Implications for Pathogenesis and Treatment. Blood 2018, 132, 1779. [Google Scholar] [CrossRef]
- Martinelli, G.; Mancini, M.; De Benedittis, C.; Rondoni, M.; Papayannidis, C.; Manfrini, M.; Meggendorfer, M.; Calogero, R.; Guadagnuolo, V.; Fontana, M.C.; et al. SETD2 and histone H3 lysine 36 methylation deficiency in advanced systemic mastocytosis. Leukemia 2018, 32, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, D.D. Mast cells and mastocytosis. Blood 2008, 112, 946–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, M.C.; Metcalfe, D.D.; Komarow, H.D. Mastocytosis. Allergy Clin. N. Am. 2014, 34, 181–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valent, P.; Akin, C.; Escribano, L.; Födinger, M.; Hartmann, K.; Brockow, K.; Castells, M.; Sperr, W.R.; Kluin-Nelemans, J.C.; Hamdy, N.A.T.; et al. Standards and standardization in mastocytosis: Consensus statements on diagnostics, treatment recommendations and response criteria. Eur. J. Clin. Investig. 2007, 37, 435–453. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Sotlar, K.; Horny, H.-P.; Metzgeroth, G.; Müller, N.; Schneider, S.; Naumann, N.; Walz, C.; et al. Molecular profiling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifies KIT D816V as a distinct and late event. Leukemia 2015, 29, 1115–1122. [Google Scholar] [CrossRef]
- Sotlar, K.; Colak, S.; Bache, A.; Berezowska, S.; Krokowski, M.; Bültmann, B.; Valent, P.; Horny, H.P. Variable presence of KITD816V in clonal haematological non-mast cell lineage diseases associated with systemicmastocytosis (SM-AHNMD). J. Pathol. 2010, 220, 586–595. [Google Scholar] [CrossRef]
- Warner, S.L.; Bearss, D.J.; Han, H.; Von Hoff, D.D. Targeting Aurora-2 kinase in cancer. Mol. Cancer Ther. 2003, 2, 589–595. [Google Scholar]
- Peter, B.V.; Gleixner, K.; Cerny-Reiterer, S.; Herrmann, H.; Winter, V.; Hadzijusufovic, E.; Ferenc, V.; Schuch, K.; Mirkina, I.; Horny, H.P.; et al. Polo-like kinase-1 as a novel target in neoplastic mast cells: Demonstration of growth-inhibitory effects of small interfering RNA and the Polo-like kinase-1 targeting drug BI 2536. Haematologica 2011, 96, 672–680. [Google Scholar] [CrossRef] [Green Version]
- Kawai, M.; Nakashima, A.; Kamada, S.; Kikkawa, U. Midostaurin preferentially attenuates proliferation of triple-negative breast cancer cell lines through inhibition of Aurora kinase family. J. Biomed. Sci. 2015, 22, 48. [Google Scholar] [CrossRef] [Green Version]
- Ammendola, M.; Sacco, R.; Sammarco, G.; Luposella, M.; Patruno, R.; Gadaleta, C.D.; De Sarro, G.; Ranieri, G. Mast Cell-Targeted Strategies in Cancer Therapy. Transfus. Med. Hemotherapy 2016, 43, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Arock, M.; Wedeh, G.; Hoermann, G.; Bibi, S.; Akin, C.; Peter, B.; Gleixner, K.V.; Hartmann, K.; Butterfield, J.H.; Metcalfe, D.D.; et al. Preclinical human models and emerging therapeutics for advanced systemic mastocytosis. Haematologica 2018, 103, 1760–1771. [Google Scholar] [CrossRef] [PubMed]
- Leo, E.; Mancini, M.; Aluigi, M.; Luatti, S.; Castagnetti, F.; Testoni, N.; Soverini, S.; Santucci, M.A.; Martinelli, G. BCR-ABL1-associated reduction of beta catenin antagonist Chibby1 in chronic myeloid leukemia. PLoS ONE 2013, 8, e81425. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, M.; Leo, E.; Campi, V.; Castagnetti, F.; Zazzeroni, L.; Gugliotta, G.; Santucci, M.A.; Martinelli, G. A calpain-cleaved fragment of β-catenin promotes BCRABL1+ cell survival evoked by autophagy induction in response to imatinib. Cell. Signal. 2014, 26, 1690–1697. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DRUGS | Dose (µM) | Fa | CI |
|---|---|---|---|
| AZD1775 | 1 | 0.20 | NA |
| MIDOSTAURIN | 1 | 0.38 | NA |
| AZD1775 + MIDOSTAURIN | 1 + 1 | 0.76 | 0.172 |
| DRUGS | Dose (µM) | Fa | CI |
|---|---|---|---|
| AZD1775 | 1 | 0.12 | NA |
| MIDOSTAURIN | 1 | 0.36 | NA |
| AZD1775 + MIDOSTAURIN | 1 + 1 | 0.50 | 0.905 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancini, M.; Monaldi, C.; De Santis, S.; Rondoni, M.; Papayannidis, C.; Sartor, C.; Curti, A.; Bruno, S.; Cavo, M.; Soverini, S. Combined Inhibition of Polo-Like Kinase-1 and Wee1 as a New Therapeutic Strategy to Induce Apoptotic Cell Death in Neoplastic Mast Cells. Cancers 2022, 14, 738. https://doi.org/10.3390/cancers14030738
Mancini M, Monaldi C, De Santis S, Rondoni M, Papayannidis C, Sartor C, Curti A, Bruno S, Cavo M, Soverini S. Combined Inhibition of Polo-Like Kinase-1 and Wee1 as a New Therapeutic Strategy to Induce Apoptotic Cell Death in Neoplastic Mast Cells. Cancers. 2022; 14(3):738. https://doi.org/10.3390/cancers14030738
Chicago/Turabian StyleMancini, Manuela, Cecilia Monaldi, Sara De Santis, Michela Rondoni, Cristina Papayannidis, Chiara Sartor, Antonio Curti, Samantha Bruno, Michele Cavo, and Simona Soverini. 2022. "Combined Inhibition of Polo-Like Kinase-1 and Wee1 as a New Therapeutic Strategy to Induce Apoptotic Cell Death in Neoplastic Mast Cells" Cancers 14, no. 3: 738. https://doi.org/10.3390/cancers14030738
APA StyleMancini, M., Monaldi, C., De Santis, S., Rondoni, M., Papayannidis, C., Sartor, C., Curti, A., Bruno, S., Cavo, M., & Soverini, S. (2022). Combined Inhibition of Polo-Like Kinase-1 and Wee1 as a New Therapeutic Strategy to Induce Apoptotic Cell Death in Neoplastic Mast Cells. Cancers, 14(3), 738. https://doi.org/10.3390/cancers14030738