
Constitutive Activation of p62/Sequestosome-1-Mediated Proteaphagy Regulates Proteolysis and Impairs Cell Death in Bortezomib-Resistant Mantle Cell Lymphoma

 , , ,
, , ,  , ,
, ,  ,
,  and
and
Abstract
:Simple Summary
Abstract
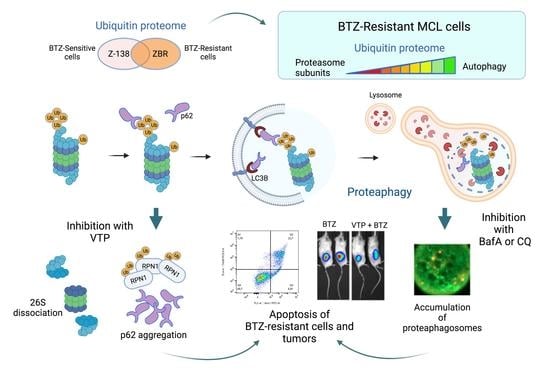
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Reduction of UPS Is Compensated by ALS Factors in BTZ-Resistant MCL Cells
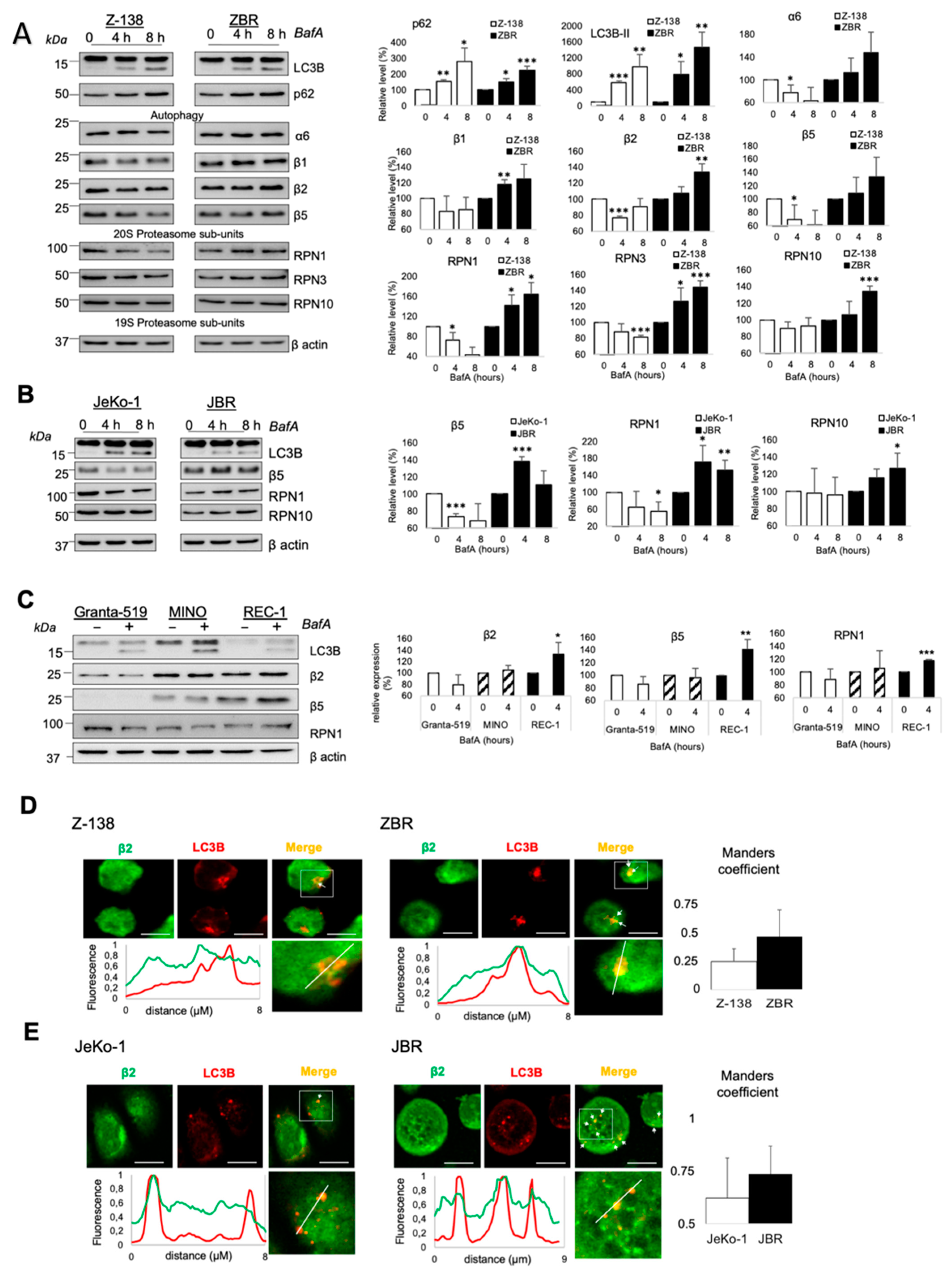
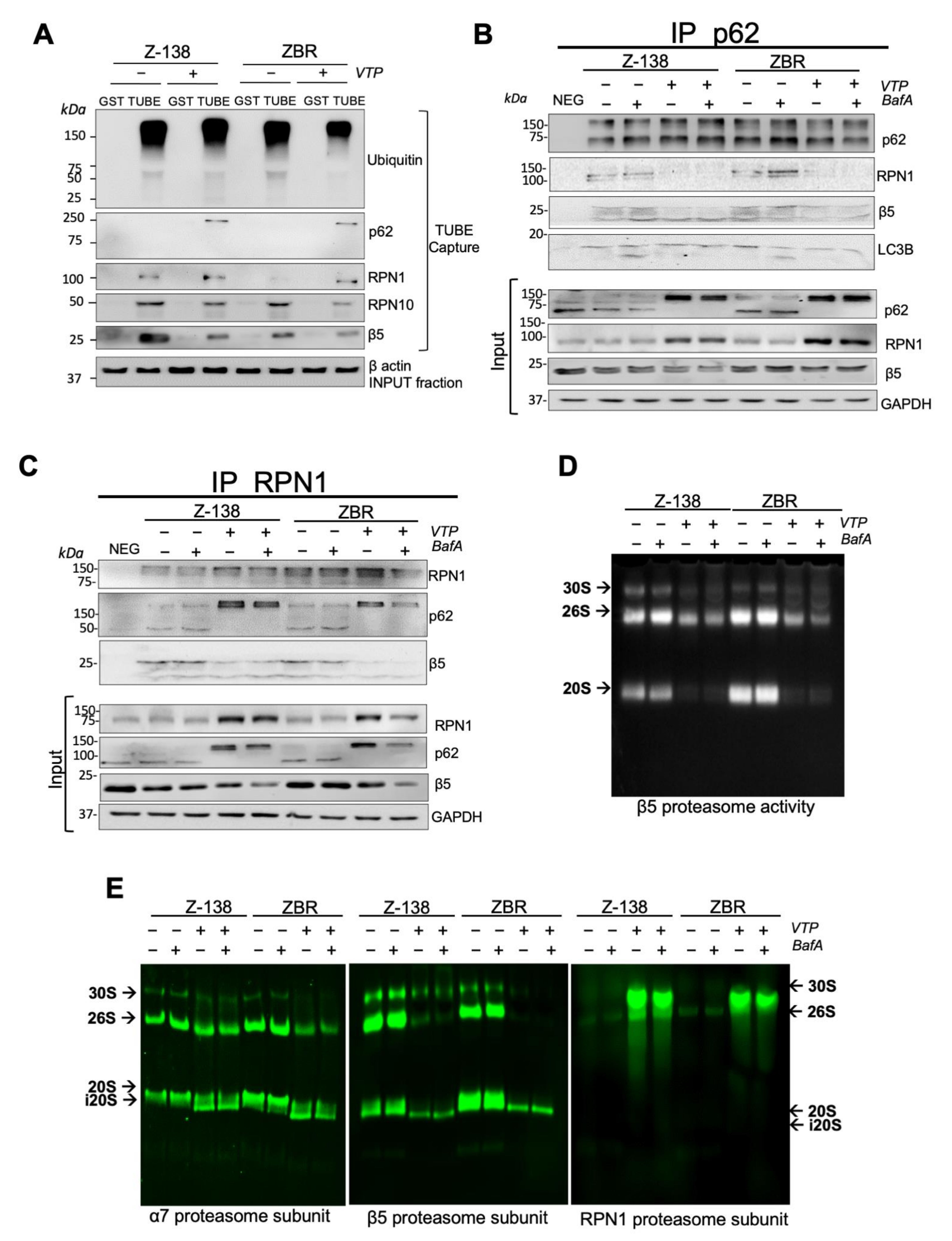
2.2. Proteaphagy Is Constitutively Active in BTZ-Resistant MCL Cells
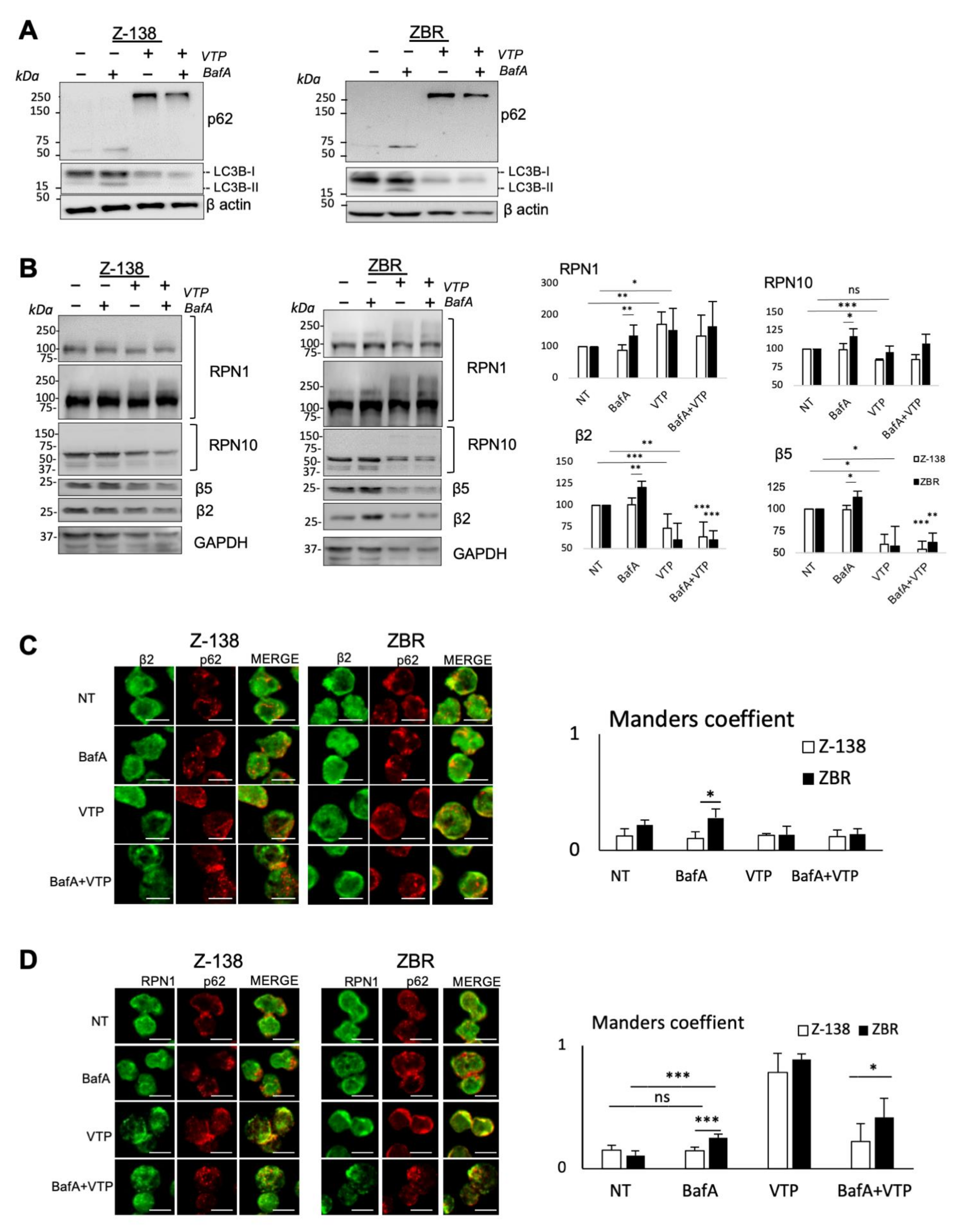
2.3. p62/Sequestosome-1 Coordinates Proteaphagosome Assembly
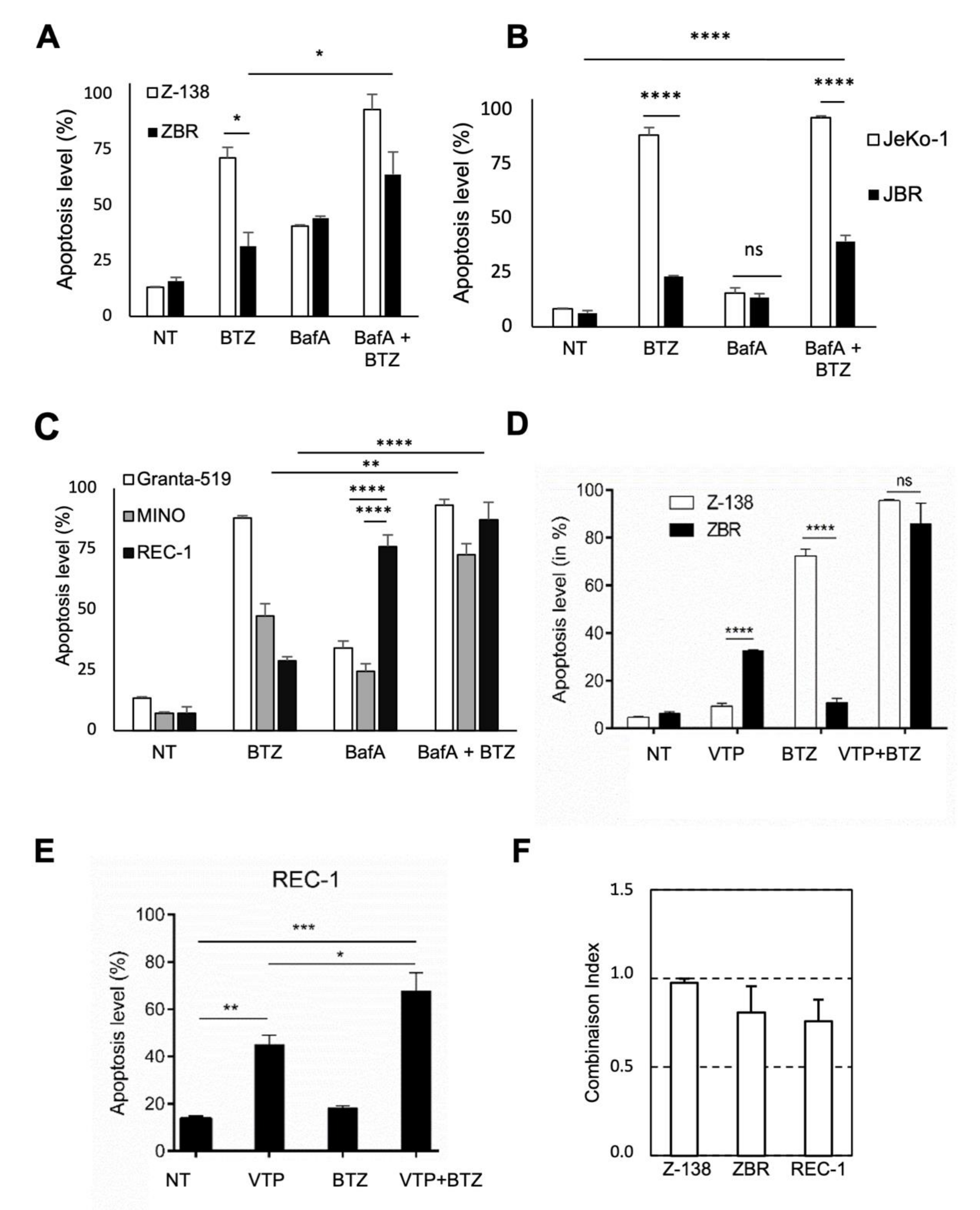
2.4. Autophagy Inhibition Increases Apoptosis in BTZ-Resistant MCL Cells
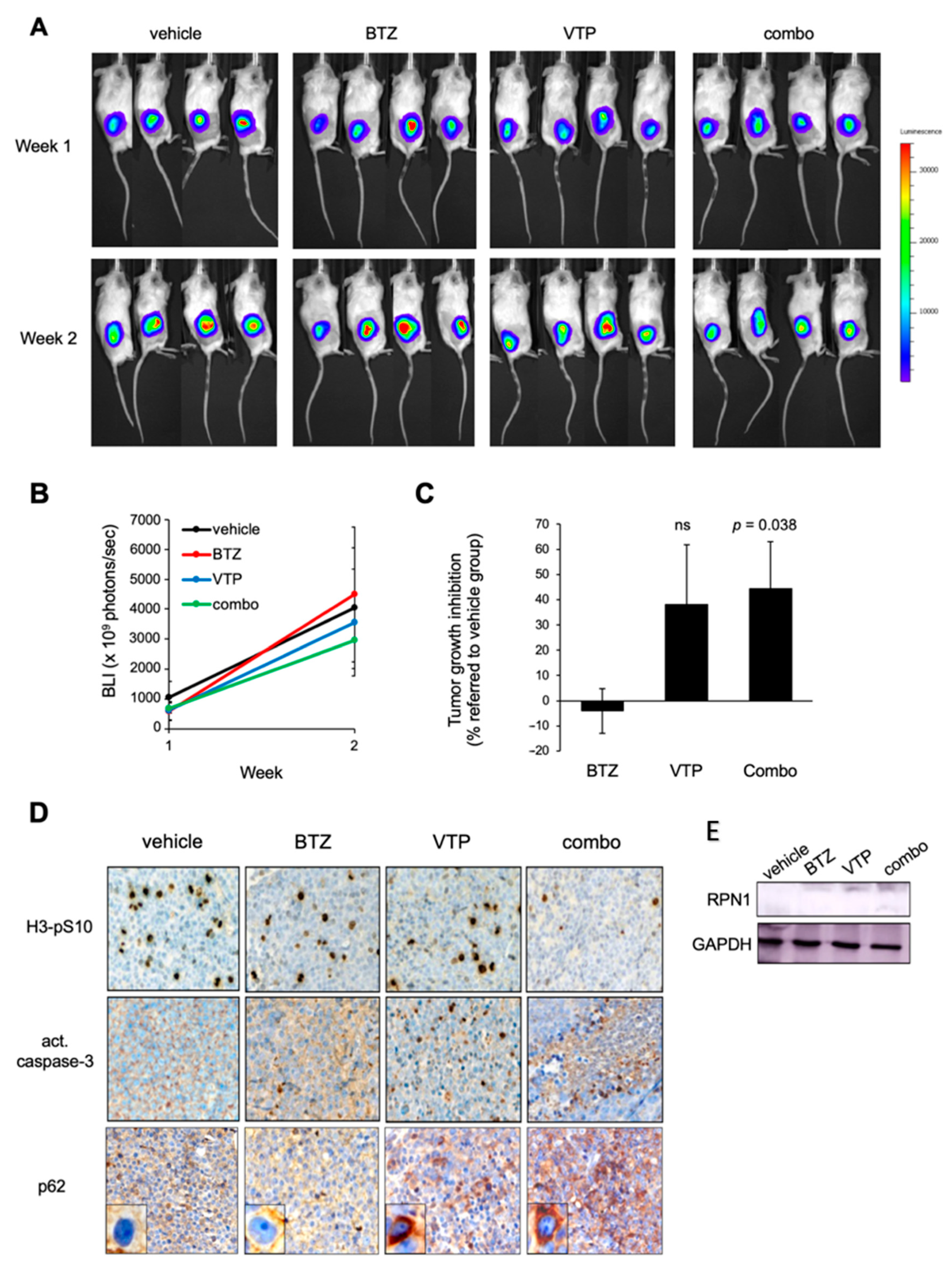
2.5. Inhibition of p62 Reduces the Growth of BTZ-Resistant MCL Tumours In Vivo
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Reagents
4.3. Cell Culture
4.4. TUBEs Capture
4.5. Mass Spectrometry Analysis
4.6. IPA Analysis
4.7. Database Search
4.8. Bioinformatics Analysis
4.9. Western Blotting
4.10. Immunoprecipitation
4.11. Immunofluorescence Microscopy
4.12. Native Gel Electrophoresis and In Gel Proteasomal Activity Assay
4.13. Flow Cytometry
4.14. In Vivo Experiments
4.15. Quantification and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Epperla, N.; Hamadani, M.; Fenske, T.S.; Costa, L.J. Incidence and survival trends in mantle cell lymphoma. Br. J. Haematol. 2018, 181, 703–706. [Google Scholar] [CrossRef] [Green Version]
- Rule, S. The modern approach to mantle cell lymphoma. Hematol. Oncol. 2019, 37, 66–69. [Google Scholar] [CrossRef] [Green Version]
- Diamond, B.; Kumar, A. Mantle Cell lymphoma: Current and emerging treatment strategies and unanswered questions. Hematol. Oncol. Clin. N. Am. 2019, 33, 613–626. [Google Scholar] [CrossRef]
- Roué, G.; Sola, B. Management of drug resistance in mantle cell lymphoma. Cancers 2020, 12, 1565. [Google Scholar] [CrossRef]
- Hambley, B.; Caimi, P.F.; William, B.M. Bortezomib for the treatment of mantle cell lymphoma: An update. Ther. Adv. Hematol. 2016, 7, 196–208. [Google Scholar] [CrossRef]
- Niewerth, D.; Jansen, G.; Assaraf, Y.G.; Zweegman, S.; Kaspers, G.J.L.; Cloos, J. Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist. Updat. 2015, 18, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Santamarta, M.; Quinet, G.; Reyes-Garau, D.; Sola, B.; Roué, G.; Rodriguez, M.S. Resistance to the proteasome inhibitors: Lessons from multiple myeloma and mantle cell lymphoma. Adv. Exp. Med. Biol. 2020, 1233, 153–174. [Google Scholar] [PubMed]
- Barrio, S.; Stühmer, T.; Da-Viá, M.; Barrio-Garcia, C.; Lehners, N.; Besse, A.; Cuenca, I.; Garitano-Trojaola, A.; Fink, S.; Leich, E.; et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia 2019, 33, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Weniger, M.A.; Rizzatti, E.G.; Pérez-Galán, P.; Liu, D.; Wang, Q.; Munson, P.J.; Raghavachari, N.; White, T.; Tweito, M.M.; Dunleavy, K.; et al. Treatment-induced oxidative stress and cellular antioxidant capacity determine response to bortezomib in mantle cell lymphoma. Clin. Cancer Res. 2011, 17, 5101–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roué, G.; Pérez-Galán, P.; Mozos, A.; López-Guerra, M.; Xargay-Torrent, S.; Rosich, L.; Saborit-Villarroya, I.; Normant, E.; Campo, E.; Colomer, D. The Hsp90 inhibitor IPI-504 overcomes bortezomib resistance in mantle cell lymphoma in vitro and in vivo by down-regulation of the prosurvival ER chaperone BiP/Grp78. Blood 2011, 117, 1270–1279. [Google Scholar] [CrossRef] [Green Version]
- Moros, A.; Rodríguez, V.; Saborit-Villarroya, I.; Montraveta, A.; Balsas, P.; Sandy, P.; Martínez, A.; Wiestner, A.; Normant, E.; Campo, E.; et al. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia 2014, 28, 2049–2059. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.T.; Ciechanover, A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Collins, G.A.; Goldberg, A.L. The logic of the 26S proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coux, O.; Zieba, B.A.; Meiners, S. The proteasome system in health and disease. Adv. Exp. Med. Biol. 2020, 1233, 55–100. [Google Scholar] [PubMed]
- Marshall, R.S.; Vierstra, R.D. Eat or be eaten: The autophagic plight of inactive 26S proteasomes. Autophagy 2015, 11, 1927–1928. [Google Scholar] [CrossRef] [Green Version]
- Marshall, R.S.; McLoughlin, F.; Vierstra, R.D. Autophagic turnover of inactive 26S proteasomes in yeast is directed by the ubiquitin receptor Cue5 and the Hsp42 chaperone. Cell Rep. 2016, 16, 1717–1732. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Fabre, B.; Ziv, T.; Kwon, Y.T.; Ciechanover, A. p62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, E7490–E7499. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Kaplan, V.; Ciechanover, A.; Livneh, I. Stress-induced polyubiquitination of proteasomal ubiquitin receptors targets the proteolytic complex for autophagic degradation. Autophagy 2017, 13, 759–760. [Google Scholar] [CrossRef] [Green Version]
- Quinet, G.; Gonzalez-Santamarta, M.; Louche, C.; Rodriguez, M.S. Mechanisms regulating the UPS-ALS crosstalk: The role of proteaphagy. Molecules 2020, 25, 2352. [Google Scholar] [CrossRef]
- Aillet, F.; Lopitz-Otsoa, F.; Hjerpe, R.; Torres-Ramos, M.; Lang, V.; Rodríguez, M.S. Isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. Methods Mol. Biol. 2012, 832, 173–183. [Google Scholar] [PubMed]
- Hjerpe, R.; Aillet, F.; Lopitz-Otsoa, F.; Lang, V.; England, P.; Rodriguez, M.S. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 2009, 10, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattern, M.; Sutherland, J.; Kadimisetty, K.; Barrio, R.; Rodriguez, M.S. Using ubiquitin binders to decipher the ubiquitin code. Trends Biochem. Sci. 2019, 44, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Accardi, F.; Toscani, D.; Bolzoni, M.; Dalla Palma, B.; Aversa, F.; Giuliani, N. Mechanism of action of bortezomib and the new proteasome inhibitors on myeloma cells and the bone microenvironment: Impact on myeloma-induced alterations of bone remodeling. Biomed. Res. Int. 2015, 2015, 172458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mata-Cantero, L.; Azkargorta, M.; Aillet, F.; Xolalpa, W.; LaFuente, M.J.; Elortza, F.; Carvalho, A.S.; Martin-Plaza, J.; Matthiesen, R.; Rodriguez, M.S. New insights into host-parasite ubiquitin proteome dynamics in P. falciparum infected red blood cells using a TUBEs-MS approach. J. Proteom. 2016, 139, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Lopitz-Otsoa, F.; Rodriguez-Suarez, E.; Aillet, F.; Casado-Vela, J.; Lang, V.; Matthiesen, R.; Elortza, F.; Rodriguez, M.S. Integrative analysis of the ubiquitin proteome isolated using Tandem Ubiquitin Binding Entities (TUBEs). J. Proteom. 2012, 75, 2998–3014. [Google Scholar] [CrossRef] [PubMed]
- Xolalpa, W.; Mata-Cantero, L.; Aillet, F.; Rodriguez, M.S. Isolation of the Ubiquitin-Proteome from Tumor Cell Lines and Primary Cells Using TUBEs. Methods Mol. Biol. 2016, 1449, 161–175. [Google Scholar]
- Marshall, R.S.; Li, F.; Gemperline, D.C.; Book, A.J.; Vierstra, R.D. Autophagic degradation of the 26S proteasome is mediated by the dual ATG8/ubiquitin receptor RPN10 in Arabidopsis. Mol. Cell. 2015, 58, 1053–1066. [Google Scholar] [CrossRef] [Green Version]
- Milan, E.; Perini, T.; Resnati, M.; Orfanelli, U.; Oliva, L.; Raimondi, A.; Cascio, P.; Bachi, A.; Marcatti, M.; Ciceri, F.; et al. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy 2015, 11, 1161–1178. [Google Scholar] [CrossRef]
- Donohue, E.; Balgi, A.D.; Komatsu, M.; Roberge, M. Induction of covalently crosslinked p62 oligomers with reduced binding to polyubiquitinated proteins by the autophagy inhibitor verteporfin. PLoS ONE 2014, 9, e114964. [Google Scholar] [CrossRef]
- Donohue, E.; Tovey, A.; Vogl, A.W.; Arns, S.; Sternberg, E.; Young, R.N.; Roberge, M. Inhibition of autophagosome formation by the benzoporphyrin derivative verteporfin. J. Biol. Chem. 2011, 286, 7290–7300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Reyes, R.G.; Quinet, G.; Gonzalez-Santamarta, M.; Larrue, C.; Sarry, J.-E.; Rodriguez, M.S. Inhibition of the proteasome and proteaphagy enhances apoptosis in FLT3-ITD-driven acute myeloid leukemia. FEBS Open Bio 2021, 11, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Saini, H.; Sharma, H.; Mukherjee, S.; Chowdhury, S.; Chowdhury, R. Verteporfin disrupts multiple steps of autophagy and regulates p53 to sensitize osteosarcoma cells. Cancer Cell Int. 2021, 21, 52. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Goldberg, A.L. Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Meth Enzymol. 2005, 398, 364–378. [Google Scholar]
- Tsvetkov, P.; Mendillo, M.L.; Zhao, J.; Carette, J.E.; Merrill, P.H.; Cikes, D.; Varadarajan, M.; van Diemen, F.R.; Penninger, J.M.; Goldberg, A.L.; et al. Compromising the 19S proteasome complex protects cells from reduced flux through the proteasome. eLife 2015, 4, e08467. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Alvear, D.; Cho, M.Y.; Wild, T.; Buchholz, T.J.; Lerner, A.G.; Simakova, O.; Korde, N.; Landgren, O.; Maric, I.; Choudhary, C.; et al. Paradoxical resistance of multiple myeloma to proteasome inhibitors by decreased levels of 19S proteasomal subunits. eLife 2015, 4, e08153. [Google Scholar] [CrossRef]
- Beà, S.; Valdés-Mas, R.; Navarro, A.; Salaverria, I.; Martín-Garcia, D.; Jares, P.; Giné, E.; Pinyol, M.; Royo, C.; Nadeu, F.; et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18250–18255. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Konstantinou, E.K.; Notomi, S.; Kosmidou, C.; Brodowska, K.; Al-Moujahed, A.; Nicolaou, F.; Tsoka, P.; Gragoudas, E.; Miller, J.W.; Young, L.H.; et al. Verteporfin-induced formation of protein cross-linked oligomers and high molecular weight complexes is mediated by light and leads to cell toxicity. Sci. Rep. 2017, 7, 46581. [Google Scholar] [CrossRef]
- Grune, T.; Catalgol, B.; Licht, A.; Ermak, G.; Pickering, A.M.; Ngo, J.K.; Davies, K.J.A. HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radic. Biol. Med. 2011, 51, 1355–1364. [Google Scholar] [CrossRef] [Green Version]
- Riz, I.; Hawley, T.S.; Marsal, J.W.; Hawley, R.G. Noncanonical SQSTM1/p62-Nrf2 pathway activation mediates proteasome inhibitor resistance in multiple myeloma cells via redox, metabolic and translational reprogramming. Oncotarget 2016, 7, 66360–66385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamik, J.; Silbermann, R.; Marino, S.; Sun, Q.; Anderson, J.L.; Zhou, D.; Xie, X.-Q.; Roodman, G.D.; Galson, D.L. XRK3F2 inhibition of p62-ZZ Domain signaling rescues myeloma-induced GFI1-driven epigenetic repression of the Runx2 gene in pre-osteoblasts to overcome differentiation suppression. Front. Endocrinol. 2018, 9, 344. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Petrusca, D.N.; Silberman, R.; Toscani, D.; Anderson, J.L.; Giuliani, N.; Xie, X.-Q.; Noriyoshi Kurihara, D.D.S.; Roodman, G.D. Inhibition of p62-ZZ domain-mediated signaling overcomes bortezomib resistance in multiple myeloma cells independent of their p53 status. Blood 2017, 130 (Suppl. S1), 4421. [Google Scholar]
- Silbermann, R.; Zhou, D.; Teramachi, J.; Xie, X.-Q.; Roodman, G.D.; Kurihara, N. The p62-ZZ domain inhibitor XRK3F2 alters myeloma-induced suppression of osteoblast differentiation and is highly cytotoxic to myeloma cells in combination with bortezomib. Blood 2014, 124, 2083. [Google Scholar] [CrossRef]
- Azkargorta, M.; Escobes, I.; Elortza, F.; Matthiesen, R.; Rodríguez, M.S. TUBEs-mass spectrometry for identification and analysis of the ubiquitin-proteome. Methods Mol. Biol. 2016, 1449, 177–192. [Google Scholar]
- Carvalho, A.S.; Ribeiro, H.; Voabil, P.; Penque, D.; Jensen, O.N.; Molina, H.; Mattheisen, R. Global mass spectrometry and transcriptomics array based drug profiling provides novel insight into glucosamine induced endoplasmic reticulum stress. Mol. Cell. Proteom. 2014, 13, 3294–3307. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Bunkenborg, J.; García, G.E.; Paz, M.I.P.; Andersen, J.S.; Molina, H. The minotaur proteome: Avoiding cross-species identifications deriving from bovine serum in cell culture models. Proteomics 2010, 10, 3040–3044. [Google Scholar] [CrossRef]
- Matthiesen, R.; Prieto, G.; Amorim, A.; Aloria, K.; Fullaondo, A.; Carvalho, A.S.; Arizmendi, J.M. SIR: Deterministic protein inference from peptides assigned to MS data. J. Proteom. 2012, 75, 4176–4183. [Google Scholar] [CrossRef]
- GO.db. Bioconductor. 2018. Available online: http://bioconductor.org/packages/GO.db/ (accessed on 29 October 2018).
- VennDiagram: Generate High-Resolution Venn and Euler Plots Version 1.6.20 from CRAN. 2018. Available online: https://rdrr.io/cran/VennDiagram/ (accessed on 29 October 2018).
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Elsasser, S.; Schmidt, M.; Finley, D. Characterization of the proteasome using native gel electrophoresis. Methods Enzymol. 2005, 398, 353–363. [Google Scholar] [PubMed]
- Body, S.; Esteve-Arenys, A.; Recasens-Zorzo, C.; Troussard, X.; Roué, G.; Sola, B. A mouse model of disseminated mantle cell lymphoma highlights a lack of activity of estrogen receptor β agonists toward tumor burden. Leuk. Lymphoma 2018, 59, 1726–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteve-Arenys, A.; Valero, J.G.; Chamorro-Jorganes, A.; Gonzalez, D.; Rodriguez, V.; Dlouhy, I.; Salaverria, I.; Campo, E.; Colomer, D.; Martinez, A.; et al. The BET bromodomain inhibitor CPI203 overcomes resistance to ABT-199 (venetoclax) by downregulation of BFL-1/A1 in in vitro and in vivo models of MYC+/BCL2+ double hit lymphoma. Oncogene 2018, 37, 1830–1844. [Google Scholar] [CrossRef]
- Yong, K.; Gonzalez-McQuire, S.; Szabo, Z.; Schoen, P.; Hajek, R. The start of a new wave: Developments in proteasome inhibition in multiple myeloma. Eur. J. Haematol. 2018, 101, 220–236. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quinet, G.; Xolalpa, W.; Reyes-Garau, D.; Profitós-Pelejà, N.; Azkargorta, M.; Ceccato, L.; Gonzalez-Santamarta, M.; Marsal, M.; Andilla, J.; Aillet, F.; et al. Constitutive Activation of p62/Sequestosome-1-Mediated Proteaphagy Regulates Proteolysis and Impairs Cell Death in Bortezomib-Resistant Mantle Cell Lymphoma. Cancers 2022, 14, 923. https://doi.org/10.3390/cancers14040923
Quinet G, Xolalpa W, Reyes-Garau D, Profitós-Pelejà N, Azkargorta M, Ceccato L, Gonzalez-Santamarta M, Marsal M, Andilla J, Aillet F, et al. Constitutive Activation of p62/Sequestosome-1-Mediated Proteaphagy Regulates Proteolysis and Impairs Cell Death in Bortezomib-Resistant Mantle Cell Lymphoma. Cancers. 2022; 14(4):923. https://doi.org/10.3390/cancers14040923
Chicago/Turabian StyleQuinet, Grégoire, Wendy Xolalpa, Diana Reyes-Garau, Núria Profitós-Pelejà, Mikel Azkargorta, Laurie Ceccato, Maria Gonzalez-Santamarta, Maria Marsal, Jordi Andilla, Fabienne Aillet, and et al. 2022. "Constitutive Activation of p62/Sequestosome-1-Mediated Proteaphagy Regulates Proteolysis and Impairs Cell Death in Bortezomib-Resistant Mantle Cell Lymphoma" Cancers 14, no. 4: 923. https://doi.org/10.3390/cancers14040923
APA StyleQuinet, G., Xolalpa, W., Reyes-Garau, D., Profitós-Pelejà, N., Azkargorta, M., Ceccato, L., Gonzalez-Santamarta, M., Marsal, M., Andilla, J., Aillet, F., Bosch, F., Elortza, F., Loza-Alvarez, P., Sola, B., Coux, O., Matthiesen, R., Roué, G., & Rodriguez, M. S. (2022). Constitutive Activation of p62/Sequestosome-1-Mediated Proteaphagy Regulates Proteolysis and Impairs Cell Death in Bortezomib-Resistant Mantle Cell Lymphoma. Cancers, 14(4), 923. https://doi.org/10.3390/cancers14040923