1. Introduction
Ovarian cancer is the fifth leading cause of cancer-related mortality and the 11th most common cancer in women. In developing countries, the mortality rate in ovarian cancer patients is even higher due to challenges in early detection, poor prognosis, frequent relapses, lack of medical infrastructure, and limited awareness [
1]. As compared to other malignancies, early-stage ovarian cancer is often asymptomatic, and therefore spreads throughout the abdominal cavity with most patients presenting later with an advanced stage ovarian cancer. Histologically, epithelial ovarian cancer is a heterogenous disease of five pathologically distinct subtypes such as (1) high-grade serous ovarian carcinoma (HGSOC), (2) low-grade serous (LGS), (3) endometrioid, (4) clear cell, and (5) mucinous carcinoma subtypes. Among these subtypes, HGSOC is the most prevalent subtype representing approximately 75% of cases, while the remaining 25% are made up of low-grade serous (LGS), endometrioid, clear cell, and mucinous carcinoma [
2]. Therefore, the majority of research is focused on cell lines particularly of serous ovarian carcinoma origin, and the other histologic subtypes of ovarian cancer are poorly represented in the available list of ovarian cancer cell lines [
2]. The most commonly used cell lines for serous ovarian cancer are SKOV-3, OVCAR-3, OVCAR-4, CAOV3, HEYA8, and IGROV1. Unfortunately, some of these cancer cell lines, which have been used for many years, are misidentified, or misclassified histologically [
2]. Another concern pertaining to the use of these cell lines in ovarian cancer research arose due to the misrepresentation of the status of p53 and BRCA1 as wild type or in its mutated form [
3].
In contrast to other cancers, peritoneal seeding is considered as the primary mechanism of metastatic spreading, while metastasis through the hematogenous mode is much more limited [
4]. Accumulation of ascites is also a common feature in ovarian cancer, which consequently worsens the prognostic outcome of ovarian cancer. Notably, various growth factors, cytokines, chemokines, extracellular matrix proteins, and proteolytic enzymes promote the growth and progression of ovarian cancer by activating varieties of oncogenic pathways in tumor cells [
5]. Ovarian cancer metastasis relies on the process where tumor cells from ovary shed into the peritoneum, then aggregate as freely floating spheroids which get implanted on various peritoneal organs [
6,
7]. As compared to monolayers, tumor spheroids derived from monolayer cells demonstrate resistance to cytotoxic drugs often due to hinderance of drug penetration, induction of cellular efflux pumps, and aberrant activation of oncogenic signaling pathways in tumor spheroids [
8,
9,
10]. Therefore, ovarian cancer cell lines, which form tumor spheroids spontaneously when confluent will have enormous value for studying the mechanism of transition from adherent form to tumor spheroids, drug resistance, anoikis, and metabolic changes. Thus, there is an urgent need to identify signaling mechanisms distinct in tumor spheroids, which are more aggressive in nature for identifying suitable targeted therapy to be used alone or in combination with chemotherapeutic agents.
Cancer cells with aggressive characteristics such as high-proliferative capacity, metastatic features, or chemoresistance rely on a precise signaling mechanism through autocrine or paracrine cues as an oncogenic signaling addiction. Therefore, we sought to characterize the precise signaling that promotes cancer stemness and the chemoresistance mechanism in one of the common subtypes of ovarian cancer endometrioid histologic subtype. Endometrioid ovarian carcinoma constitutes about 20% of epithelial ovarian carcinoma among women in the United States and commonly arises from endometriosis of the ovary [
11]. Patients with endometrioid carcinoma of the ovary are often diagnosed at a comparatively younger age with early-stage disease and have relatively better 5-year survival outcome (80.3%) than serous ovarian carcinoma (18.4%). By establishing an endometrioid subtype cell line, we uncovered the mechanism required for spontaneous transition of monolayer cells to tumor spheroids and chemoresistance in endometrioid subtype cancer cells.
2. Material and Methods
2.1. Patients and Human Ethics Statement
Written informed consent was obtained from the patient according to the Institutional Review Board (IRB) of the Medical College of Wisconsin, Milwaukee, WI, USA. A 70-year-old female presented with elevated CA-125 of 81.8 units/mL, and a complex right adnexal mass. She underwent staging surgery including exploratory laparotomy, total abdominal hysterectomy, bilateral salpingo-oophorectomy, omentectomy, diaphragm cytology, appendectomy, pelvic and para-aortic lymph node dissection. The patient was untreated at the time of surgery. On the histopathology examination, the right ovary was positive for adenocarcinoma, endometrioid type (FIGO stage 1A, grade 1) with extensive necrosis. The left ovary and fallopian tube were negative for carcinoma. All lymph nodes (left and right pelvic and para-aortic lymph nodes) were negative for carcinoma.
2.2. Isolation and Culture of Ovarian Tumor-Derived Cells
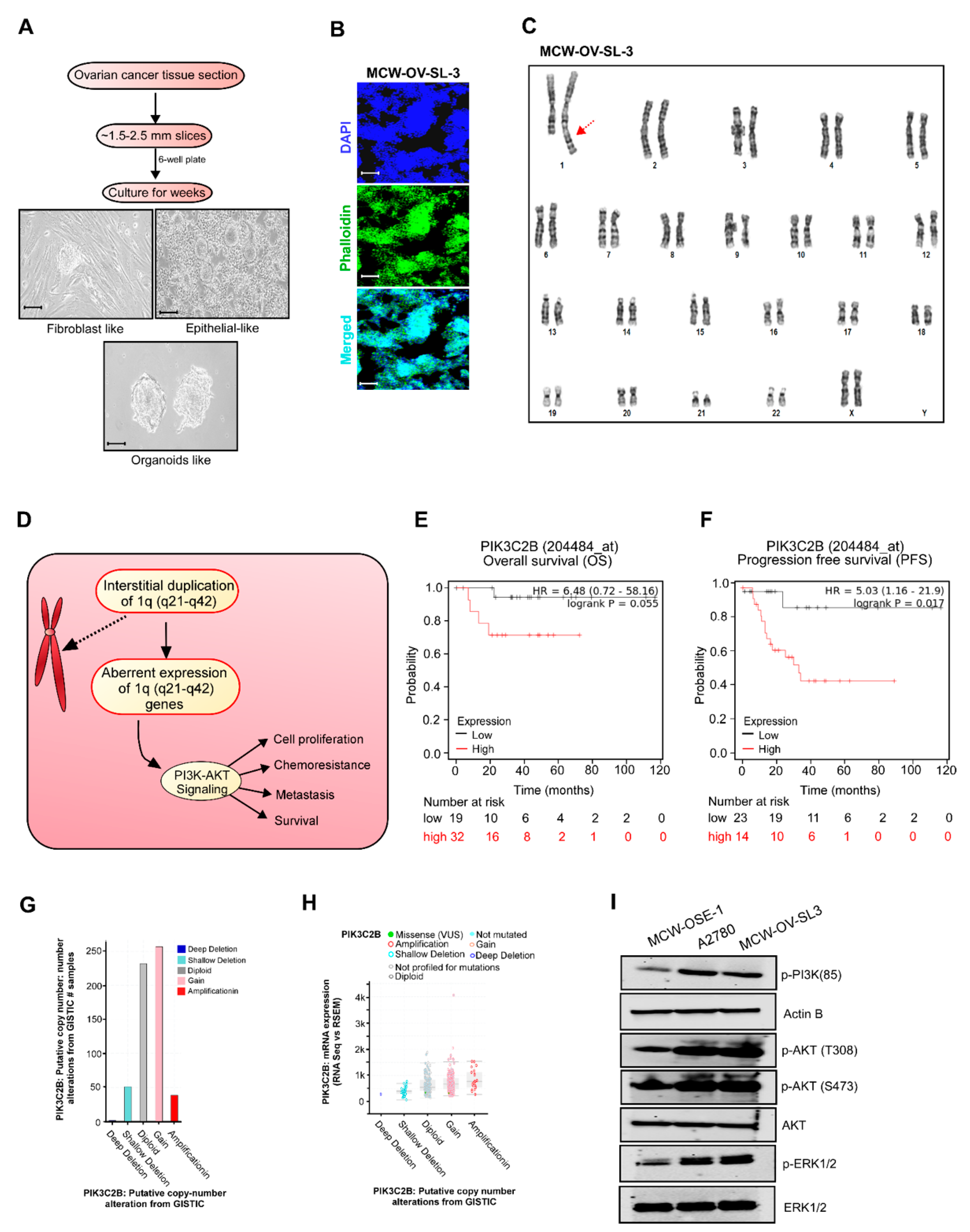
Tumor tissue obtained was minced into approximately 1.5–2.5 mm
3 pieces, washed with 1X PBS (Ca
2+ and Mg
2+ free), then digested using 0.1% Collagenase Type IV (Gibco, ThermoFisher Scientific, Waltham, MA, USA) in DMEM for 30 min at 37 °C with occasional shaking at low speed in a 15 mL centrifuge tube (Corning Inc., Corning, NY, USA). The cell suspension was filtered by 45 µm cell strainers to remove any large tissue fragments and centrifuged at 1000 rpm for 3 min. Cells were resuspended in advanced DMEM supplemented with 10% fetal bovine serum (FBS, R&D Systems, Inc., Minneapolis, MN, USA), 50 units/mL penicillin, and 50 μg/mL streptomycin (Gibco, ThermoFisher Scientific, Waltham, MA, USA), 2 mM L-glutamine (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), and cultured in 24-well cell culture plates at 37 °C in a humidified 5% CO
2 atmosphere. Continuous culture resulted in adhesion to the culture dishes and an outgrowth of cells attached to the tissue and the plate was observed daily (
Figure 1). Two types of cells, epithelial-like cuboidal cells and fibroblast-like elongated cells were visible for the few weeks. Fibroblast cells were eliminated by differential trypsinization method. Later, epithelial-like cells started making dome-like colonies. These cell domes were isolated, picked, and transferred to a fresh tissue culture flask. These cells were continuously passaged 40 times for about 6 months, and a tumor cell clone was manually selected to establish the stable MCW-OV-SL-3 cell line. The cells were frozen in 5% DMSO in fetal bovine serum (FBS) solution in liquid nitrogen for further experiment. Karyotyping was performed after the 15th passage for characterization. These cells were regularly passaged and expanded in complete DMEM media for characterization. During early steps of establishment, this cell line grew rapidly in the presence of 2% heat inactivated autologous human ascitic filtrate. Once the cell line was established the filtrate was no longer added. Short tandem repeat (STR) characterization was performed at IDEXX Bioanalytics Services (Columbia, MO, USA) for cell authentication.
2.3. Isolation and Culture of Normal Ovarian Surface Epithelial (OSE) Cells
Normal ovarian surface epithelium cells (OSE) were obtained by scraping the surface of healthy ovaries from non-cancerous patient from the Department of Obstetrics and Gynecology, Froedtert Hospital, Medical College of Wisconsin. All human samples were collected with written informed consents from patients under an IRB of the Medical College of Wisconsin-approved protocol in accordance with recognized ethical guidelines of the declaration of Helsinki.
Histological examination confirmed that ovaries were grossly normal, and no pathological lesions were observed. The normal ovarian tissue obtained was minced into approximately 1.0–2.0 mm3 pieces, then washed with 1X PBS. The OSE cells were scraped with a surgical blade after collagenase digestion under aseptic conditions using 0.1% Collagenase Type IV (Gibco) in DMEM for 30 min at 37 °C with occasional shaking at low speed in a 15 mL centrifuge tube (Corning Inc., Corning, NY, USA). Then, cell viability was checked by the trypan blue dye exclusion assay, and it was observed that 95% cells were viable. OSE cells isolated were cultured with Medium 199/MCDB105 (1:1, Sigma, St. Louis, MO, USA). supplemented with 15% FBS, 1% pen-strep, 10 ng/mL human epidermal growth factor (Life Technologies, Carlsbad, MA, USA), 0.5 μg/mL hydrocortisone (Sigma St. Louis, MO, USA)., 5 μg/mL bovine insulin (Cell Applications, CA, USA), 34 μg protein/mL bovine pituitary extract (Life Technologies, Carlsbad, MA, USA) at 37 °C in a humidified 5% CO2 atmosphere. Cells were detached from the dish with 0.125% trypsin and 0.11% ethylenediamine tetra acetic acid (EDTA) and split in 1:2 ratios in a new culture dish when confluent.
Karyotyping was performed after the 5th passage for characterization and STR profiling was performed at IDEXX Bioanalytics Services (Columbia, MO, USA) to confirm that the identity of the cell line we established was unique and was not contaminated with other established cell lines.
2.4. Cell Lines
A2780 parental and cisplatin-resistant A2780-cisR cells were obtained from Millipore Sigma (St. Louis, MO, USA). The OVCAR-4 cell line was purchased from NCI-DCTD repository. The HeyA8 cell line was received from the Characterized Cell Line core at M.D. Anderson Cancer center, Texas, USA. All cell lines were cultured in Dulbecco’s DMEM high glucose (4.5 g/L) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and antibiotics (60 mg/L penicillin, 100 mg/L streptomycin sulfate (PenStrep)) at 37 °C under 5% CO2. A2780-cisR cells were maintained in the presence of 2 μM of cisplatin. Cell line authentication was performed by short tandem repeat profiling at the IDEXX Bioanalytic Laboratories Inc. and tested as Mycoplasma negative by PCR detection kit (Mycosenser Mycoplasma assay kit, Agilent, Santa Clara, CA, USA) as recent as two months prior to the previous experiments.
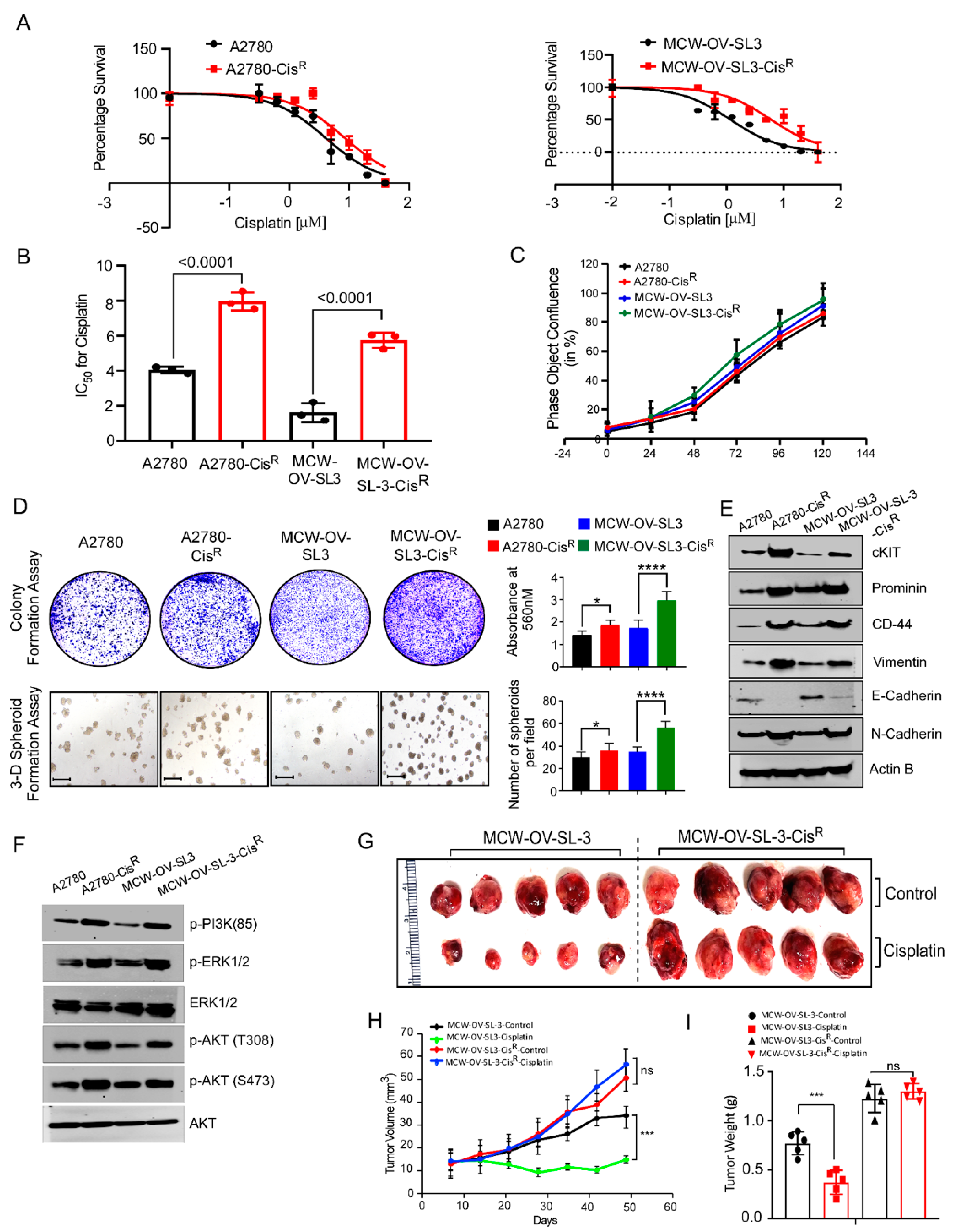
Cisplatin-resistant (CisR) variants of the MCW-OV-SL-3 cell line were derived in vitro from their original parental (PT) cell line by continuous exposure to cisplatin (Selleckchem, Houston, TX, USA) before intraperitoneal injection. Initially, the MCW-OV-SL-3-CisR subline was treated with cisplatin (IC50 of 2.5 uM) for 72 h. To recover the cells, the old media with cisplatin was changed with complete DMEM without cisplatin and cells were left for a further 72 h. This development period continued with increasing doses of cisplatin for approximately 12 months, after which the IC50 concentrations were re-assessed in the resistant cell line. The resistant cells were maintained with new IC-50.
2.5. Karyotype Analysis
Karyotyping was performed on MCW-OSE-1 (ovarian surface epithelial cells from normal ovary of healthy control) and MCW-OV-SL-3 cell lines to determine the chromosomal abnormalities as described before [
12]. Cells in the logarithmic growth phase were cultured in DMEM until 80% confluency and were sent to Wisconsin Diagnostic Laboratories Cytogenetics for karyotyping.
Metaphase spreads were prepared following standard cytogenetic procedures. Briefly, cells were harvested by trypsinization and complete DMEM was added to inactivate trypsin. After centrifugation at 1000 rpm for 4 min, cells were initially exposed to 50 µL of EtBr (0.33 mg/mL) for 1 h, followed by 100 µL of 1.11 µg/mL colcemid for 20 min and fixed in Carnoy’s fixative (1:3 acetic acid: methanol) twice at room temperature for 30 min. Slides were air-dried and stained with Giemsa stain for 20 min. Twenty metaphase cells from each cell line were analyzed and 3–4 metaphase spreads were karyotyped according to the International System for Human Cytogenetic Nomenclature (ISCN 2020) [
13,
14]. The karyotyping was performed after analyzing 20 proliferating (metaphases) cells with 400–550 band resolution in MCW-OSE-1 and 400–425 band resolution in MCW-OV-SL-3 cells.
2.6. Drugs
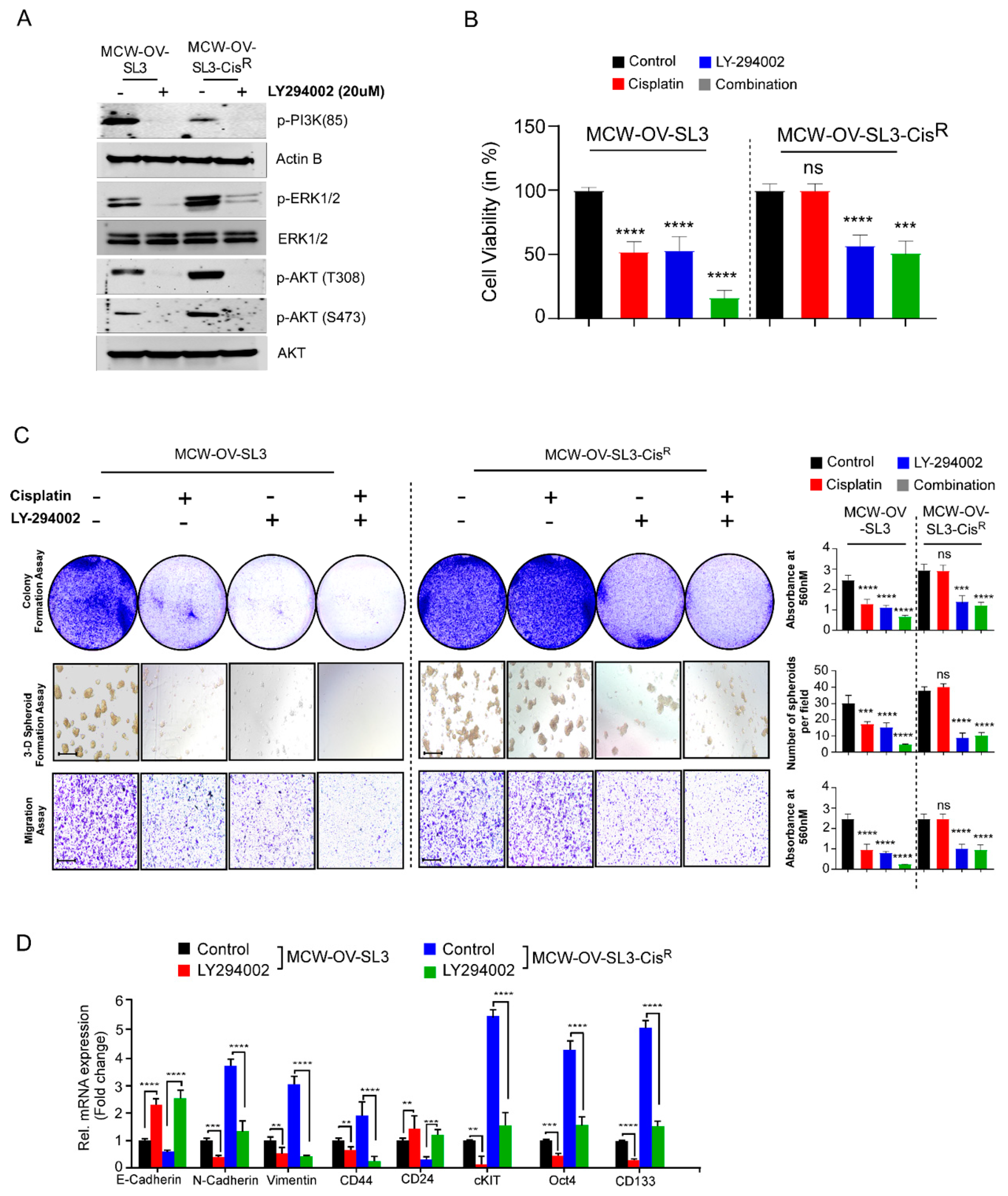
Cisplatin (NSC 119875, Cisplatinum, cis-diamminedichloroplatinum II, CDDP, cis DDP, DDP) was purchased from Selleckchem and a 5 mM stock solution was prepared. LY294002 (S1105, NSC 697286) was purchased from Selleckchem. Aliquots of cisplatin and LY294002 were stored at −20 °C for up to a maximum of three months and thawed immediately before use.
2.7. Morphological Examination of MCW-OSE-1 and MCW-OV-SL-3
MCW-OSE-1 and MCW-OV-SL-3 cells were seeded in a 6-well tissue culture plate and incubated at 37 °C in a humidified 5% CO2 incubator for 2 weeks. Cells were observed under phase contrast microscope daily to check general morphology.
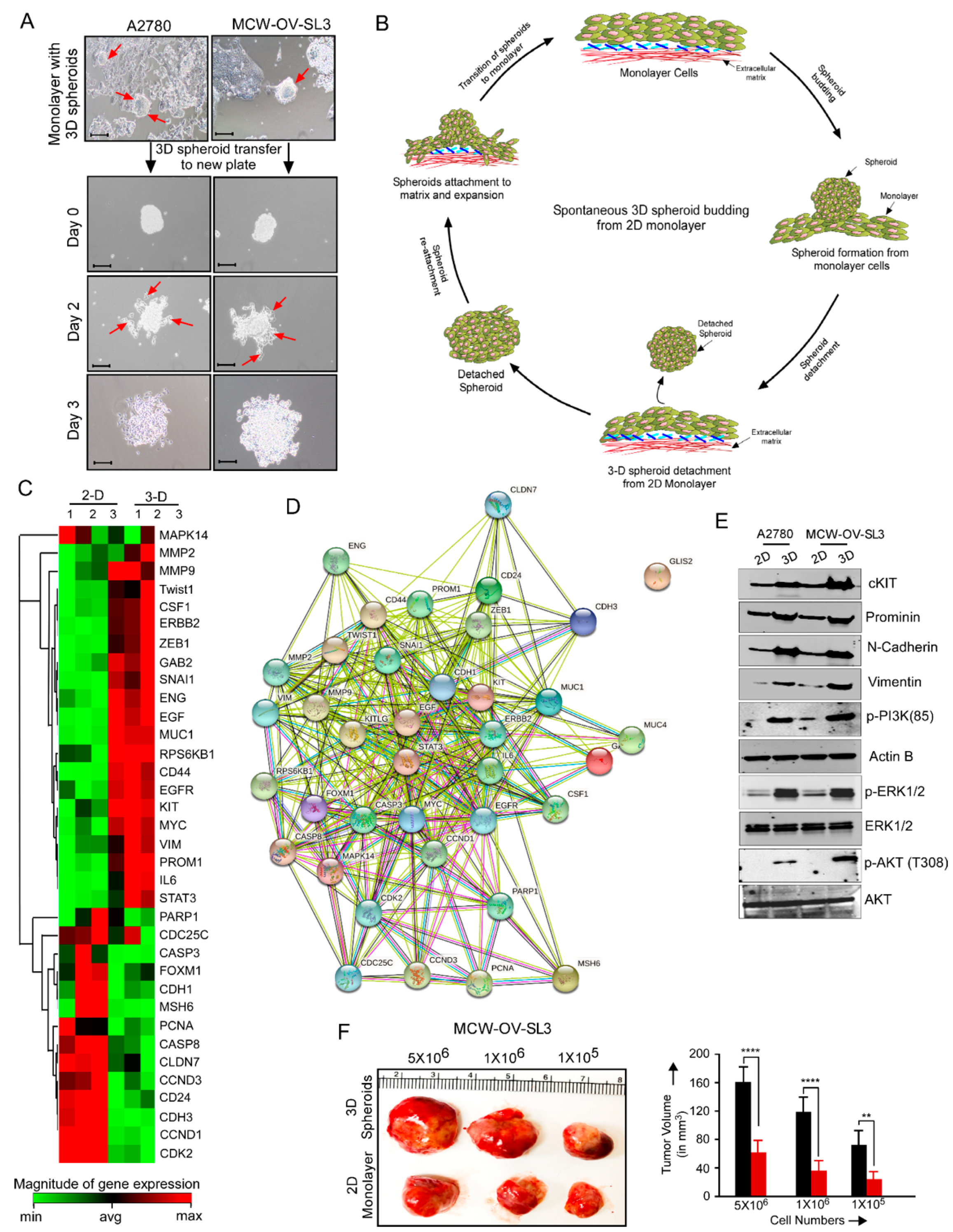
2.8. 3D Spheroid Formation Assay
Spheroid formation (3D culture) with 3000 cells per spheroid was performed as described previously [
15]. In brief, a cell monolayer was washed with 1X PBS and treated with trypsin–EDTA solution for 2 min at 37 °C. Trypsin was then neutralized by adding complete growth medium, and the cell suspension was centrifuged at 1000 rpm for 4 min. Supernatant was removed and 3000 cells per spheroid were resuspended in 500 μL of complete DMEM containing 5% growth factor reduced Matrigel (BD Biosciences, Bedford, MA, USA) in each well of 24-well, growth factor reduced Matrigel-coated non-adherent plates for 7 days. In parallel, cells were also seeded on cell culture plates (2D culture) and cultivated in the same medium and incubated at 37 °C in a 5% CO
2 incubator. Cultures were treated with different inhibitors (either cisplatin (2.2 and 6.2 µM), LY-294002 (5 and 10 µM) or their combination (either cisplatin 1.1 and 3.1 µM), LY-294002 (2.5 and 5 µM) for 48 h. Morphology of the monolayer and spheroids was evaluated using a phase contrast microscope and number of spheroids was determined.
2.9. Cell Viability Assay
To measure the cell viability, MCW-OSE-1, MCW-OV-SL3, OVCAR 8, OVCAR 4, SKOV3, and A2780 cells were seeded at the density of 5 × 103 cells/well with five replicates in 96-well plates. The next day, after reaching 80% confluency, the cells were treated with cisplatin, PI3K inhibitor (LY294002), or a combination of cisplatin and LY294002. Cell viability was measured using 3-(4,5-dimethylthia-zol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent (Sigma Aldrich, St. Louis, MO, USA). MTT reagent (5 mg/mL) was added into each well of 96-well plates and cells were incubated at 37 °C for 3 h. Formazan crystals thus formed were dissolved with acidic isopropanol, and absorbance was measured at 560 nm in a microplate reader (Tecan, Mannedorf, Switzerland). Cell viability at 24, 48, and 72 h was calculated by taking the ratio to control cells from the day 0 reading to account for plating unevenness.
2.10. Cellular Proliferation and Colony Formation Assay
To study the cellular proliferation, 1 × 105 cells were seeded in tetraplicates in a 6-well plate. Cell number was counted manually with a hemocytometer at three different time points after seeding for 24, 48, and 72 h. For the colony formation assay, 1000 cells per well were seeded in a 6-well plate in triplicates. Cell were also treated with cisplatin, LY294002, or a combination of cisplatin and LY294002 for 48 h before seeding and incubated at 37 °C in a 5% CO2 incubator. After 14 days, cells were rinsed with 1X PBS, fixed in 5% glutaraldehyde for 20 min, and stained with 0.5% crystal violet (Sigma Aldrich, St. Louis, MO, USA) for 20 min. Plates were washed with water and dried before scanning. Crystal Violet was solubilized with 10% acetic acid and absorbance was measured at 450 nm in a microplate reader (Tecan, Mannedorf, Switzerland).
2.11. Cell Migration, Invasion, and Wound Healing Assay
The cellular motility was analyzed by carrying out cell migration, invasion, and wound healing assay as described earlier [
10]. Cells were also treated with cisplatin, LY294002, or a combination of both for 48 h before migration, invasion, and wound healing assays. In brief, for the migration assay, 1 × 10
5 cells were seeded on the upper chamber of the 8.0 µm pore trans-well inserts (BD Biosciences, Bedford, MA, USA). Growth medium containing 10% FBS was the chemoattractant in the lower chamber.
For the invasion assay inserts were coated with 5 mg/mL Matrigel (BD Biosciences, Bedford, MA, USA) and 2 × 105 cells were seeded similarly as for the migration assay. Cells were incubated at 37 °C for 12 h for migration and invasion assays and fixed with 0.5% glutaraldehyde for 20 min. Non-migrated cells were removed using a cotton swab and culture inserts were washed and stained with 0.5% crystal violet in 10% methanol. Migrated and invaded cells were imaged using a phase contrast microscope. Moreover, stained membranes were also dissolved in 10% acetic acid and absorbance was measured in a microplate reader at 560 nm.
For the wound healing assay, 2 × 10
6 cells were seeded in a 35 mm Petri dish. After 12 h of incubation, a wound was mechanically created using an aerosol P200 pipette tip, and cells were photomicrographed at various time points using a phase contrast micro-scope (Nikon, Fukok, Japan). Images were analyzed using Image J software (
https://imagej.nih.gov/ij/download.html accessed on 20 August 2021).
2.12. RNA Isolation, cDNA Synthesis, and Real-Time PCR Analysis
Total RNA from cancer cells and normal ovarian cells were extracted using RNeasy Plus kit (QIAGEN, Valencia, CA, USA) according to the manufacturer’s protocol. mRNA level of various genes was determined in a Bio-Rad CFX Connect using SYBR Green Supermix (Bio-Rad, Hercules, CA, USA). Primers for real-time PCR were designed using primer3 software (
https://bioinfo.ut.ee/primer3-0.4.0/ accessed on 30 April 2021) and listed (
Supplementary Table S1). PCR was performed as follows: hot start for 2 min at 95 °C, denaturation for 10 s at 95 °C, annealing for 15 s according to the Tm of each primer, and extension for 10 s at 72 °C for 15–30 cycles. Relative mRNA level was quantitated using β-actin or GAPDH as an endogenous control using the ΔΔCt algorithm. The experiment was performed in technical and experimental triplicates. RT2 Profiler PCR Array for genes associated with tumor cell proliferation, EMT, and cancer stemness (catalog no., CAPA9696-12: CLAH36595) was purchased from Qiagen.
2.13. Immunoblot Analysis
Immunoblotting was performed as described earlier with some modification [
16,
17,
18]. Briefly, whole-cell lysates were prepared from 2D or 3D cultures. Cells and spheroids were washed with cold 1X PBS and lysed using RIPA lysis buffer (50 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% TritonX-100, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM Phenylmethylsulfonyl fluoride (PMSF), 4 μg/mL aprotinin, 4 μg/mL leupeptin, 0.6 μg/mL benzamidinchloride, 20 μg/mL trypsin inhibitor) for 15 min at 4 °C. Lysates were centrifuged at 4 °C for 15 min at 13,000 rpm to remove insoluble debris. Protein concentration in the lysate was estimated using bicinchoninic acid (BCA method) as described by the manufacturer (Thermo Fisher Scientific Inc., Waltham, MA, USA). Proteins were then resolved on a 10% SDS-PAGE gel and transferred to a polyvinylidene difluoride membrane (Millipore Corporation, Burlington, MA, USA). The PVDF membrane was blocked with 5% nonfat dry milk and was incubated with primary antibody (1:1000 dilution) overnight at 4 °C followed by incubation with horseradish peroxidase-conjugated secondary antibodies (Bio-Rad, Hercules, CA, USA). β-actin was used as a loading control. The expression of specific proteins was detected using chemiluminescence in iBright Western Blot Imaging Systems (Thermofisher scientific, Waltham, MA, USA). For antibodies, see
Table S2.
2.14. Animal Study
All experiments on mice were performed in accordance with the Medical College of Wisconsin institutional Animal Care and Use Committee. All mice were housed and cared for according to the Institutional Animal Care and Use Committee (IACUC) at the Medical College of Wisconsin with institution guidelines. Animal health was monitored daily, and animal weights measured at least weekly.
Subcutaneous Tumor Cell Inoculation
MCW-OV-SL-3 or A2780 cells were trypsinized, washed, and resuspended in Hanks’ balanced salt solution (HBSS, GIBCO, Carlsbad, CA, USA) and a 50 µL cell suspension containing 1 × 106 cells was injected into each mouse (n = 6) subcutaneously into the left flank region. We purchased 4–6-week-old female athymic nude mice (CrTac: NCr-Foxn1nu) from Taconic Laboratories and they were housed in pathogen free conditions. Tumor-bearing mice were randomly divided into four groups (n = 6/group) after tumors had grown to an average of 100 mm3. Mice were treated weekly with intraperitoneal doses of PBS control, cisplatin (7 mg/kg body weight) at room temperature beginning on day 10 post-inoculation. Tumor volume was calculated according to the formula V = (length × width 2)½. Treatment was continued for 7 weeks, at which point, all mice were sacrificed, necropsied, and tumors were harvested. Subcutaneous tumor measurement was performed weekly in mice exhibiting palpable subcutaneous tumors until 6 weeks or humane endpoints. Tumor tissue was prepared as snap frozen for RNA and protein isolation or fixed in 10% formalin for immunohistochemistry.
2.15. Bioinformatic Analysis
A heatmap representing fold change of genes in monolayer versus spheroids was prepared by the Qiagen RT-PCR Profiler Software (
https://dataanalysis2.qiagen.com/pcr accessed on 5 March 2021) by converting the individual normalized ΔΔCt values of “monolayer” and “spheroid” population to 2
−ΔΔCt. The experiment was performed in technical triplicates as recommended by the Qiagen RT-PCR Profiler Analysis program. Genes with ≥ 1.4-fold change in expression with a
p < 0.05 were selected. Five housekeeping genes (B2M, HPRT1, RPLP0, GAPDH, and ACTB) were used for normalizing the data.
Clinical Data Analysis was performed by cBioPortal
http://www.cbioportal.org/ accessed on 8 April 2021 and GISTIC2 (Genomic Identification of Significant Targets in Cancer, version 2) analysis using Firehose-suggested parameters. Protein–protein interaction was analyzed by String software (Search Tool for the Retrieval of Interacting Genes/Proteins,
https://string-db.org/ accessed on 15 March 2021 ).
2.16. Statistical Analysis
All assays were performed in at least triplicate or more as indicated in the figure legends. Data are represented as means ±SE. Statistical comparisons were performed using unpaired two-tailed Student’s t tests or by ANOVA, where appropriate, with a probability value of 0.05 considered significant using GraphPad Software
https://www.graphpad.com/scientificsoftware/prism/ accessed on 19 March 2020.
4. Discussion
Dissecting the genomic alterations and transcriptomic landscapes of well characterized cell lines of a histological subtype will allow for the study of the molecular mechanisms underlying the progression and chemoresistance in ovarian cancer. In this regard, we established an endometrioid ovarian cancer cell line that forms tumor spheroids when confluent as floating spheroids, which occurs in ovarian cancer patients during metastasis. Recently, we demonstrated a mechanism that underlies the transition from a monolayer of adherent cells to non-adherent 3D spheroids, where we found that in HGSOC cell lines the adherent cells are highly proliferative and rely on the FOXM1 transcription factor for cell adhesion and proliferation [
23]. In contrast, we also found that the non-adherent spheroids in HGSOC cell lines rely on EGFR or ERBB2 signaling for their survival and growth [
23]. However, the exact mechanism that facilitates spheroid formation in conjunction with cancer stemness and chemoresistance is poorly studied in the endometrioid subtype of ovarian cancer. Therefore, we established an MCW-OV-SL3 cell line to characterize the underlying mechanism in endometrioid ovarian cancer and determined the role of chromosomal alteration in chemoresistance and cancer cell aggressiveness.
Chromosomal abnormalities are common phenomena in development and progression of many human cancers including ovarian carcinoma [
24]. Previously, we have demonstrated that several genes including both protein coding and non-coding genes are aberrantly expressed due to copy number gain in ovarian cancer and in breast cancer [
17,
24,
25,
26,
27,
28]. Moreover, chromosomal abnormalities in 1q21 such as amplifications, rearrangements, and translocations, have been reported in several hematological malignancies and solid tumors including endometroid ovarian carcinoma [
29,
30,
31]. In conjunction, we identified that chromosomal abnormalities in the 1q (q21–q42) locus in MCW-OV-SL3 are an important contributor to cancer stemness in highly aggressive tumors. Importantly, the interstitial duplication in the 1q21–q42 locus resulted in the activation of PI3K/AKT signaling, which is likely to be a major reason for chemoresistance. Thus, the genes located in the 1q21–q42 locus could serve as reliable indicators to predict the out-come of chemotherapy as well as the need for PI3K inhibitors for cancer therapy. We also observed that PIK3C2, PIP5K1B, and AKT3 are highly frequently amplified in the TCGA dataset of ovarian cancer.
Among the genes located in 1q (21–42) are protein kinase B (AKT 1–3), PIK3CA, and PTEN, which are frequently hyperactivated or deleted in nearly 12–20% of patients with endometroid ovarian carcinoma [
32]. It is well known that PIK3C2, PIP5K1B, AKT3 are either amplified or mutated in many cancers including ovarian cancer [
2]. The PIK3CA gene encodes the catalytic subunit of phosphatidylinositol 3-kinase (PI3K-p110α), and AKT is the downstream effector in the PI3K pathway. PI3K proteins are heterodimers composed of a catalytic p110 subunit (PIK3CA) and a regulatory p85 subunit (PIK3R) that mediate receptor binding and activation. PI3K directly binds to phosphotyrosine residues of growth factor receptors or adaptors through pleckstrin homology domains leading to allosteric activation of the catalytic p110 subunit, and the phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) converts it to the active second messenger, PIP3. As a result, the PI3K complex is recruited to the plasma membrane and activates the pyruvate dehydrogenase kinase 1 (PDK1) and Akt proteins [
33]. The PI3K/AKT pathway plays a significant role in the pathogenesis of ovarian cancer growth, survival, metabolic programing, autophagy, transcription regulation, and angiogenesis [
34,
35,
36].
We noticed that MCW-OV-SL-3 and A2780 cells exhibited similar signaling mechanisms and dependency on PI3K/Akt signaling for spheroid formation and cancer stemness. It has also been observed that these cells do not adhere firmly to plastic and glass surfaces as is common with other cell lines and can be detached easily unlike other ovarian cancer cell lines, suggesting that these are aggressive cell lines and likely to be highly metastatic through peritoneal spreading, which is a hallmark of ovarian cancer metastasis [
12].
In this study, we observed that a hyperactive PI3K/Akt cascade is associated with the spheroid formation, cancer stemness, chemoresistance, and epithelial to mesenchymal transition in the MCW-OV-SL3 cell line, and aggressive growth of the cisplatin-resistant versions of MCW-OV-SL3 cells. The mechanism of chemoresistance is multifactorial and is proposed to arise from a physical barrier to drug penetration, induction of genes or signaling pathways that enhance survival of drug-resistant cancer stem cell subpopulations, or due to chromosomal alterations and epigenetic disposition [
37,
38]. In this study, we found that the treatment of parental and cisplatin-resistant MCW-OV-SL3 with PI3K-Akt inhibitor LY294002 decreased PI3K/AKT/MEK signaling. We also found that LY294002 inhibitor decreased cell viability, migration, invasion, and spheroid forming ability of chemo-resistant MCW-OV-SL3-CisR. Taken together, our data support the notion that the cell line we developed is clinically relevant to study the pathophysiology and drug resistance mechanism in ovarian cancer. Studies like ours that use the primary cell lines established from patient samples for testing of chromosomal aberrations and the associated mechanism of tumor progression and chemoresistance through tumor spheroid formation ability will identify appropriate regiments for personalized therapy for ovarian cancer.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




