
Toward More Comprehensive Homologous Recombination Deficiency Assays in Ovarian Cancer Part 2: Medical Perspectives




Abstract
:Simple Summary
Abstract
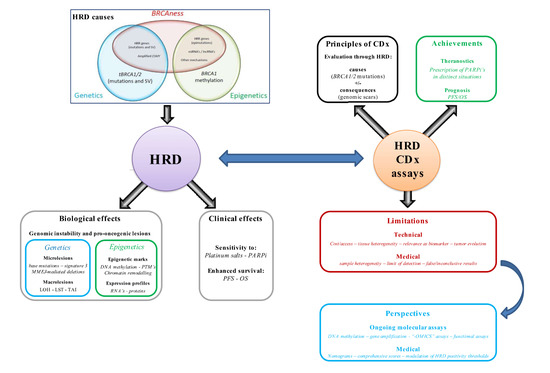
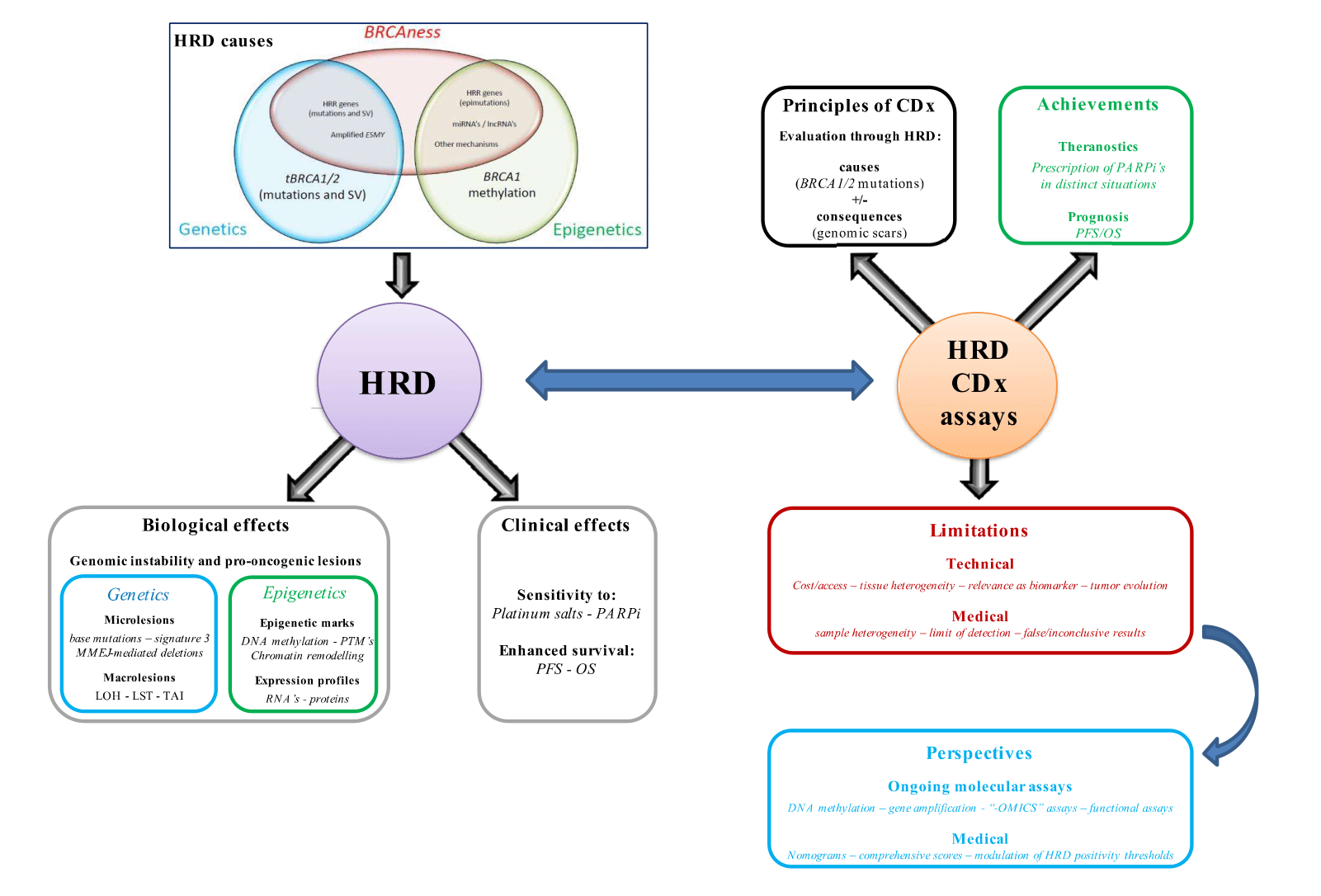
1. Introduction
2. HRD Companion Assays in Clinical Practice
2.1. Introduction to CDx Assays
2.2. Assay Achievements in Clinical Practice: The PARPi Era
2.2.1. Introduction to Clinical Trials Assessing PARPis
2.2.2. Newly Diagnosed Advanced Epithelial Ovarian Cancer: Frontline Treatment
2.2.3. Recurrent Epithelial Ovarian Cancer: Second-Line Maintenance
2.2.4. Recurrent Epithelial Ovarian Cancer: Third Line and Beyond
3. HRD Evaluation in Clinics: Current Limitations
3.1. General Considerations
3.2. A Matter of Tissues
3.3. Relevance of HRD Status as a Predictive Biomarker
3.4. An Evolutionary Perspective on HRD Status
4. Emerging Strategies and Perspectives for Accurate and Dynamic Assessment of HRD
4.1. Introduction
4.2. Frontline Maintenance Perspectives
4.3. Recurrent Epithelial Ovarian Cancer Perspectives
5. Synthesis and Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Prat, J. Ovarian Carcinomas: Five Distinct Diseases with Different Origins, Genetic Alterations, and Clinicopathological Features. Virchows Arch. 2012, 460, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Salazar, C.; Campbell, I.G.; Gorringe, K.L. When Is “Type I” Ovarian Cancer Not “Type I”? Indications of an Out-Dated Dichotomy. Front. Oncol. 2018, 8, 654. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, I. Mechanisms of High-Grade Serous Carcinogenesis in the Fallopian Tube and Ovary: Current Hypotheses, Etiologic Factors, and Molecular Alterations. Int. J. Mol. Sci. 2021, 22, 4409. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial Ovarian Cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef] [Green Version]
- Matz, M.; Coleman, M.P.; Carreira, H.; Salmeron, D.; Chirlaque, M.D.; Allemani, C.; Concord Working Group. Worldwide Comparison of Ovarian Cancer Survival: Histological Group and Stage at Diagnosis (CONCORD-2). Gynecol. Oncol. 2017, 144, 396–404. [Google Scholar] [CrossRef]
- Matz, M.; Coleman, M.P.; Sant, M.; Chirlaque, M.D.; Visser, O.; Gore, M.; Allemani, C.; Bouzbid, S.; Hamdi-Chérif, M.; Zaidi, Z.; et al. The Histology of Ovarian Cancer: Worldwide Distribution and Implications for International Survival Comparisons (CONCORD-2). Gynecol. Oncol. 2017, 144, 405–413. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Hilpert, F.; Weber, B.; Reuss, A.; Poveda, A.; Kristensen, G.; Sorio, R.; Vergote, I.; Witteveen, P.; Bamias, A.; et al. Bevacizumab Combined with Chemotherapy for Platinum-Resistant Recurrent Ovarian Cancer: The AURELIA Open-Label Randomized Phase III Trial. J. Clin. Oncol. 2014, 32, 1302–1308. [Google Scholar] [CrossRef]
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; et al. Standard Chemotherapy with or without Bevacizumab for Women with Newly Diagnosed Ovarian Cancer (ICON7): Overall Survival Results of a Phase 3 Randomised Trial. Lancet Oncol. 2015, 16, 928–936. [Google Scholar] [CrossRef]
- Aghajanian, C.; Blank, S.V.; Goff, B.A.; Judson, P.L.; Teneriello, M.G.; Husain, A.; Sovak, M.A.; Yi, J.; Nycum, L.R. OCEANS: A Randomized, Double-Blind, Placebo-Controlled Phase III Trial of Chemotherapy with or without Bevacizumab in Patients with Platinum-Sensitive Recurrent Epithelial Ovarian, Primary Peritoneal, or Fallopian Tube Cancer. J. Clin. Oncol. 2012, 30, 2039–2045. [Google Scholar] [CrossRef] [Green Version]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.; Sonke, G.S.; Colombo, N.; Špaček, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib Combined with Chemotherapy for Recurrent Platinum-Sensitive Ovarian Cancer: A Randomised Phase 2 Trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef]
- Tewari, K.S.; Burger, R.A.; Enserro, D.; Norquist, B.M.; Swisher, E.M.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Huang, H.; Homesley, H.D.; et al. Final Overall Survival of a Randomized Trial of Bevacizumab for Primary Treatment of Ovarian Cancer. J. Clin. Oncol. 2019, 37, 2317–2328. [Google Scholar] [CrossRef] [PubMed]
- Gogineni, V.; Morand, S.; Staats, H.; Royfman, R.; Devanaboyina, M.; Einloth, K.; Dever, D.; Stanbery, L.; Aaron, P.; Manning, L.; et al. Current Ovarian Cancer Maintenance Strategies and Promising New Developments. J. Cancer 2021, 12, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Foo, T.; George, A.; Banerjee, S. PARP Inhibitors in Ovarian Cancer: An Overview of the Practice-Changing Trials. Genes Chromosomes Cancer 2021, 60, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; George, A.; Kaye, S.B.; Banerjee, S. BRCA Somatic Mutations and Epigenetic BRCA Modifications in Serous Ovarian Cancer. Ann. Oncol. 2016, 27, 1449–1455. [Google Scholar] [CrossRef]
- Kalachand, R.D.; Stordal, B.; Madden, S.; Chandler, B.; Cunningham, J.; Goode, E.L.; Ruscito, I.; Braicu, E.I.; Sehouli, J.; Ignatov, A.; et al. BRCA1 Promoter Methylation and Clinical Outcomes in Ovarian Cancer: An Individual Patient Data Meta-Analysis. J. Natl. Cancer Inst. 2020, 112, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Ewing, A.; Meynert, A.; Churchman, M.; Grimes, G.R.; Hollis, R.L.; Herrington, C.S.; Rye, T.; Bartos, C.; Croy, I.; Ferguson, M.; et al. Structural Variants at the BRCA1/2 Loci Are a Common Source of Homologous Repair Deficiency in High-Grade Serous Ovarian Carcinoma. Clin. Cancer Res. 2021, 27, 3201–3214. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness Revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and Somatic Mutations in Homologous Recombination Genes Predict Platinum Response and Survival in Ovarian, Fallopian Tube, and Peritoneal Carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Riaz, N.; Blecua, P.; Lim, R.S.; Shen, R.; Higginson, D.S.; Weinhold, N.; Norton, L.; Weigelt, B.; Powell, S.N.; Reis-Filho, J.S. Pan-Cancer Analysis of Bi-Allelic Alterations in Homologous Recombination DNA Repair Genes. Nat. Commun. 2017, 8, 857. [Google Scholar] [CrossRef]
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate Biomarkers of PARP Inhibitor Sensitivity in Ovarian Cancer beyond the BRCA Genes. Br. J. Cancer 2018, 119, 1401–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes-Davies, L.; Huntsman, D.; Ruas, M.; Fuks, F.; Bye, J.; Chin, S.-F.; Milner, J.; Brown, L.A.; Hsu, F.; Gilks, B.; et al. EMSY Links the BRCA2 Pathway to Sporadic Breast and Ovarian Cancer. Cell 2003, 115, 523–535. [Google Scholar] [CrossRef] [Green Version]
- Hollis, R.L.; Churchman, M.; Michie, C.O.; Rye, T.; Knight, L.; McCavigan, A.; Perren, T.; Williams, A.R.W.; McCluggage, W.G.; Kaplan, R.S.; et al. High EMSY Expression Defines a BRCA-like Subgroup of High-Grade Serous Ovarian Carcinoma with Prolonged Survival and Hypersensitivity to Platinum. Cancer 2019, 125, 2772–2781. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.M.; Cicek, M.S.; Larson, N.B.; Davila, J.; Wang, C.; Larson, M.C.; Song, H.; Dicks, E.M.; Harrington, P.; Wick, M.; et al. Clinical Characteristics of Ovarian Cancer Classified by BRCA1, BRCA2, and RAD51C Status. Sci. Rep. 2014, 4, 4026. [Google Scholar] [CrossRef]
- Potapova, A.; Hoffman, A.M.; Godwin, A.K.; Al-Saleem, T.; Cairns, P. Promoter Hypermethylation of the PALB2 Susceptibility Gene in Inherited and Sporadic Breast and Ovarian Cancer. Cancer Res. 2008, 68, 998–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasty, P.; Montagna, C. Chromosomal Rearrangements in Cancer: Detection and Potential Causal Mechanisms. Mol. Cell. Oncol. 2014, 1, e29904. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering Signatures of Mutational Processes Operative in Human Cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.A.; Drew, Y.; Kristeleit, R.S. Homologous Recombination Deficiency and Ovarian Cancer. Eur. J. Cancer 2016, 60, 49–58. [Google Scholar] [CrossRef]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of Genomic Loss of Heterozygosity Predict Homologous Recombination Repair Defects in Epithelial Ovarian Cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric Allelic Imbalance Indicates Defective DNA Repair and Sensitivity to DNA-Damaging Agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popova, T.; Manie, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and Large-Scale Genomic Instability Consistently Identify Basal-like Breast Carcinomas with BRCA1/2 Inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 Defects with Genomic Scores Predictive of DNA Damage Repair Deficiency among Breast Cancer Subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef] [Green Version]
- Setton, J.; Zinda, M.; Riaz, N.; Durocher, D.; Zimmermann, M.; Koehler, M.; Reis-Filho, J.S.; Powell, S.N. Synthetic Lethality in Cancer Therapeutics: The Next Generation. Cancer Discov. 2021, 11, 1626–1635. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Plummer, E.R.; Elattar, A.; Soohoo, S.; Uzir, B.; Quinn, J.E.; McCluggage, W.G.; Maxwell, P.; Aneke, H.; Curtin, N.J.; et al. Clinicopathological Features of Homologous Recombination-Deficient Epithelial Ovarian Cancers: Sensitivity to PARP Inhibitors, Platinum, and Survival. Cancer Res. 2012, 72, 5675–5682. [Google Scholar] [CrossRef] [Green Version]
- Milanesio, M.C.; Giordano, S.; Valabrega, G. Clinical Implications of DNA Repair Defects in High-Grade Serous Ovarian Carcinomas. Cancers 2020, 12, 1315. [Google Scholar] [CrossRef]
- List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools). Available online: https://www.fda.gov/medical-devices/in-vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-in-vitro-and-imaging-tools (accessed on 12 December 2021).
- BRACAnalysis CDx® Technical Information. Available online: https://s3.amazonaws.com/myriad-web/BRACAnalysisCDxTS.pdf (accessed on 1 October 2021).
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO Recommendations on Predictive Biomarker Testing for Homologous Recombination Deficiency and PARP Inhibitor Benefit in Ovarian Cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef]
- Haunschild, C.E.; Tewari, K.S. The Current Landscape of Molecular Profiling in the Treatment of Epithelial Ovarian Cancer. Gynecol. Oncol. 2021, 160, 333–345. [Google Scholar] [CrossRef]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [Green Version]
- Ford, L.; Wolford, J.E.; Brown, S.M.; Randall, L.M. A Profile on the FoundationFocus CDxBRCA Tests. Expert Rev. Mol. Diagn. 2020, 20, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, R.; Li, M.; Hughes, J.; Delfosse, D.; Skoletsky, J.; Ma, P.; Meng, W.; Dewal, N.; Milbury, C.; Clark, T.; et al. Clinical and Analytical Validation of FoundationOne Liquid CDx, a Novel 324-Gene CfDNA-Based Comprehensive Genomic Profiling Assay for Cancers of Solid Tumor Origin. PLoS ONE 2020, 15, e0237802. [Google Scholar] [CrossRef] [PubMed]
- Tew, W.P.; Lacchetti, C.; Ellis, A.; Maxian, K.; Banerjee, S.; Bookman, M.; Jones, M.B.; Lee, J.M.; Lheureux, S.; Liu, J.F.; et al. PARP Inhibitors in the Management of Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 3468–3493. [Google Scholar] [CrossRef]
- Vergote, I.; González-Martín, A.; Ray-Coquard, I.; Harter, P.; Colombo, N.; Pujol, P.; Lorusso, D.; Mirza, M.R.; Brasiuniene, B.; Madry, R.; et al. European Experts Consensus: BRCA/Homologous Recombination Deficiency Testing in First-Line Ovarian Cancer. Ann. Oncol. 2021, 33, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific Killing of BRCA2-Deficient Tumours with Inhibitors of Poly(ADP-Ribose) Polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Banerjee, S.; Moore, K.N.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; et al. Maintenance Olaparib for Patients with Newly Diagnosed Advanced Ovarian Cancer and a BRCA Mutation (SOLO1/GOG 3004): 5-Year Follow-up of a Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2021, 22, 1721–1731. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Colombo, N.; Sessa, C.; du Bois, A.; Ledermann, J.; McCluggage, W.G.; McNeish, I.; Morice, P.; Pignata, S.; Ray-Coquard, I.; Vergote, I.; et al. ESMO-ESGO Consensus Conference Recommendations on Ovarian Cancer: Pathology and Molecular Biology, Early and Advanced Stages, Borderline Tumours and Recurrent Diseasedagger. Ann. Oncol. 2019, 30, 672–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pothuri, B.; O’Cearbhaill, R.; Eskander, R.; Armstrong, D. Frontline PARP Inhibitor Maintenance Therapy in Ovarian Cancer: A Society of Gynecologic Oncology Practice Statement. Gynecol. Oncol. 2020, 159, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Pignata, S.; Cecere, S.C.; Du Bois, A.; Harter, P.; Heitz, F. Treatment of Recurrent Ovarian Cancer. Ann. Oncol. 2017, 28, viii51–viii56. [Google Scholar] [CrossRef]
- Mirza, M.R.; Coleman, R.L.; González-Martín, A.; Moore, K.N.; Colombo, N.; Ray-Coquard, I.; Pignata, S. The Forefront of Ovarian Cancer Therapy: Update on PARP Inhibitors. Ann. Oncol. 2020, 31, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib Maintenance Treatment for Recurrent Ovarian Carcinoma after Response to Platinum Therapy (ARIEL3): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- FoundationOne®CDx—Summary of Safety and Effectiveness Data (SSED). Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf17/P170019S017B.pdf (accessed on 1 October 2021).
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib Tablets as Maintenance Therapy in Patients with Platinum-Sensitive, Relapsed Ovarian Cancer and a BRCA1/2 Mutation (SOLO2/ENGOT-Ov21): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [Green Version]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in Relapsed, Platinum-Sensitive High-Grade Ovarian Carcinoma (ARIEL2 Part 1): An International, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Balasubramaniam, S.; Beaver, J.A.; Horton, S.; Fernandes, L.L.; Tang, S.; Horne, H.N.; Liu, J.; Liu, C.; Schrieber, S.J.; Yu, J.; et al. FDA Approval Summary: Rucaparib for the Treatment of Patients with Deleterious BRCA Mutation-Associated Advanced Ovarian Cancer. Clin. Cancer Res. 2017, 23, 7165–7170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib Monotherapy for Late-Line Treatment of Ovarian Cancer (QUADRA): A Multicentre, Open-Label, Single-Arm, Phase 2 Trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Schwarz, R.F.; Ng, C.K.; Cooke, S.L.; Newman, S.; Temple, J.; Piskorz, A.M.; Gale, D.; Sayal, K.; Murtaza, M.; Baldwin, P.J.; et al. Spatial and Temporal Heterogeneity in High-Grade Serous Ovarian Cancer: A Phylogenetic Analysis. PLoS Med. 2015, 12, e1001789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Yoon, J.K.; Kim, B.; Kim, S.; Kim, M.A.; Lim, H.; Bang, D.; Song, Y.S. Tumor Evolution and Intratumor Heterogeneity of an Epithelial Ovarian Cancer Investigated Using Next-Generation Sequencing. BMC Cancer 2015, 15, 85. [Google Scholar] [CrossRef] [Green Version]
- Hunt, A.L.; Bateman, N.W.; Barakat, W.; Makohon-Moore, S.; Hood, B.L.; Conrads, K.A.; Zhou, M.; Calvert, V.; Pierobon, M.; Loffredo, J.; et al. Extensive Three-Dimensional Intratumor Proteomic Heterogeneity Revealed by Multiregion Sampling in High-Grade Serous Ovarian Tumor Specimens. iScience 2021, 24, 102757. [Google Scholar] [CrossRef]
- Matei, D.; Nephew, K.P. Epigenetic Attire in Ovarian Cancer: The Emperor’s New Clothes. Cancer Res. 2020, 80, 3775–3785. [Google Scholar] [CrossRef]
- Barnes, D.R.; Rookus, M.A.; McGuffog, L.; Leslie, G.; Mooij, T.M.; Dennis, J.; Mavaddat, N.; Adlard, J.; Ahmed, M.; Aittomaki, K.; et al. Polygenic Risk Scores and Breast and Epithelial Ovarian Cancer Risks for Carriers of BRCA1 and BRCA2 Pathogenic Variants. Genet. Med. 2020, 22, 1653–1666. [Google Scholar] [CrossRef]
- Turinetto, M.; Scotto, G.; Tuninetti, V.; Giannone, G.; Valabrega, G. The Role of PARP Inhibitors in the Ovarian Cancer Microenvironment: Moving Forward From Synthetic Lethality. Front. Oncol. 2021, 11, 689829. [Google Scholar] [CrossRef]
- Lecuelle, J.; Boidot, R.; Mananet, H.; Derangere, V.; Albuisson, J.; Goussot, V.; Arnould, L.; Tharin, Z.; Ray Coquard, I.; Ghiringhelli, F.; et al. TCR Clonality and Genomic Instability Signatures as Prognostic Biomarkers in High Grade Serous Ovarian Cancer. Cancers 2021, 13, 4394. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Olivier, T.; Rodrigues, M.; Ferraioli, D.; Derbel, O.; Bodmer, A.; Petignat, P.; Rak, B.; Chopin, N.; Tredan, O.; et al. Location of Mutation in BRCA2 Gene and Survival in Patients with Ovarian Cancer. Clin. Cancer Res. 2018, 24, 326–333. [Google Scholar] [CrossRef] [Green Version]
- Lheureux, S.; Lai, Z.; Dougherty, B.A.; Runswick, S.; Hodgson, D.R.; Timms, K.M.; Lanchbury, J.S.; Kaye, S.; Gourley, C.; Bowtell, D.; et al. Long-Term Responders on Olaparib Maintenance in High-Grade Serous Ovarian Cancer: Clinical and Molecular Characterization. Clin. Cancer Res. 2017, 23, 4086–4094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngoi, N.Y.L.; Tan, D.S.P. The Role of Homologous Recombination Deficiency Testing in Ovarian Cancer and Its Clinical Implications: Do We Need It? ESMO Open 2021, 6, 100144. [Google Scholar] [CrossRef] [PubMed]
- Washington, C.R.; Moore, K.N. PARP Inhibitors in the Treatment of Ovarian Cancer: A Review. Curr. Opin. Obstet. Gynecol. 2021, 33, 1–6. [Google Scholar] [CrossRef]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demeny, M.A.; Virag, L. The PARP Enzyme Family and the Hallmarks of Cancer Part 2: Hallmarks Related to Cancer Host Interactions. Cancers 2021, 13, 2057. [Google Scholar] [CrossRef]
- Nath, A.; Cosgrove, P.A.; Mirsafian, H.; Christie, E.L.; Pflieger, L.; Copeland, B.; Majumdar, S.; Cristea, M.C.; Han, E.S.; Lee, S.J.; et al. Evolution of Core Archetypal Phenotypes in Progressive High Grade Serous Ovarian Cancer. Nat. Commun. 2021, 12, 3039. [Google Scholar] [CrossRef]
- Masoodi, T.; Siraj, S.; Siraj, A.K.; Azam, S.; Qadri, Z.; Parvathareddy, S.K.; Tulbah, A.; Al-Dayel, F.; AlHusaini, H.; AlOmar, O.; et al. Genetic Heterogeneity and Evolutionary History of High-Grade Ovarian Carcinoma and Matched Distant Metastases. Br. J. Cancer 2020, 122, 1219–1230. [Google Scholar] [CrossRef] [Green Version]
- McPherson, A.; Roth, A.; Laks, E.; Masud, T.; Bashashati, A.; Zhang, A.W.; Ha, G.; Biele, J.; Yap, D.; Wan, A.; et al. Divergent Modes of Clonal Spread and Intraperitoneal Mixing in High-Grade Serous Ovarian Cancer. Nat. Genet. 2016, 48, 758–767. [Google Scholar] [CrossRef]
- Lambrechts, S.; Smeets, D.; Moisse, M.; Braicu, E.I.; Vanderstichele, A.; Zhao, H.; Van Nieuwenhuysen, E.; Berns, E.; Sehouli, J.; Zeillinger, R.; et al. Genetic Heterogeneity after First-Line Chemotherapy in High-Grade Serous Ovarian Cancer. Eur. J. Cancer 2016, 53, 51–64. [Google Scholar] [CrossRef]
- Bashashati, A.; Ha, G.; Tone, A.; Ding, J.; Prentice, L.M.; Roth, A.; Rosner, J.; Shumansky, K.; Kalloger, S.; Senz, J.; et al. Distinct Evolutionary Trajectories of Primary High-Grade Serous Ovarian Cancers Revealed through Spatial Mutational Profiling. J. Pathol. 2013, 231, 21–34. [Google Scholar] [CrossRef]
- Cooke, S.L.; Ng, C.K.; Melnyk, N.; Garcia, M.J.; Hardcastle, T.; Temple, J.; Langdon, S.; Huntsman, D.; Brenton, J.D. Genomic Analysis of Genetic Heterogeneity and Evolution in High-Grade Serous Ovarian Carcinoma. Oncogene 2010, 29, 4905–4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klotz, D.M.; Wimberger, P. Overcoming PARP Inhibitor Resistance in Ovarian Cancer: What Are the Most Promising Strategies? Arch. Gynecol. Obstet. 2020, 302, 1087–1102. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Z.Y.; Wu, N.; Chen, Y.C.; Cheng, Q.; Wang, J. PARP Inhibitor Resistance: The Underlying Mechanisms and Clinical Implications. Mol. Cancer 2020, 19, 107. [Google Scholar] [CrossRef] [PubMed]
- Tobalina, L.; Armenia, J.; Irving, E.; O’Connor, M.J.; Forment, J.V. A Meta-Analysis of Reversion Mutations in BRCA Genes Identifies Signatures of DNA End-Joining Repair Mechanisms Driving Therapy Resistance. Ann. Oncol. 2021, 32, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Mohyuddin, G.R.; Aziz, M.; Britt, A.; Wade, L.; Sun, W.; Baranda, J.; Al-Rajabi, R.; Saeed, A.; Kasi, A. Similar Response Rates and Survival with PARP Inhibitors for Patients with Solid Tumors Harboring Somatic versus Germline BRCA Mutations: A Meta-Analysis and Systematic Review. BMC Cancer 2020, 20, 507. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-Ribose Polymerase Inhibition: Frequent Durable Responses in BRCA Carrier Ovarian Cancer Correlating with Platinum-Free Interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral Poly(ADP-Ribose) Polymerase Inhibitor Olaparib in Patients with BRCA1 or BRCA2 Mutations and Recurrent Ovarian Cancer: A Proof-of-Concept Trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Belotserkovskaya, R.; Raga Gil, E.; Lawrence, N.; Butler, R.; Clifford, G.; Wilson, M.D.; Jackson, S.P. PALB2 Chromatin Recruitment Restores Homologous Recombination in BRCA1-Deficient Cells Depleted of 53BP1. Nat. Commun. 2020, 11, 819. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 Counteracts DNA Double-Strand Break Resection and Affects PARP Inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Drean, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-Wide and High-Density CRISPR-Cas9 Screens Identify Point Mutations in PARP1 Causing PARP Inhibitor Resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Meghani, K.; Fuchs, W.; Detappe, A.; Drane, P.; Gogola, E.; Rottenberg, S.; Jonkers, J.; Matulonis, U.; Swisher, E.M.; Konstantinopoulos, P.A.; et al. Multifaceted Impact of MicroRNA 493-5p on Genome-Stabilizing Pathways Induces Platinum and PARP Inhibitor Resistance in BRCA2-Mutated Carcinomas. Cell Rep. 2018, 23, 100–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, D.; Stenzinger, A. Homologous Recombination Repair Deficiency (HRD): From Biology to Clinical Exploitation. Genes Chromosomes Cancer 2021, 60, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; O’Connor, K.W.; Mouw, K.W.; Li, A.Y.; Matulonis, U.A.; D’Andrea, A.D.; Konstantinopoulos, P.A. A Unique Subset of Epithelial Ovarian Cancers with Platinum Sensitivity and PARP Inhibitor Resistance. Cancer Res. 2015, 75, 628–634. [Google Scholar] [CrossRef] [Green Version]
- McCormick, A.; Donoghue, P.; Dixon, M.; O’Sullivan, R.; O’Donnell, R.L.; Murray, J.; Kaufmann, A.; Curtin, N.J.; Edmondson, R.J. Ovarian Cancers Harbor Defects in Nonhomologous End Joining Resulting in Resistance to Rucaparib. Clin. Cancer Res. 2017, 23, 2050–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderstichele, A.; Busschaert, P.; Olbrecht, S.; Lambrechts, D.; Vergote, I. Genomic Signatures as Predictive Biomarkers of Homologous Recombination Deficiency in Ovarian Cancer. Eur. J. Cancer 2017, 86, 5–14. [Google Scholar] [CrossRef]
- Lee, C.K.; Simes, R.J.; Brown, C.; Lord, S.; Wagner, U.; Plante, M.; Vergote, I.; Pisano, C.; Parma, G.; Burges, A.; et al. Prognostic Nomogram to Predict Progression-Free Survival in Patients with Platinum-Sensitive Recurrent Ovarian Cancer. Br. J. Cancer 2011, 105, 1144–1150. [Google Scholar] [CrossRef] [Green Version]
- Previs, R.A.; Bevis, K.S.; Huh, W.; Tillmanns, T.; Perry, L.; Moore, K.; Chapman, J.; McClung, C.; Kiet, T.; Java, J.; et al. A Prognostic Nomogram to Predict Overall Survival in Women with Recurrent Ovarian Cancer Treated with Bevacizumab and Chemotherapy. Gynecol. Oncol. 2014, 132, 531–536. [Google Scholar] [CrossRef]
- Paik, E.S.; Sohn, I.; Baek, S.Y.; Shim, M.; Choi, H.J.; Kim, T.J.; Choi, C.H.; Lee, J.W.; Kim, B.G.; Lee, Y.Y.; et al. Nomograms Predicting Platinum Sensitivity, Progression-Free Survival, and Overall Survival Using Pretreatment Complete Blood Cell Counts in Epithelial Ovarian Cancer. Cancer Res. Treat. 2017, 49, 635–642. [Google Scholar] [CrossRef] [Green Version]
- Rose, P.G.; Java, J.J.; Salani, R.; Geller, M.A.; Secord, A.A.; Tewari, K.S.; Bender, D.P.; Mutch, D.G.; Friedlander, M.L.; Van Le, L.; et al. Nomogram for Predicting Individual Survival After Recurrence of Advanced-Stage, High-Grade Ovarian Carcinoma. Obstet. Gynecol. 2019, 133, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Song, M.; Hwangbo, S.; Lee, S.; Cho, U.; Kim, J.H.; Lee, M.; Kim, H.S.; Chung, H.H.; Suh, D.S.; et al. Development of Web-Based Nomograms to Predict Treatment Response and Prognosis of Epithelial Ovarian Cancer. Cancer Res. Treat. 2019, 51, 1144–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjokrowidjaja, A.; Friedlander, M.; Lord, S.J.; Asher, R.; Rodrigues, M.; Ledermann, J.A.; Matulonis, U.A.; Oza, A.M.; Bruchim, I.; Huzarski, T.; et al. Prognostic Nomogram for Progression-Free Survival in Patients with BRCA Mutations and Platinum-Sensitive Recurrent Ovarian Cancer on Maintenance Olaparib Therapy Following Response to Chemotherapy. Eur. J. Cancer 2021, 154, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Leffers, N.; Gooden, M.J.; de Jong, R.A.; Hoogeboom, B.N.; ten Hoor, K.A.; Hollema, H.; Boezen, H.M.; van der Zee, A.G.; Daemen, T.; Nijman, H.W. Prognostic Significance of Tumor-Infiltrating T-Lymphocytes in Primary and Metastatic Lesions of Advanced Stage Ovarian Cancer. Cancer Immunol. Immunother. 2009, 58, 449–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, J.; Yu, H.; Zhang, T.; An, R.; Xue, Y. Prognostic Impact of Tumor-Infiltrating Lymphocytes in High Grade Serous Ovarian Cancer: A Systematic Review and Meta-Analysis. Ther. Adv. Med. Oncol. 2020, 12, 1758835920967241. [Google Scholar] [CrossRef]
- Laumont, C.M.; Wouters, M.C.A.; Smazynski, J.; Gierc, N.S.; Chavez, E.A.; Chong, L.C.; Thornton, S.; Milne, K.; Webb, J.R.; Steidl, C.; et al. Single-Cell Profiles and Prognostic Impact of Tumor-Infiltrating Lymphocytes Coexpressing CD39, CD103, and PD-1 in Ovarian Cancer. Clin. Cancer Res. 2021, 27, 4089–4100. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, J.N.; Porter, H.; Kobel, M.; Nelson, B.H.; Prentice, L.M.; Kalloger, S.E.; Senz, J.; Milne, K.; Ding, J.; Shah, S.P.; et al. BRCA1 and BRCA2 Mutations Correlate with TP53 Abnormalities and Presence of Immune Cell Infiltrates in Ovarian High-Grade Serous Carcinoma. Mod. Pathol. 2012, 25, 740–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwangbo, S.; Kim, S.I.; Kim, J.H.; Eoh, K.J.; Lee, C.; Kim, Y.T.; Suh, D.S.; Park, T.; Song, Y.S. Development of Machine Learning Models to Predict Platinum Sensitivity of High-Grade Serous Ovarian Carcinoma. Cancers 2021, 13, 1875. [Google Scholar] [CrossRef]
- Nougaret, S.; McCague, C.; Tibermacine, H.; Vargas, H.A.; Rizzo, S.; Sala, E. Radiomics and Radiogenomics in Ovarian Cancer: A Literature Review. Abdom. Radiol. 2021, 46, 2308–2322. [Google Scholar] [CrossRef]
- Pujol, P.; Barberis, M.; Beer, P.; Friedman, E.; Piulats, J.M.; Capoluongo, E.D.; Garcia Foncillas, J.; Ray-Coquard, I.; Penault-Llorca, F.; Foulkes, W.D.; et al. Clinical Practice Guidelines for BRCA1 and BRCA2 Genetic Testing. Eur. J. Cancer 2021, 146, 30–47. [Google Scholar] [CrossRef]
- How, J.A.; Jazaeri, A.A.; Fellman, B.; Daniels, M.S.; Penn, S.; Solimeno, C.; Yuan, Y.; Schmeler, K.; Lanchbury, J.S.; Timms, K.; et al. Modification of Homologous Recombination Deficiency Score Threshold and Association with Long-Term Survival in Epithelial Ovarian Cancer. Cancers 2021, 13, 946. [Google Scholar] [CrossRef] [PubMed]
- Stronach, E.A.; Paul, J.; Timms, K.M.; Hughes, E.; Brown, K.; Neff, C.; Perry, M.; Gutin, A.; El-Bahrawy, M.; Steel, J.H.; et al. Biomarker Assessment of HR Deficiency, Tumor BRCA1/2 Mutations, and CCNE1 Copy Number in Ovarian Cancer: Associations with Clinical Outcome Following Platinum Monotherapy. Mol. Cancer Res. 2018, 16, 1103–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorski, J.W.; Ueland, F.R.; Kolesar, J.M. CCNE1 Amplification as a Predictive Biomarker of Chemotherapy Resistance in Epithelial Ovarian Cancer. Diagnostics 2020, 10, 279. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, X.; Gao, Y.; Shang, C.; Yu, B.; Wang, T.; Su, J.; Huang, C.; Wu, Y.; Guo, H.; et al. Development of a Genomic Signatures-Based Predictor of Initial Platinum-Resistance in Advanced High-Grade Serous Ovarian Cancer Patients. Front. Oncol. 2020, 10, 625866. [Google Scholar] [CrossRef] [PubMed]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutierrez-Enriquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzman, M.; et al. A RAD51 Assay Feasible in Routine Tumor Samples Calls PARP Inhibitor Response beyond BRCA Mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef]
- Ni, J.; Cheng, X.; Zhou, R.; Zhao, Q.; Xu, X.; Guo, W.; Gu, H.; Chen, C.; Chen, X. Adverse Events as a Potential Clinical Marker of Antitumor Efficacy in Ovarian Cancer Patients Treated With Poly ADP-Ribose Polymerase Inhibitor. Front. Oncol. 2021, 11, 724620. [Google Scholar] [CrossRef]
- Vanacker, H.; Harter, P.; Labidi-Galy, S.I.; Banerjee, S.; Oaknin, A.; Lorusso, D.; Ray-Coquard, I. PARP-Inhibitors in Epithelial Ovarian Cancer: Actual Positioning and Future Expectations. Cancer Treat. Rev. 2021, 99, 102255. [Google Scholar] [CrossRef]

{kind=link}
| Molecular Status 1 | Advanced Epithelial Ovarian Cancer in Complete/Partial Response to Platinum First-Line Maintenance Monotherapy 2 | Recurrent Epithelial Ovarian Cancer in Complete/Partial Response to Platinum Second-Line Maintenance Monotherapy 2 | Recurrent Epithelial Ovarian Cancer ≥Third-Line (3L) Monotherapy |
|---|---|---|---|
| gBRCA* | Olaparib (FDA 2018/EMA 2019; SOLO-1)—BA-CDx | Olaparib after ≥3L—regardless of platinum sensitivity 3 (FDA 2014; STUDY-42)—BA-CDx | |
| tBRCA* | Olaparib (FDA 2018/EMA 2019; SOLO-1)—F1-CDx | Rucaparib after ≥2L—regardless of platinum sensitivity 4 (FDA 2016/EMA 2019; ARIEL-2/STUDY-10)—F1-CDx | |
| HRD+ | Olaparib + bevacizumab (FDA 2020/EMA 2020; PAOLA-1)—MC-CDx | Niraparib after ≥3L—potentially platinum sensitive 5 (FDA 2019; QUADRA)—MC-CDx | |
| Biomarker agnostic | Niraparib (FDA 2020/EMA 2020; PRIMA) | Niraparib (FDA 2017/EMA 2017; NOVA) 6 Olaparib (FDA 2017/EMA 2018; SOLO-2/STUDY-19) Rucaparib (FDA 2018/EMA 2019; ARIEL-3)—F1-CDx 7 |
| General Consideration | Distinct CDx Are Not Interchangeable |
|---|---|
| Preanalytical considerations | ≈5–10% of specimens are inadequate Sample heterogeneity:
|
| Analytical considerations | Limit of detection (BRCA1/2):
|
| Post-analytical considerations | ≈5–10% of results are inconclusive GIS positivity thresholds:
|
| General Consideration | Cost and Access to HRD Assays Currently Restricted to Private Companies |
|---|---|
| Tissue heterogeneity of HGSOC | Sample heterogeneity:
Cellular microenvironment effect |
| Relevance as a biomarker (PARPi sensitivity) | Patient selection and clinical context (e.g., platinum-sensitivity status) Timing of analysis Limited predictivity of GIS:
|
| Evolutionary perspective of HGSOC | PARPi resistance: reverse mutations, HRD-unrelated mechanisms Genomic scars are irreversible Iterative analysis and PARPi rechallenge |
| Advanced Epithelial Ovarian Cancer —First-Line Maintenance | Recurrent Epithelial Ovarian Cancer | |
|---|---|---|
| Shown to be associated with PARPi sensitivity | Molecular assays (e.g., HRDetect, RAD51C methylation, gene amplification, SVs, non-coding RNAs, transcriptomics, proteomics) Functional assays (e.g., RAD51 foci) | Reverse mutations (e.g., in BRCA1/2) Nomogram |
| Research and future strategies | Stepwise approach (i.e., integrate other biomarkers in inconclusive cases) Deeper refinement of HRD status (e.g., type of tBRCA*, GIS thresholds, tumor heterogeneity) Comprehensive PARPi sensitivity score Integrating clinical and biopathological data Through-treatment dynamic markers | More accurate and comprehensive evaluation, such as:
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quesada, S.; Fabbro, M.; Solassol, J. Toward More Comprehensive Homologous Recombination Deficiency Assays in Ovarian Cancer Part 2: Medical Perspectives. Cancers 2022, 14, 1098. https://doi.org/10.3390/cancers14041098
Quesada S, Fabbro M, Solassol J. Toward More Comprehensive Homologous Recombination Deficiency Assays in Ovarian Cancer Part 2: Medical Perspectives. Cancers. 2022; 14(4):1098. https://doi.org/10.3390/cancers14041098
Chicago/Turabian StyleQuesada, Stanislas, Michel Fabbro, and Jérôme Solassol. 2022. "Toward More Comprehensive Homologous Recombination Deficiency Assays in Ovarian Cancer Part 2: Medical Perspectives" Cancers 14, no. 4: 1098. https://doi.org/10.3390/cancers14041098
APA StyleQuesada, S., Fabbro, M., & Solassol, J. (2022). Toward More Comprehensive Homologous Recombination Deficiency Assays in Ovarian Cancer Part 2: Medical Perspectives. Cancers, 14(4), 1098. https://doi.org/10.3390/cancers14041098