
Current Status of CRISPR/Cas9 Application in Clinical Cancer Research: Opportunities and Challenges


Abstract
:Simple Summary
Abstract
1. Introduction
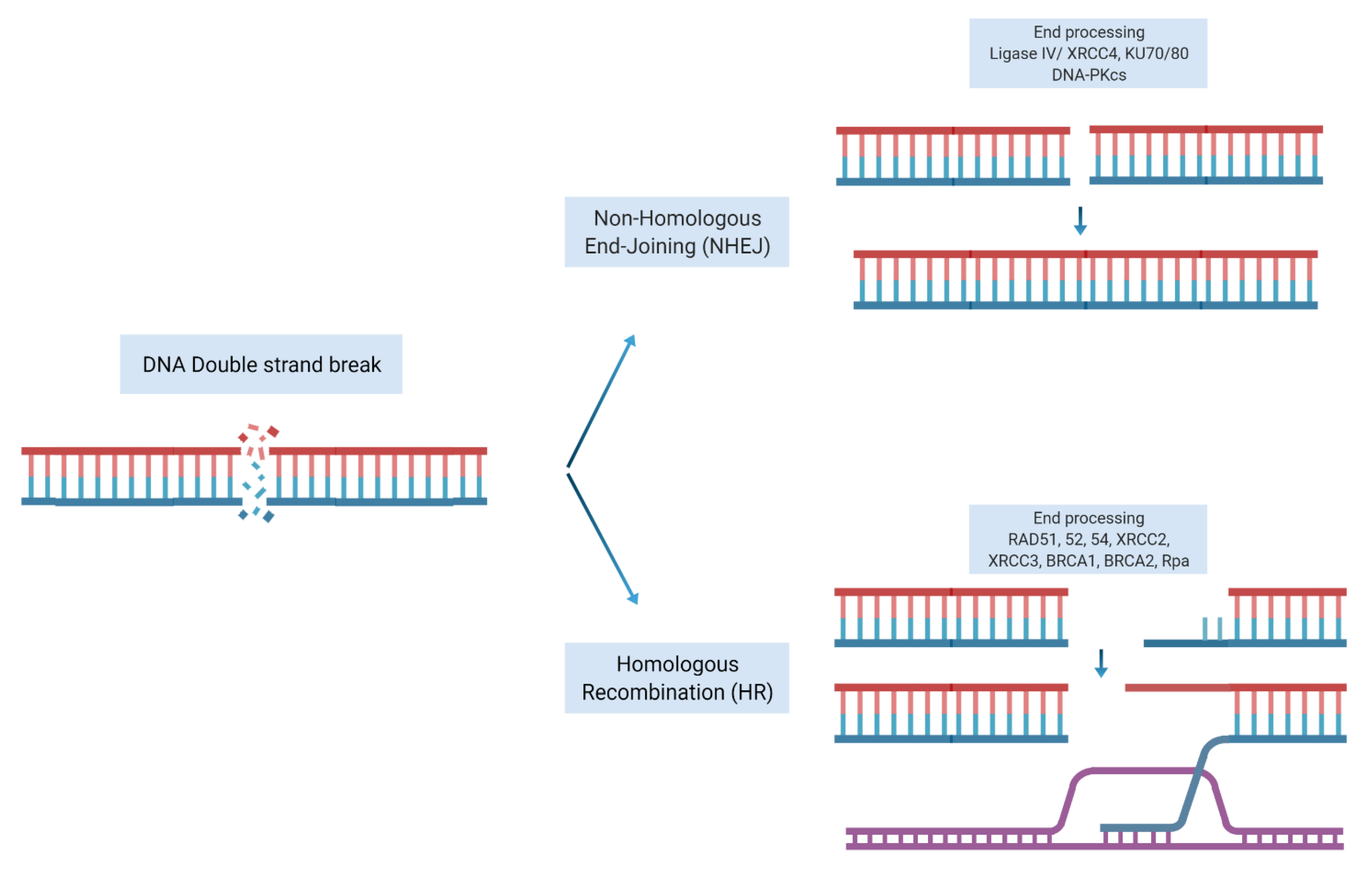
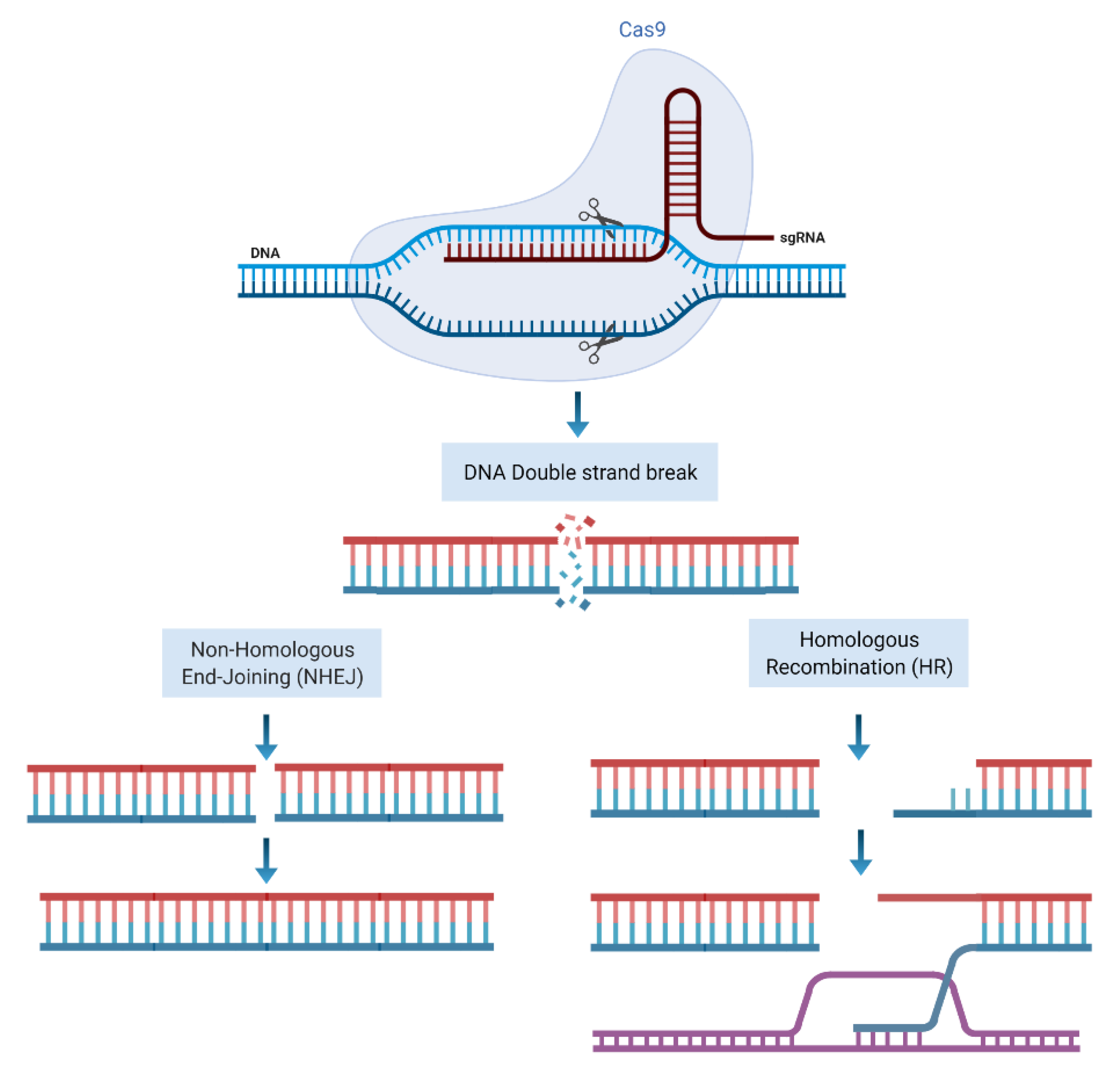
What Is CRISPR/Cas9?
2. How Can CRISPR Technology Be Used in Cancer Research?
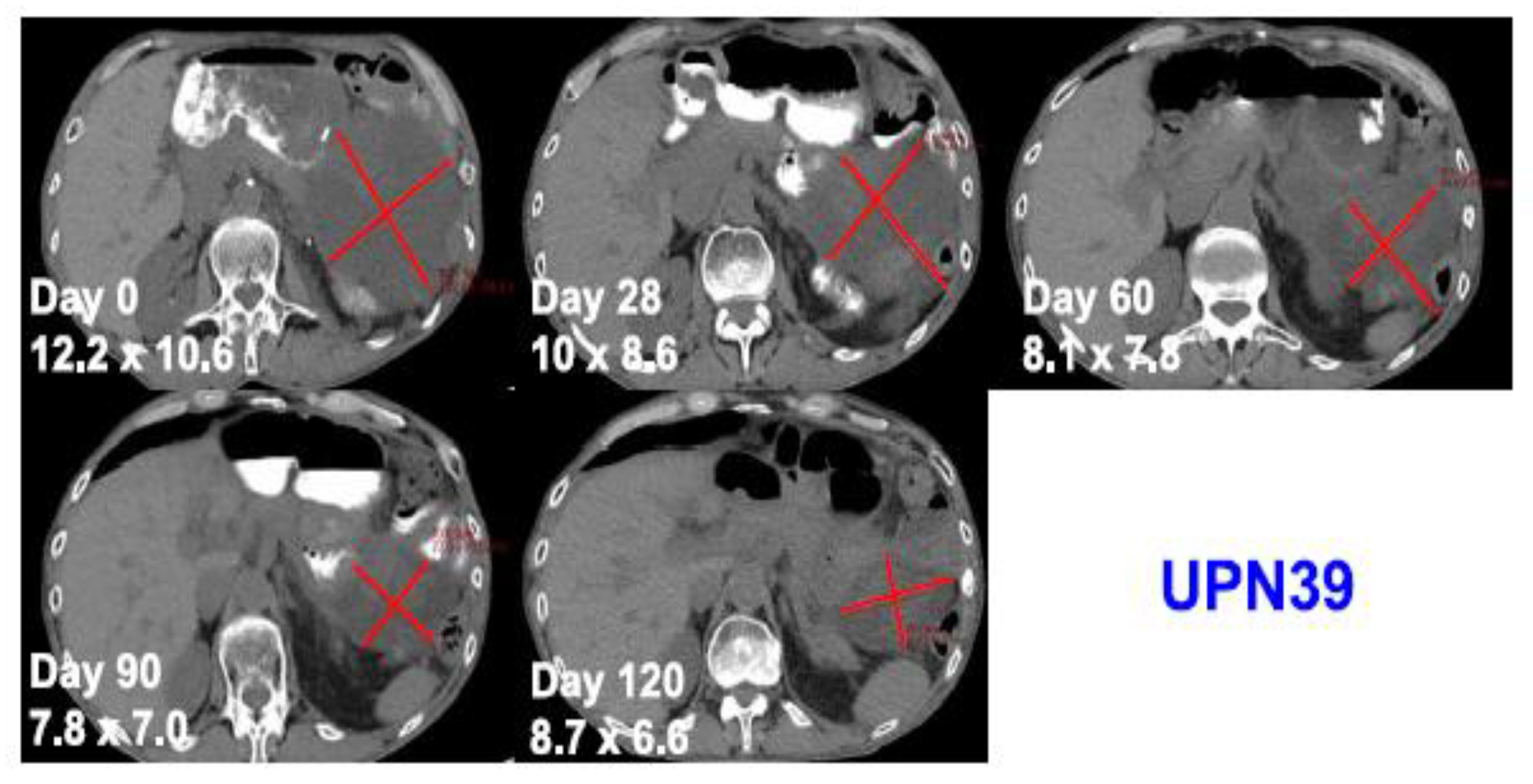
3. What Is the Current Status of Using CRISPR/Cas9 in Cancer Treatment?
3.1. CRISPR/Cas9 and CAR-T Cell Therapies
3.2. CRISPR/Cas9 and Adaptive T Cell Therapies
4. Challenges of Using CRISPR Technology in Cancer Therapy
4.1. Methods of Delivery of CRISPR-Cas9 into Cells
4.2. Target Specification
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rafii, S.; Kaye, S.; Banerjee, S. Current Status and Future Direction of PARP Inhibition in Cancer Therapy. In Targeted Therapy in Translational Cancer Research; Tsimberidou, A.-M., Kurzrock, R., Anderson, K.C., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 325–340. [Google Scholar]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Ates, I.; Rathbone, T.; Stuart, C.; Bridges, P.H.; Cottle, R.N. Delivery Approaches for Therapeutic Genome Editing and Challenges. Genes 2020, 11, 1113. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.H. Genome-Editing Technologies: Concept, Pros, and Cons of Various Genome-Editing Techniques and Bioethical Concerns for Clinical Application. Mol. Ther. Nucleic Acids 2019, 16, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holkers, M.; Maggio, I.; Henriques, S.F.D.; Janssen, J.M.; Cathomen, T.; Goncalves, M. Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat. Methods 2014, 11, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 Endonucleases Reveal RNA-Mediated Conformational Activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleditzsch, D.; Pausch, P.; Esparza, H.M.; Özcan, A.; Guo, X.; Bange, G.; Randau, L. PAM identification by CRISPR-Cas effector complexes: Diversified mechanisms and structures. RNA Biol. 2018, 16, 504–517. [Google Scholar] [CrossRef]
- Janik, E.; Niemcewicz, M.; Ceremuga, M.; Krzowski, L.; Saluk-Bijak, J.; Bijak, M. Various Aspects of a Gene Editing System—CRISPR–Cas9. Int. J. Mol. Sci. 2020, 21, 9604. [Google Scholar] [CrossRef]
- Yi, L.; Li, J. CRISPR-Cas9 therapeutics in cancer: Promising strategies and present challenges. Biochim. Biophys. Acta 2016, 1866, 197–207. [Google Scholar] [CrossRef]
- Zuckermann, M.; Hovestadt, V.; Knobbe-Thomsen, C.B.; Zapatka, M.; Northcott, P.A.; Schramm, K.; Belic, J.; Jones, D.T.W.; Tschida, B.R.; Moriarity, B.S.; et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat. Commun. 2015, 6, 7391. [Google Scholar] [CrossRef]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9–mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Zhan, T.; Ambrosi, G.; Wandmacher, A.M.; Rauscher, B.; Betge, J.; Rindtorff, N.; Häussler, R.S.; Hinsenkamp, I.; Bamberg, L.; Hessling, B.; et al. MEK inhibitors activate Wnt signalling and induce stem cell plasticity in colorectal cancer. Nat. Commun. 2019, 10, 2197. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, W. CRISPR-mediated targeting of HER2 inhibits cell proliferation through a dominant negative mutation. Cancer Lett. 2016, 385, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Moses, C.; Nugent, F.; Waryah, C.B.; Garcia-Bloj, B.; Harvey, A.R.; Blancafort, P. Activating PTEN Tumor Suppressor Expression with the CRISPR/dCas9 System. Mol. Ther.-Nucleic Acids 2018, 14, 287–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, S.R.; Cui, Y.; Lubecka, K.; Stefanska, B.; Irudayaraj, J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 2016, 7, 46545–46556. [Google Scholar] [CrossRef] [Green Version]
- Batır, M.B.; Şahin, E.; Çam, F.S. Evaluation of the CRISPR/Cas9 directed mutant TP53 gene repairing effect in human prostate cancer cell line PC-3. Mol. Biol. Rep. 2019, 46, 6471–6484. [Google Scholar] [CrossRef] [PubMed]
- Floc’H, N.; Martin, M.J.; Riess, J.W.; Orme, J.P.; Staniszewska, A.; Ménard, L.; Cuomo, M.E.; O’Neill, D.J.; Ward, R.A.; Finlay, M.R.V.; et al. Antitumor Activity of Osimertinib, an Irreversible Mutant-Selective EGFR Tyrosine Kinase Inhibitor, in NSCLC Harboring EGFR Exon 20 Insertions. Mol. Cancer Ther. 2018, 17, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zhou, J.; Xu, F.; Bai, W.; Zhang, W. High expression of Aurora-B is correlated with poor prognosis and drug resistance in non-small cell lung cancer. Int. J. Biol. Markers 2018, 33, 215–221. [Google Scholar] [CrossRef]
- Bahreini, A.; Li, Z.; Wang, P.; Levine, K.M.; Tasdemir, N.; Cao, L.; Weir, H.M.; Puhalla, S.L.; Davidson, N.E.; Stern, A.M.; et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res. 2017, 19, 60. [Google Scholar] [CrossRef] [Green Version]
- Harrod, A.; Fulton, J.; Van Nguyen, T.M.; Periyasamy, M.; Ramos-Garcia, L.; Lai, C.-F.; Metodieva, G.; de Giorgio, A.; Williams, R.L.; Santos, D.B.; et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 2017, 36, 2286–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Liu, C.; Lu, H.; Yin, M.; Shao, C.; Hu, X.; Wu, J.; Wang, Y. The expression of APE1 in triple-negative breast cancer and its effect on drug sensitivity of Olaparib. Tumor Biol. 2017, 39, 1010428317713390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.M.; Yang, Y.; Oh, S.J.; Hong, Y.; Seo, M.; Jang, M. Cancer-derived exosomes as a delivery platform of CRISPR/Cas9 confer cancer cell tropism-dependent targeting. J. Control. Release 2017, 266, 8–16. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, N.M.; Dörrie, J.; Schaft, N. CARs: Beyond T Cells and T Cell-Derived Signaling Domains. Int. J. Mol. Sci. 2020, 21, 3525. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benmebarek, M.-R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 17, 147–167. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Holstein, S.A.; Lunning, M.A. CAR T-Cell Therapy in Hematologic Malignancies: A Voyage in Progress. Clin. Pharmacol. Ther. 2019, 107, 112–122. [Google Scholar] [CrossRef]
- Boyiadzis, M.M.; Dhodapkar, M.V.; Brentjens, R.J.; Kochenderfer, J.N.; Neelapu, S.S.; Maus, M.V.; Porter, D.L.; Maloney, D.G.; Grupp, S.A.; Mackall, C.L.; et al. Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: Clinical perspective and significance. J. Immunother. Cancer 2018, 6, 137. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, Y.J. Adoptive Cell Therapy Targeting Neoantigens: A Frontier for Cancer Research. Front. Immunol. 2020, 11, 176. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Rietberg, S.P.; Sotillo, E.; Dong, R.; Vachharajani, V.T.; Labanieh, L.; Myklebust, J.H.; Kadapakkam, M.; Weber, E.W.; Tousley, A.M.; et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov. 2020, 10, 702–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priceman, S.J.; Gerdts, E.A.; Tilakawardane, D.; Kennewick, K.T.; Murad, J.P.; Park, A.K.; Jeang, B.; Yamaguchi, Y.; Yang, X.; Urak, R.; et al. Co-stimulatory signaling determines tumor antigen sensitivity and persistence of CAR T cells targeting PSCA+ metastatic prostate cancer. OncoImmunology 2018, 7, e1380764. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Yarmarkovich, M.; Marshall, Q.F.; Warrington, J.M.; Premaratne, R.; Farrel, A.; Groff, D.; Li, W.; di Marco, M.; Runbeck, E.; Truong, H.; et al. Cross-HLA targeting of intracellular oncoproteins with peptide-centric CARs. Nature 2021, 599, 477–484. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Kren, N.P.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 38, 448–459. [Google Scholar] [CrossRef]
- Shifrut, E.; Carnevale, J.; Tobin, V.; Roth, T.L.; Woo, J.M.; Bui, C.T.; Li, P.J.; Diolaiti, M.E.; Ashworth, A.; Marson, A. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 2018, 175, 1958–1971. [Google Scholar] [CrossRef] [Green Version]
- Van der Woude, L.L.; Gorris, M.; Halilovic, A.; Figdor, C.G.; de Vries, I.J.M. Migrating into the Tumor: A Roadmap for T Cells. Trends Cancer 2017, 3, 797–808. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.; Wickman, E.; DeRenzo, C.; Gottschalk, S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol. Ther. 2020, 28, 2320–2339. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Cheng, C.; Cheng, A.; Zhang, X.; Li, N.; Xia, C.; Wei, X.; Liu, X.; Wang, H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2016, 27, 154–157. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Adamow, M.; Ginsberg, B.A.; Rasalan, T.S.; Ritter, E.; Gallardo, H.F.; Xu, Y.; Pogoriler, E.; Terzulli, S.L.; Kuk, D.; et al. Integrated NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc. Natl. Acad. Sci. USA 2011, 108, 16723–16728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, 7365. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]
- Senís, E.; Fatouros, C.; Große, S.; Wiedtke, E.; Niopek, D.; Mueller, A.-K.; Börner, K.; Grimm, D. CRISPR/Cas9-mediated genome engineering: An adeno-associated viral (AAV) vector toolbox. Biotechnol. J. 2014, 9, 1402–1412. [Google Scholar] [CrossRef]
- Hayashi, H.; Kubo, Y.; Izumida, M.; Matsuyama, T. Efficient viral delivery of Cas9 into human safe harbor. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Rosenblum, D.; Gutkin, A.; Kedmi, R.; Ramishetti, S.; Veiga, N.; Jacobi, A.M.; Schubert, M.S.; Friedmann-Morvinski, D.; Cohen, Z.R.; Behlke, M.A.; et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 2020, 6, eabc9450. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, J.; Ge, S.; Lai, L. CRISPR/Cas: Advances, Limitations, and Applications for Precision Cancer Research. Front. Med. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Behr, M.; Zhou, J.; Xu, B.; Zhang, H. In vivo delivery of CRISPR-Cas9 therapeutics: Progress and challenges. Acta Pharm. Sin. B 2021, 11, 2150–2171. [Google Scholar] [CrossRef]
- Petek, L.M.; Russell, D.W.; Miller, D.G. Frequent Endonuclease Cleavage at Off-target Locations In Vivo. Mol. Ther. 2010, 18, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Gkazi, S.A. Quantifying CRISPR off-target effects. Emerg. Top. Life Sci. 2019, 3, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Giannoukos, G.; Ciulla, D.M.; Marco, E.; Abdulkerim, H.S.; Barrera, L.A.; Bothmer, A.; Dhanapal, V.; Gloskowski, S.W.; Jayaram, H.; Maeder, M.L.; et al. UDiTaS™, a genome editing detection method for indels and genome rearrangements. BMC Genom. 2018, 19, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Barbosa, K.; Cheng, K.; Leiserson, M.D.M.; Jain, P.; Deshpande, A.; Wilson, D.M.; Ryan, B.M.; Luo, J.; Ronai, Z.A.; et al. A systematic genome-wide mapping of oncogenic mutation selection during CRISPR-Cas9 genome editing. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Enache, O.M.; Rendo, V.; Abdusamad, M.; Lam, D.; Davison, D.; Pal, S.; Currimjee, N.; Hess, J.; Pantel, S.; Nag, A.; et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 2020, 52, 662–668. [Google Scholar] [CrossRef]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef]
- Kuscu, C.; Parlak, M.; Tufan, T.; Yang, J.; Szlachta, K.; Wei, X.; Mammadov, R.; Adli, M. CRISPR-STOP: Gene silencing through base-editing-induced nonsense mutations. Nat. Methods 2017, 14, 710–712. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Agent | Clinical Trial * | Sponsor | n †, Population, Age | Primary Outcomes Measures |
|---|---|---|---|---|
| Neoantigen-specific TIL edited with CRISPR/Cas-9 to inhibit intracellular immune checkpoint CISH | NCT04426669 Phase 1/2, non-randomized, sequential assignment Recruiting | Intima Bioscience, Inc., New York, NY, USA | n = 20 Metastatic gastrointestinal epithelial cancer with progressive disease following at least one first-line standard therapy 18–70 yr | MTD Tumor diameter AE |
| CD19-specific CAR-T cells edited with CRISPR guide RNA to disrupt expression of endogenous HPK1 (XYF19 CAR-T cell) | NCT04037566 Phase 1, non-randomized, single group assignment Recruiting | Xijing Hospital, China | n = 40 Relapsed/refractory CD19+ B-cell leukemia or lymphoma 18–55 yr | AE MTD/DLT |
| NY-ESO-1 redirected autologous T cells and edited with CRISPR guide RNA to disrupt expression of TCR and PD-1 (NYCE T Cells) | NCT03399448 Phase 1, non-randomized, parallel assignment Terminated | University of Pennsylvania, USA | n = 3 Relapsed/refractory multiple myeloma, melanoma, synovial sarcoma, or myxoid/round cell liposarcoma ≥18 yr | AE Manufacturing feasibility |
| CD34+ hematopoietic stem/progenitor cells with CRISPR/Cas9 disruption of CCR5 | NCT03164135 Phase 1, non-randomized, single group assignment Unknown | Peking University Affiliated Hospital to Academy of Military Medical Sciences, China | n = 5 HIV-infected hematologic malignancies 18–60 yr | Persistence of CCR5 gene disruption in engrafted cells |
| Mesothelin-directed CAR-T cells with CRISPR/Cas9 mediated PD-1 and TCR knock out | NCT03545815 Phase 1, non-randomized, single group assignment Recruiting | Chinese PLA General Hospital, China | n = 10 Mesothelin positive solid tumors with failure of at least one prior standard of care chemotherapy for advanced stage disease 18–70 yr | AE Disease control rate |
| CD19-specific CAR-T cells with chRDNA integrated CD19-CAR at TRAC and PD-1 knock out (CB-010) | NCT04637763 (CB010A) Phase 1, non-randomized, sequential assignment Recruiting | Caribou Biosciences, Inc., Berkeley, CA, USA | n = 50 Relapsed/refractory NHL after prior standard of care ≥18 yr | DLT Objective response rate |
| CD19-specific CAR-T cells with CRISPR/Cas9 disruption of B2M, CIITA, and TRAC (PACE CART19) | NCT05037669 Phase 1, non-randomized, sequential assignment Not yet recruiting | University of Pennsylvania, USA | n = 36 Relapsed/refractory ALL, CLL, NHL ≥18 yr | Recommended expansion dose |
| TALEN and CRISP/Cas9 disrupted HPV 16/18 E6/E7 | NCT03057912 Phase 1, non-randomized, parallel assignment Unknown | First Affiliated Hospital, Sun Yat-Sen University, China | n = 60 Women with HPV16 or HPV18 infection at risk of HPV-related cervical intraepithelial neoplasia 18–50 yr | AE |
| BCMA-directed T-cell immunotherapy modified ex vivo using CRISPR/Cas9 (CTX120) | NCT04244656 Phase 1, non-randomized, sequential assignment Recruiting | CRISPR Therapeutics AG, Switzerland/USA | n = 80 Relapse/refractory multiple myeloma ≥18 yr | AE Objective response rate |
| CD70-directed T-cell immunotherapy modified ex vivo using CRISPR/Cas9 (CTX130) | NCT04438083 (COBALT-RCC) Phase 1, non-randomized, sequential assignment Recruiting | CRISPR Therapeutics AG, Switzerland/USA | n = 107 Unresectable or metastatic renal cell carcinoma that has exploited standard of care treatment ≥18 yr | AE Objective response rate |
| CD70-directed T-cell immunotherapy comprised of allogeneic T cells genetically modified ex vivo using CRISPR-Cas9 gene editing components | NCT04502446 (COBALT-LYM) Phase 1, non-randomized, sequential assignment Recruiting | CRISPR Therapeutics AG, Switzerland/USA | n = 45 T cell malignancy or DLBCL ≥18 yr | AE Objective response rate |
| CD19-specific CAR-T cells with CRISPR/Cas9 edited CD52 and TRAC (PBLTT52CAR19) | NCT04557436 Phase 1, non-randomized, single group assignment Recruiting | Great Ormond Street Hospital for Children NHS Foundation Trust, UK | n = 10 Relapsed/refractory CD19+ B-cell ALL 6 months-18 yr | Remission |
| Mesothelin-directed CAR-T cells with CRISPR/Cas9 mediated PD-1 knock out | NCT03747965 Phase 1, non-randomized, single group assignment Unknown | Chinese PLA General Hospital, China | n = 10 Mesothelin positive solid tumors (especially pancreatic cancer, cholangiocarcinoma, ovarian cancer) with failure of at least one prior standard of care chemotherapy for advanced stage disease 18–70 yr | AE Disease control rate |
| CD19 and CD20 or CD22-specific CRISPR/Cas9 edited CAR-T cells | NCT03747965 Phase 1/2, non-randomized, single group assignment Recruiting | Chinese PLA General Hospital, China | n = 80 Relapsed/refractory B-cell leukemia or lymphoma 12–70 yr | AE MTD CAR copies |
| CD19-directed T-cell immunotherapy modified ex vivo using CRISPR/Cas9 (CTX110) | NCT04035434 (CARBON) Phase 1, non-randomized, sequential assignment Recruiting | CRISPR Therapeutics AG, Switzerland/USA | n = 143 Relapsed/refractory B-cell ALL or NHL ≥18 yr | AE Objective response rate |
| CD19-specific CRISPR/Cas9 edited CAR-T cells(UCART019) | NCT03166878 Phase 1/2, non-randomized, single group assignment Recruiting | Chinese PLA General Hospital, China | n = 80 Relapsed/refractory CD19+ B-cell leukemia or lymphoma 12–75 yr | AE DLT UCAR019 copies |
| T cells with CRISPR/Cas9 PD-1 knock out (PD-1 Knockout EBV-CTL) | NCT03044743 Phase 1/2, non-randomized, single group assignment Recruiting | Yan Yang, China | n = 20 EBV positive stage IV gastric carcinoma, nasopharyngeal carcinoma and lymphoma 18–75 yr | AE |
| T cells with CRISPR/Cas9 PD-1 knock out | NCT02793856 Phase 1, non-randomized, parallel assignment Completed | Sichuan University, China | n = 12 Stage IV non-small-cell lung cancer 18–70 yr | AE |
| T cells with CRISPR/Cas9 PD-1 knock out combined with transcatheter arterial chemoembolization | NCT04417764 Phase 1, non-randomized, single group assignment Recruiting | Central South University, China | n = 10 Unresectable hepatocellular carcinoma 18–70 yr | AE |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rafii, S.; Tashkandi, E.; Bukhari, N.; Al-Shamsi, H.O. Current Status of CRISPR/Cas9 Application in Clinical Cancer Research: Opportunities and Challenges. Cancers 2022, 14, 947. https://doi.org/10.3390/cancers14040947
Rafii S, Tashkandi E, Bukhari N, Al-Shamsi HO. Current Status of CRISPR/Cas9 Application in Clinical Cancer Research: Opportunities and Challenges. Cancers. 2022; 14(4):947. https://doi.org/10.3390/cancers14040947
Chicago/Turabian StyleRafii, Saeed, Emad Tashkandi, Nedal Bukhari, and Humaid O. Al-Shamsi. 2022. "Current Status of CRISPR/Cas9 Application in Clinical Cancer Research: Opportunities and Challenges" Cancers 14, no. 4: 947. https://doi.org/10.3390/cancers14040947
APA StyleRafii, S., Tashkandi, E., Bukhari, N., & Al-Shamsi, H. O. (2022). Current Status of CRISPR/Cas9 Application in Clinical Cancer Research: Opportunities and Challenges. Cancers, 14(4), 947. https://doi.org/10.3390/cancers14040947