
DNA Methylation in Lung Cancer: Mechanisms and Associations with Histological Subtypes, Molecular Alterations, and Major Epidemiological Factors


Abstract
:Simple Summary
Abstract
1. Introduction
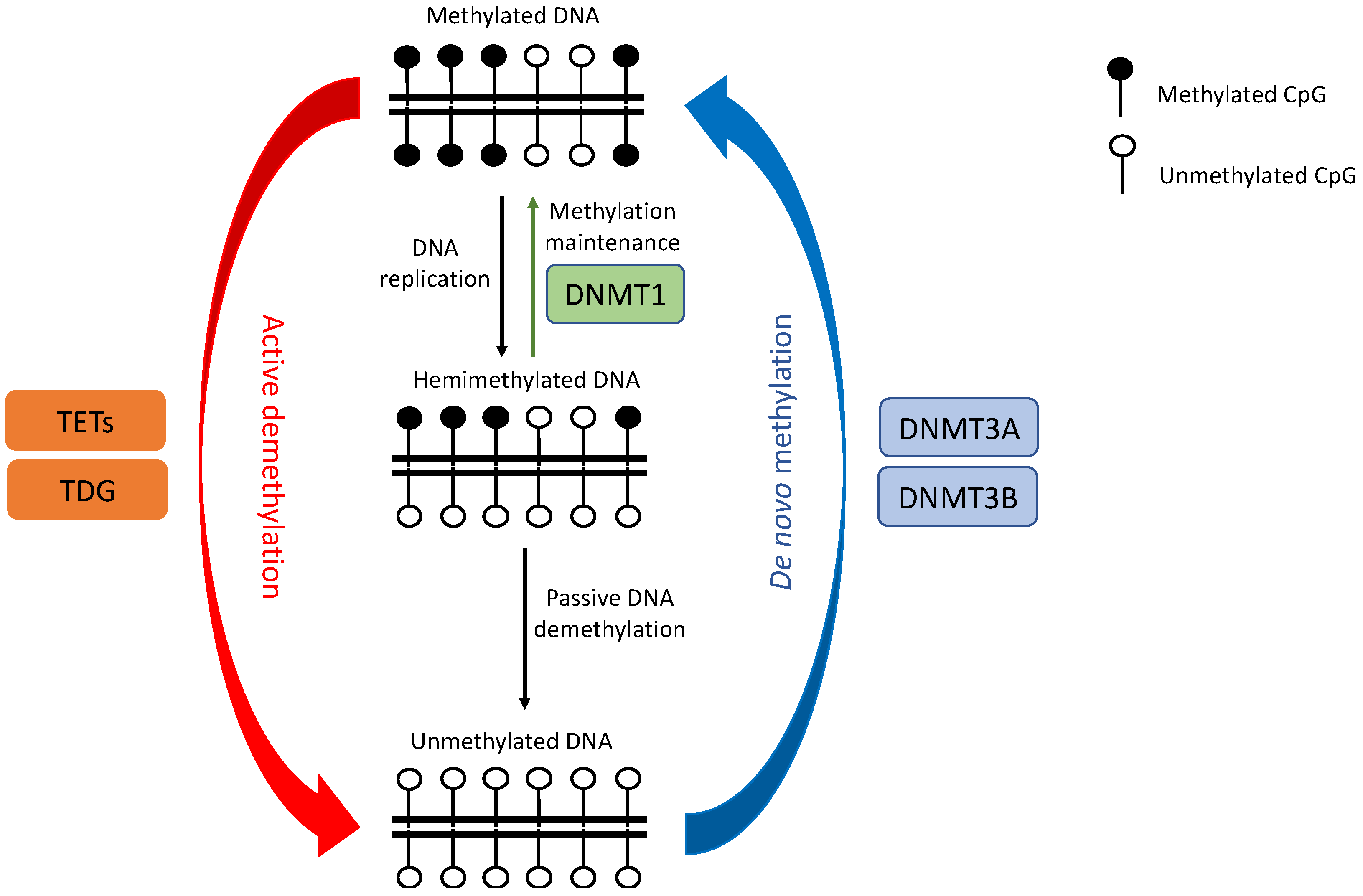
2. DNA Methylation Dysregulation in Lung Cancer
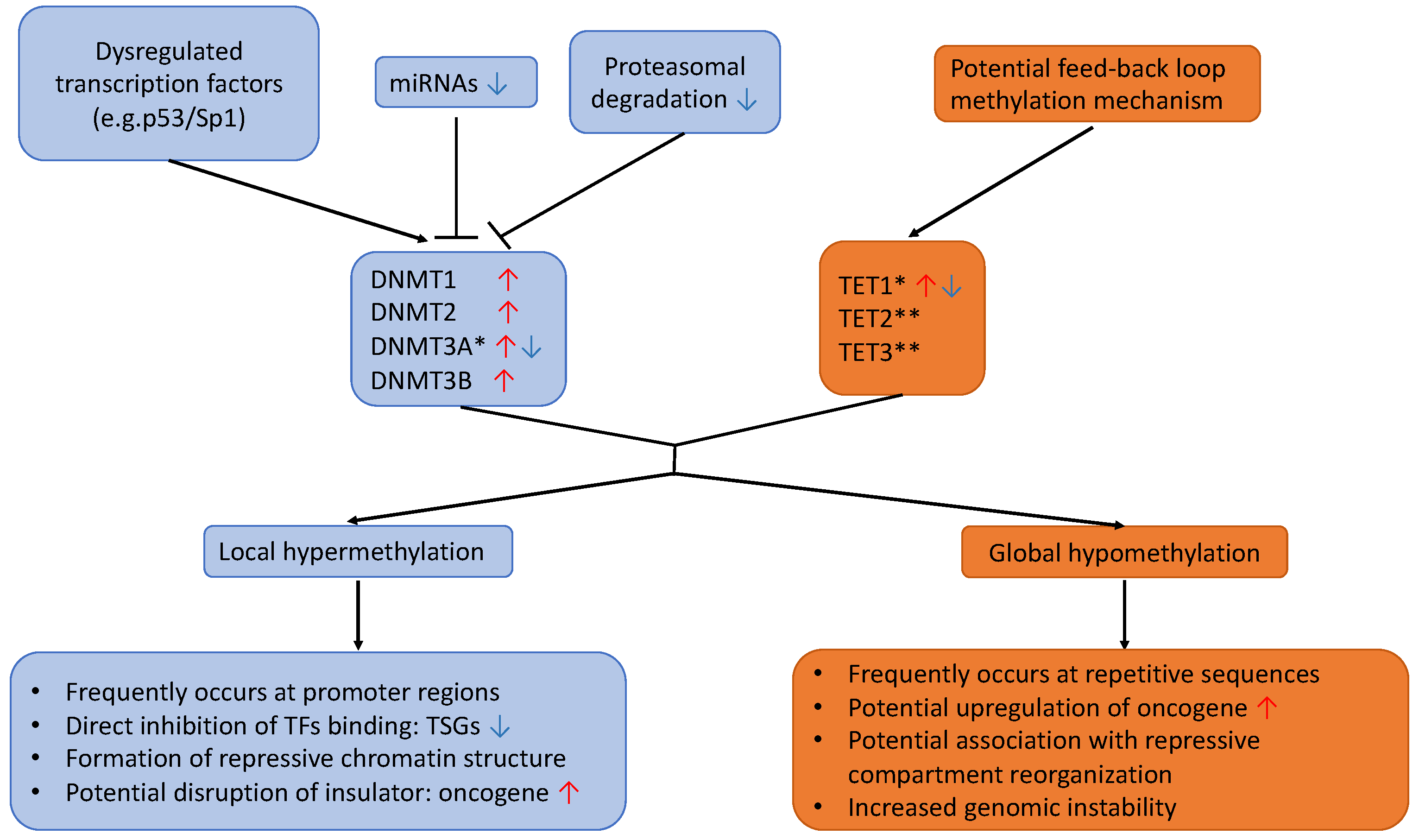
2.1. DNMTs Dysregulation
2.2. TETs Dysregulation
2.3. Hypomethylation
2.4. Hypermethylation

{kind=link}
{kind=link}
{kind=link}
| Methylation Changes in Tumors | Gene | Pathways | Histological Type Reported | Reference |
|---|---|---|---|---|
| Hypermethylation | APC | Cell proliferation, migration, and cell adhesion | NSCL, SCLC | [109,110] |
| CASP8 | Apoptosis | SCLC | [90] | |
| CDH1 | Cell adhesion | NSCL, SCLC | [111,112] | |
| CDH13 | Cell adhesion | NSCLC, SCLC | [113] | |
| CDKN2A/p16 | Cell cycle regulation | NSCLC, SCLC | [55,95] | |
| DAPK | Apoptosis | NSCLC | [92] | |
| FHIT | Cell proliferation and apoptosis | NSCLC, SCLC | [114,115] | |
| GSTP1 | Detoxification | NSCLC, SCLC | [116,117] | |
| MGMT | DNA repair | NSCLC, SCLC | [116,118] | |
| MLH1 | DNA repair | NSCLC | [98] | |
| MSH2 | DNA repair | NSCLC | [98] | |
| PTEN | Cell cycle regulation | NSCLC | [119] | |
| RARβ | Cell differentiation and proliferation | NSCLC, SCLC | [99] | |
| RASSF1A | Cell cycle regulation, genomic-stability maintenance, apoptosis, cell migration and invasion | NSCLC, SCLC | [120,121] | |
| RUNX3 | TGF-β/Wnt signaling pathway | NSCLC, SCLC | [100] | |
| SEMA3B | Cell adhesion | NSCLC, SCLC | [122,123] | |
| SHOX2 | Cell differentiation and proliferation | NSCLC, SCLC | [102] | |
| TERT | Immortalization of cancer cells | Lung cancer | [106] | |
| TGFBR2 | Signaling | NSCLC | [124] | |
| TNFRSF6/Fas | Apoptosis | SCLC | [90] | |
| TRAIL-R1/DR4 | Apoptosis | SCLC | [90] | |
| TSLC1 | Cell adhesion | NSCLC, SCLC | [125] | |
| Hypomethylation | MAGE | Transcriptional regulation, cancer development and progression | NSCLC | [78] |
| SNCG | Cell migration and invasion | Lung cancer | [76] |
3. DNA Methylation in Different Histological Subtypes
3.1. Non-Small Cell and Small Cell Lung Cancer
3.2. Lung Adenocarcinoma and Squamous Cell Carcinoma
4. Smoking and DNA Methylation
5. Molecular Status (KRAS, EGFR, TP53 Mutations) and Methylation
6. Race/Ethnicity and Sex
| Gene | Groups with Higher Methylated Frequency | Ethnic/Racial/Geographical Difference Reported | Sex Difference Reported |
|---|---|---|---|
| CDH13 | Females | [167] | |
| ERα | Males | [193] | |
| ESR1 | Females | [167] | |
| GATA5 | Females | [167] | |
| GSTP1 | USA/Australia higher than Japan/Taiwan | [14] | |
| KCNH5 | Females | [138] | |
| KCNH8 | Females | [138] | |
| MGMT | Conflicting reports, USA/Australia higher than Japan/Taiwan | [14] | [162] |
| PAX6 | Females | [167] | |
| RARβ | Conflicting reports | [17,138] | |
| RASSF1 | Conflicting reports | [17,160] |
7. Conclusions and Perspectives
- The dysregulation of DNMTs, TETs, and other related proteins (e.g., Polycomb protein EZH2) play a major role in altering DNAm patterns in lung cancer, creating a window of opportunity for targeted drug development and treatment. However, as highlighted in our review, further studies are required to reconcile and elucidate the conflicting reports regarding their specific roles (e.g., DNMT3A, TETs). We suspect that the paradox might be attributed to various factors, including mutational driver status, model systems, and histological subtypes.
- In addition, there remains a knowledge gap in how different DNMT isoforms specifically contribute to lung cancer development. For instance, DNMTB has over 30 different isoforms with ∆DNMT3B4-del being the most abundant isoform in NSCLC [194]. In this study, the overexpression of ∆DNMT3B4-del led to increased global hypomethylation, local hypermethylation, and epithelial hyperplasia. However, the expression of ∆DNMT3B4-del alone was not sufficient to transform lung epithelial cells into tumor cells in animal models. In addition, the abnormal expression of other isoforms was also observed. Therefore, a better knowledge of the role of DNMT isoforms in lung cancer is important.
- The dynamic remodeling of DNAm is essential for lung development and cell fate decisions as stem cells exit pluripotency [195]. Many studies have shown that DNA hypermethylation at key developmental genes occupied by the Polycomb complex in embryonic stem cells is a common hallmark in many tumor types, including lung cancers [128,166,196]. The current model suggests that DNA hypermethylation could result in shifting the balance towards the silencing of these developmental genes. These genes are maintained at low expression in embryonic and adult stem cells; hence, their silencing contributes to a stem-like state with upregulated oncogenic pathways (e.g., KRAS/MAPK signaling) and to sensitizing cells to malignant transformation. The dysregulation of various lineage TFs, either through DNA methylation or somatic mutations, in combination with cancer-driver-gene mutations could potentially influence the formation of different lung cancer subtypes [197]. It has been shown that the dysregulation of neuroendocrine-specifying TFs by DNAm contributes to SCLC tumorigenesis [85]. It is important to fully comprehend whether and which TFs are responsible for the development of other lung cancer subtypes through this mechanism, and how we can exploit this knowledge for therapeutic treatment and prevention.
- Most studies so far have focused on describing hypermethylated promoters and downregulated target genes. However, the notable example of TERT upregulation through DNA hypermethylation requires further investigation of similar phenomena [106]. Although the detailed biological mechanisms for such activation remain unknown, hypermethylation might regulate the binding of methylation-sensitive TFs and/or the expression of nearby genes that ultimately influence expression. Alternatively, DNA methylation might result in the disruption of genome topology, driving aberrant regulatory interactions and abnormal expression of oncogenes in cancer [107,108]; thus it should be investigated in lung cancers. In addition, further studies integrating multi-omic data are required to elucidate the roles of global hypomethylation in lung and other cancer types. How concomitant hypermethylation and hypomethylation in CpG sites within the same gene regulates the gene expression of the target gene is another research question of interest.
- Although cell lines provide a useful model to study processes that can be observed in tumors, several studies have revealed that DNAm data from cell lines might not be representative of those from primary tumors. For instance, a global analysis indicated that cell lines are much more heavily methylated compared to primary tumors [198]. In concordance, Poirier et al. observed that DNA methylation profiles in primary SCLC are distinct from those of cell lines [126]. The source of the difference is unclear, but this means researchers should be cautious, and conclusions drawn from cell line models should be validated in primary tumors.
- Although different lung cancer subtypes (NSCLC vs. SCLC, LUAD vs. LUSC) have distinct genetic and molecular profiles, there have been limited direct comparisons of the DNAm epigenome between them. Future studies with a large sample size encompassing various lung cancer pathological entities would be required to systematically characterize the differences in their DNAm landscapes. The knowledge would be essential to understand the underlying mechanisms driving tumorigenesis of different subtypes, identify biomarkers for accurate differential diagnosis, and develop effective personalized treatments.
- While in lung cancers from smokers, tobacco-smoking carcinogens can provide a “fertile ground” for oncogenic mutations that drive tumor development [166] even in the early stages of lung carcinogenesis [199]. However, which exogenous/endogenous factors (e.g., environmental pollutants, inflammation, aging, chronic cellular stress) drive the epigenetic transformation of lung cancers in the absence of smoking carcinogens is unclear and warrants further investigation. With large datasets including genomic, epigenomic, and expression data from lung cancers in never-smokers, e.g., the Sherlock-Lung study [200], many of these questions could be answered.
- Recent studies have observed congruent genomic and DNAm evolutionary trajectories in lung cancer [82,201] as well as other cancers [202,203], highlighting the potential for epigenetic changes to provide a milieu for genomic changes driving tumorigenesis. Integrating the genomic and epigenomic profiles of lung cancer in future studies is essential to better comprehend lung tumor evolution.
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, S.B.; Phil, A.; Crosbie, P.A.; Balata, H.; Chudziak, J.; Hussell, T.; Dive, C. Progress and prospects of early detection in lung cancer. Open Biol. 2017, 7, 170070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-cell lung cancer. Nat. Rev. Dis. Primers 2021, 7, 3. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- van Den Broeck, A.; Brambilla, E.; Moro-Sibilot, D.; Lantuejoul, S.; Brambilla, C.; Eymin, B.; Khochbin, S.; Gazzeri, S. Loss of Histone H4K20 Trimethylation Occurs in Preneoplasia and Influences Prognosis of Non–Small Cell Lung Cancer. Clin. Cancer Res. 2008, 14, 7237–7245. [Google Scholar] [CrossRef] [Green Version]
- Langevin, S.; Kratzke, R.A.; Kelsey, K.T. Epigenetics of lung cancer. Transl. Res. 2015, 165, 74–90. [Google Scholar] [CrossRef] [Green Version]
- Kurdyukov, S.; Bullock, M. DNA Methylation Analysis: Choosing the Right Method. Biology 2016, 5, 3. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Rauch, T.A. DNA methylation patterns in lung carcinomas. Semin. Cancer Biol. 2009, 19, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Iniesta, P.; Morán, A.; Fernandez-Marcelo, T.; Carro, J.; De Juan, C.; Pascua, I.; Head, J.; Gómez, A.; Hernando, F.; Torres, A.-J.; et al. Methylation profiling in non-small cell lung cancer: Clinical implications. Int. J. Oncol. 2011, 40, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokk, K.; Vooder, T.; Kolde, R.; Välk, K.; Võsa, U.; Roosipuu, R.; Milani, L.; Fischer, K.; Koltsina, M.; Urgard, E.; et al. Methylation Markers of Early-Stage Non-Small Cell Lung Cancer. PLoS ONE 2012, 7, e39813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, G.; Babinsky, V.N.; Ziegler, B.; Weinzierl, M.; Noll, C.; Altenberger, C.; Müllauer, L.; Dekan, G.; Grin, Y.; Lang, G.; et al. Genome-wide CpG island methylation analyses in non-small cell lung cancer patients. Carcinogenesis 2013, 34, 513–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjaanæs, M.M.; Fleischer, T.; Halvorsen, A.R.; Daunay, A.; Busato, F.; Solberg, S.; Jørgensen, L.; Kure, E.; Edvardsen, H.; Børresen-Dale, A.-L.; et al. Genome-wide DNA methylation analyses in lung adenocarcinomas: Association with EGFR, KRAS and TP53 mutation status, gene expression and prognosis. Mol. Oncol. 2016, 10, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Selamat, S.A.; Chung, B.S.; Girard, L.; Zhang, W.; Zhang, Y.; Campan, M.; Siegmund, K.D.; Koss, M.N.; Hagen, J.A.; Lam, W.L.; et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012, 22, 1197–1211. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Liu, L.; Chen, H.; Wang, Y.; Xu, Y.; Mao, H.; Li, J.; Mills, G.B.; Shu, Y.; Li, L.; et al. Comprehensive Characterization of Molecular Differences in Cancer between Male and Female Patients. Cancer Cell 2016, 29, 711–722. [Google Scholar] [CrossRef] [Green Version]
- Toyooka, S.; Maruyama, R.; Toyooka, K.O.; McLerran, D.; Feng, Z.; Fukuyama, Y.; Virmani, A.K.; Zöchbauer-Müller, S.; Tsukuda, K.; Sugio, K.; et al. Smoke exposure, histologic type and geography-related differences in the methylation profiles of non-small cell lung cancer. Int. J. Cancer 2003, 103, 153–160. [Google Scholar] [CrossRef]
- Park, S.L.; Patel, Y.M.; Loo, L.W.M.; Mullen, D.J.; Offringa, I.A.; Maunakea, A.; Stram, D.O.; Siegmund, K.; Murphy, S.E.; Tiirikainen, M.; et al. Association of internal smoking dose with blood DNA methylation in three racial/ethnic populations. Clin. Epigenet. 2018, 10, 1–12. [Google Scholar] [CrossRef]
- Brock, M.V.; Hooker, C.M.; Ota-Machida, E.; Han, Y.; Guo, M.; Ames, S.; Glöckner, S.; Piantadosi, S.; Gabrielson, E.; Pridham, G.; et al. DNA Methylation Markers and Early Recurrence in Stage I Lung Cancer. N. Engl. J. Med. 2008, 358, 1118–1128. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, J.; Mendez-Gonzalez, J.; Nadal, E.; Chen, G.; Carmona, F.J.; Sayols, S.; Moran, S.; Heyn, H.; Vizoso, M.; Gomez, A.; et al. A Prognostic DNA Methylation Signature for Stage I Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2013, 31, 4140–4147. [Google Scholar] [CrossRef]
- Karlsson, A.; Jönsson, M.; Lauss, M.; Brunnström, H.; Jönsson, P.; Borg, Å.; Jönsson, G.; Ringnér, M.; Planck, M.; Staaf, J. Genome-wide DNA Methylation Analysis of Lung Carcinoma Reveals One Neuroendocrine and Four Adenocarcinoma Epitypes Associated with Patient Outcome. Clin. Cancer Res. 2014, 20, 6127–6140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, R.; Li, X.; Li, W.; Zhu, X.; Li, C. DNA methylation in lung cancer patients: Opening a "window of life" under precision medicine. Biomed. Pharmacother. 2021, 144, 112202. [Google Scholar] [CrossRef]
- Ren, M.; Wang, C.; Sheng, D.; Shi, Y.; Jin, M.; Xu, S. Methylation analysis of SHOX2 and RASSF1A in bronchoalveolar lavage fluid for early lung cancer diagnosis. Ann. Diagn. Pathol. 2017, 27, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, W.; Wang, L.; Zhao, M.; Guo, Q.; Lv, S.; Hu, X.; Lou, J. DNA Methylation Analysis of the SHOX2 and RASSF1A Panel in Bronchoalveolar Lavage Fluid for Lung Cancer Diagnosis. J. Cancer 2017, 8, 3585–3591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponomaryova, A.A.; Rykova, E.Y.; Cherdyntseva, N.V.; Skvortsova, T.E.; Dobrodeev, A.; Zav’Yalov, A.A.; Bryzgalov, L.O.; Tuzikov, S.; Vlassov, V.; Laktionov, P.P. Potentialities of aberrantly methylated circulating DNA for diagnostics and post-treatment follow-up of lung cancer patients. Lung Cancer 2013, 81, 397–403. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Y.; Wang, L.; Xiong, J.; Wang, H.; Cui, F.; Peng, H. The performance of the SHOX2/PTGER4 methylation assay is influenced by cancer stage, age, type and differentiation. Biomark. Med. 2020, 14, 341–351. [Google Scholar] [CrossRef]
- Weiss, G.; Schlegel, A.; Kottwitz, D.; König, T.; Tetzner, R. Validation of the SHOX2/PTGER4 DNA Methylation Marker Panel for Plasma-Based Discrimination between Patients with Malignant and Nonmalignant Lung Disease. J. Thorac. Oncol. 2017, 12, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Grote, H.J.; Schmiemann, V.; Geddert, H.; Rohr, U.P.; Kappes, R.; Gabbert, H.E.; Böcking, A. Aberrant promoter methylation of p16(INK4a), RARB2 and SEMA3B in bronchial aspirates from patients with suspected lung cancer. Int. J. Cancer 2005, 116, 720–725. [Google Scholar] [CrossRef]
- Yu, J.; Bulk, E.; Ji, P.; Hascher, A.; Tang, M.; Metzger, R.; Marra, A.; Serve, H.; Berdel, W.E.; Wiewroth, R.; et al. The EPHB6 Receptor Tyrosine Kinase Is a Metastasis Suppressor That Is Frequently Silenced by Promoter DNA Hypermethylation in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2010, 16, 2275–2283. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.-A.; Kim, Y.; Hong, S.-H.; Lee, J.; Cho, Y.G.; Han, J.-Y.; Kim, Y.-H.; Han, J.; Shim, Y.M.; Lee, Y.-S.; et al. Epigenetic Inactivation of Heparan Sulfate (Glucosamine) 3-O-Sulfotransferase 2 in Lung Cancer and Its Role in Tumorigenesis. PLoS ONE 2013, 8, e79634. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Feng, N.; Yu, X.; Lin, H.; Zhang, X.; Shi, O.; Zhang, H.; Zhang, S.; Li, L.; Zheng, M.; et al. Promoter methylation of Wnt/β-Catenin signal inhibitor. Cancer Biol. Med. 2017, 14, 377–386. [Google Scholar] [PubMed] [Green Version]
- Zhang, Y.; Xu, R.; Li, G.; Xie, X.; Long, J.; Wang, H. Loss of expression of the differentially expressed in adenocarcinoma of the lung (DAL-1) protein is associated with metastasis of non-small cell lung carcinoma cells. Tumor Biol. 2012, 33, 1915–1925. [Google Scholar] [CrossRef]
- Søes, S.; Daugaard, I.; Sørensen, B.S.; Carus, A.; Mattheisen, M.; Alsner, J.; Overgaard, J.; Hager, H.; Hansen, L.L.; Kristensen, L.S. Hypomethylation and increased expression of the putative oncogene ELMO3 are associated with lung cancer development and metastases formation. Oncoscience 2014, 1, 367–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, K.; Narita, Y.; Matsushita, Y.; Miyakita, Y.; Ono, M.; Kayama, T.; Shibui, S. Methylation status of O6-methylguanine-DNA-methyl transferase promoter region in non-small-cell lung cancer patients with brain metastasis. Clin. Transl. Oncol. 2012, 14, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.-W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2015, 1856, 189–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef]
- Nunes, S.P.; Diniz, F.; Moreira-Barbosa, C.; Constâncio, V.; Silva, A.V.; Oliveira, J.; Soares, M.; Paulino, S.; Cunha, A.L.; Rodrigues, J.; et al. Subtyping Lung Cancer Using DNA Methylation in Liquid Biopsies. J. Clin. Med. 2019, 8, 1500. [Google Scholar] [CrossRef] [Green Version]
- De Cáceres, I.I.; Cortés-Sempere, M.; Moratilla, C.; Machado-Pinilla, R.; Rodriguez-Fanjul, V.; Manguan-García, C.; Cejas, P.; Lopez-Rios, F.; Paz-Ares, L.; de CastroCarpeño, J.; et al. IGFBP-3 hypermethylation-derived deficiency mediates cisplatin resistance in non-small-cell lung cancer. Oncogene 2009, 29, 1681–1690. [Google Scholar] [CrossRef] [Green Version]
- Fischer, J.R.; Ohnmacht, U.; Rieger, N.; Zemaitis, M.; Stoffregen, C.; Manegold, C.; Lahm, H. Prognostic significance of RASSF1A promoter methylation on survival of non-small cell lung cancer patients treated with gemcitabine. Lung Cancer 2007, 56, 115–123. [Google Scholar] [CrossRef]
- Hiddinga, B.I.; Pauwels, P.; Janssens, A.; van Meerbeeck, J. O 6 -Methylguanine-DNA methyltransferase (MGMT): A drugable target in lung cancer? Lung Cancer 2017, 107, 91–99. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Jurkowska, R.Z. New concepts in DNA methylation. Trends Biochem. Sci. 2014, 39, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, E. Establishment and Maintenance of DNA Methylation Patterns in Mammals. Curr. Top. Microbiol. Immunol. 2006, 301, 179–201. [Google Scholar] [CrossRef]
- Tuorto, F.; Herbst, F.; Alerasool, N.; Bender, S.; Popp, O.; Federico, G.; Reitter, S.; Liebers, R.; Stoecklin, G.; Gröne, H.-J.; et al. The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J. 2015, 34, 2350–2362. [Google Scholar] [CrossRef] [Green Version]
- Jurkowska, R.Z.; Anspach, N.; Urbanke, C.; Jia, D.; Reinhardt, R.; Nellen, W.; Cheng, X.; Jeltsch, A. Formation of nucleoprotein filaments by mammalian DNA methyltransferase Dnmt3a in complex with regulator Dnmt3L. Nucleic Acids Res. 2008, 36, 6656–6663. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kwon, Y.M.; Kim, J.S.; Han, J.; Shim, Y.M.; Park, J.; Kim, D.-H. Elevated mRNA levels of DNA methyltransferase-1 as an independent prognostic factor in primary nonsmall cell lung cancer. Cancer 2006, 107, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.-K.; Hsu, H.-S.; Chang, J.-W.; Chen, C.-Y.; Chen, J.-T.; Wang, Y.-C. Alteration of DNA methyltransferases contributes to 5′CpG methylation and poor prognosis in lung cancer. Lung Cancer 2007, 55, 205–213. [Google Scholar] [CrossRef]
- Lin, R.K.; Wu, C.Y.; Chang, J.W.; Juan, L.J.; Hsu, H.S.; Chen, C.Y.; Lu, Y.-Y.; Tang, Y.-A.; Yang, Y.-C.; Yang, P.-C.; et al. Dysregulation of p53/Sp1 control leads to DNA methyltransferase-1 overexpression in lung cancer. Cancer Res. 2010, 70, 5807–5817. [Google Scholar] [CrossRef] [Green Version]
- Belinsky, S.A.; Klinge, D.M.; Stidley, C.A.; Issa, J.-P.; Herman, J.G.; March, T.H.; Baylin, S.B. Inhibition of DNA methylation and histone deacetylation prevents murine lung cancer. Cancer Res. 2003, 63, 7089–7093. [Google Scholar]
- Kassis, E.S.; Zhao, M.; Hong, J.A.; Chen, G.A.; Nguyen, D.M.; Schrump, D.S. Depletion of DNA methyltransferase 1 and/or DNA methyltransferase 3b mediates growth arrest and apoptosis in lung and esophageal cancer and malignant pleural mesothelioma cells. J. Thorac. Cardiovasc. Surg. 2006, 131, 298–306.e2. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Shen, N.; Pang, J.; Xie, D.; Deng, B.; Molina, J.R.; Yang, P.; Liu, S. Restoration of miR-101 suppresses lung tumorigenesis through inhibition of DNMT3a-dependent DNA methylation. Cell Death Dis. 2014, 5, e1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.-K.; Wang, Y.-C. Dysregulated transcriptional and post-translational control of DNA methyltransferases in cancer. Cell Biosci. 2014, 4, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husni, R.E.; Shiba-Ishii, A.; Iiyama, S.; Shiozawa, T.; Kim, Y.; Nakagawa, T.; Sato, T.; Kano, J.; Minami, Y.; Noguchi, M. DNMT3a expression pattern and its prognostic value in lung adenocarcinoma. Lung Cancer 2016, 97, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Steine, E.J.; Barrasa, M.I.; Hockemeyer, D.; Pawlak, M.; Fu, D.; Reddy, S.; Bell, G.W.; Jaenisch, R. Deletion of the de novo DNA methyltransferase Dnmt3a promotes lung tumor progression. Proc. Natl. Acad. Sci. USA 2011, 108, 18061–18066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Horio, Y.; Sekido, Y.; Minna, J.D.; Shimokata, K.; Hasegawa, Y. The expression of DNA methyltransferases and methyl-CpG-binding proteins is not associated with the methylation status of p14ARF, p16INK4a and RASSF1A in human lung cancer cell lines. Oncogene 2002, 21, 4822–4829. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.L.; Piccolo, S.R.; Cheng, L.; Soldi, R.; Han, B.; Johnson, W.E.; Bild, A.H. Genomic pathway analysis reveals that EZH2 and HDAC4 represent mutually exclusive epigenetic pathways across human cancers. BMC Med. Genom. 2013, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Xu, W.; Wang, Q.; Xiao, W.; Xu, R. Potential of DNMT and its Epigenetic Regulation for Lung Cancer Therapy. Curr. Genom. 2009, 10, 336–352. [Google Scholar] [CrossRef] [PubMed]
- Espada, J.; Ballestar, E.; Santoro, R.; Fraga, M.; Garea, A.V.; Németh, A.; Lopez-Serra, L.; Ropero, S.; Aranda, A.; Orozco, H.; et al. Epigenetic disruption of ribosomal RNA genes and nucleolar architecture in DNA methyltransferase 1 (Dnmt1) deficient cells. Nucleic Acids Res. 2007, 35, 2191–2198. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-Y.; Chen, H.-C.; Li, W.-W.; Yan, J.-D.; Lv, R.-Y. DNMT1 promotes cell proliferation via methylating hMLH1 and hMSH2 promoters in EGFR-mutated non-small cell lung cancer. J. Biochem. 2020, 168, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Serra, L.; Ballestar, E.; Fraga, M.F.; Alaminos, M.; Setién, F.; Esteller, M. A Profile of Methyl-CpG Binding Domain Protein Occupancy of Hypermethylated Promoter CpG Islands of Tumor Suppressor Genes in Human Cancer. Cancer Res. 2006, 66, 8342–8346. [Google Scholar] [CrossRef] [Green Version]
- Mutskov, V.J.; Farrell, C.M.; Wade, P.A.; Wolffe, A.P.; Felsenfeld, G. The barrier function of an insulator couples high histone acetylation levels with specific protection of promoter DNA from methylation. Genes Dev. 2002, 16, 1540–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frigola, J.; Song, J.; Stirzaker, C.; Hinshelwood, R.A.; Peinado, M.A.; Clark, S. Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat. Genet. 2006, 38, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Youssef, E.M.; Jelinek, J.; Chen, R.; Yang, A.S.; Garcia-Manero, G.; Issa, J.-P. Effect of Cytarabine and Decitabine in Combination in Human Leukemic Cell Lines. Clin. Cancer Res. 2007, 13, 4225–4232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, B.F.; Karpenko, M.J.; Liu, Z.; Aimiuwu, J.; Villalona-Calero, M.A.; Chan, K.K.; Grever, M.R.; Otterson, G.A. Phase I study of 5-aza-2′-deoxycytidine in combination with valproic acid in non-small-cell lung cancer. Cancer Chemother. Pharmacol. 2012, 71, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Ma, J.; Hu, C.; Zou, F.; Jiang, S.; Wang, Y.; Han, C.; Zhang, Y. Decitabine reverses gefitinib resistance in PC9 lung adenocarcinoma cells by demethylation of RASSF1A and GADD45β promoter. Int. J. Clin. Exp. Pathol. 2019, 12, 4002–4010. [Google Scholar]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar] [CrossRef] [Green Version]
- Karpf, A.R.; Matsui, S.-I. Genetic Disruption of Cytosine DNA Methyltransferase Enzymes Induces Chromosomal Instability in Human Cancer Cells. Cancer Res. 2005, 65, 8635–8639. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.-Y.; Ma, S.-H.; Liu, J.; Xu, Z.-D.; Zhu, H.-G.; Ling, Z.-Q.; Ye, D.; et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-Q.; Chen, D.-J.; Li, Y.; Yuan, W.-B.; Fan, J.; Zhang, Z.; Han, F.; Jiang, X.; Chen, J.-P.; Wang, D.-D.; et al. Epigenetic silencing of TET1 mediated hydroxymethylation of base excision repair pathway during lung carcinogenesis. Environ. Pollut. 2021, 268, 115860. [Google Scholar] [CrossRef]
- Li, L.; Li, C.; Mao, H.; Du, Z.; Chan, W.Y.; Murray, P.; Luo, B.; Chan, A.T.C.; Mok, T.; Chan, F.K.; et al. Epigenetic inactivation of the CpG demethylase TET1 as a DNA methylation feedback loop in human cancers. Sci. Rep. 2016, 6, 26591. [Google Scholar] [CrossRef] [PubMed]
- Filipczak, P.T.; Leng, S.; Tellez, C.S.; Do, K.C.; Grimes, M.J.; Thomas, C.L.; Walton-Filipczak, S.R.; Picchi, M.A.; Belinsky, S.A. p53-Suppressed Oncogene TET1 Prevents Cellular Aging in Lung Cancer. Cancer Res. 2019, 79, 1758–1768. [Google Scholar] [CrossRef] [Green Version]
- Rauch, T.A.; Zhong, X.; Wu, X.; Wang, M.; Kernstine, K.H.; Wang, Z.; Riggs, A.D.; Pfeifer, G.P. High-resolution mapping of DNA hypermethylation and hypomethylation in lung cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 252–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitahara, H.; Okamoto, T.; Shimamatsu, S.; Kohno, M.; Morodomi, Y.; Tagawa, T.; Kitao, H.; Okano, S.; Oda, Y.; Maehara, Y.; et al. LINE-1 Hypomethylation Is Associated with Malignant Traits and Cell Proliferation in Lung Adenocarcinoma. Anticancer Res. 2020, 40, 5659–5666. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Shiraishi, K.; Eguchi, A.; Shibata, H.; Yoshimoto, K.; Mori, T.; Baba, Y.; Baba, H.; Suzuki, M. Long Interspersed Nucleotide Element 1 Hypomethylation Is Associated with Poor Prognosis of Lung Adenocarcinoma. Ann. Thorac. Surg. 2013, 96, 1790–1794. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, W.; Wu, Y.; Zhou, Y.; Xue, R.; Luo, C.; Wang, L.; Zhao, W.; Jiang, J.-D.; Liu, J. Loss of epigenetic control of synuclein-gamma gene as a molecular indicator of metastasis in a wide range of human cancers. Cancer Res. 2005, 65, 7635–7643. [Google Scholar] [CrossRef] [Green Version]
- Shao, T.; Song, P.; Hua, H.; Zhang, H.; Sun, X.; Kong, Q.; Wang, J.; Luo, T.; Jiang, Y. Gamma synuclein is a novel Twist1 target that promotes TGF-β-induced cancer cell migration and invasion. Cell Death Dis. 2018, 9, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.J.; Soria, J.C.; Wang, L.; Hassan, K.A.; Morice, R.C.; Walsh, G.L.; Hong, W.K.; Mao, L. Activation of melanoma antigen tumor antigens occurs early in lung carcinogenesis. Cancer Res. 2001, 61, 7959–7963. [Google Scholar] [PubMed]
- Weon, J.; Potts, P.R. The MAGE protein family and cancer. Curr. Opin. Cell Biol. 2015, 37, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vachtenheim, J.; Horáková, I.; Novotná, H. Hypomethylation of CCGG sites in the 3’ region of H-ras protooncogene is frequent and is associated with H-ras allele loss in non-small cell lung cancer. Cancer Res. 1994, 54, 1145–1148. [Google Scholar]
- Daskalos, A.; Nikolaidis, G.; Xinarianos, G.; Savvari, P.; Cassidy, A.; Zakopoulou, R.; Kotsinas, A.; Gorgoulis, V.; Field, J.K.; Liloglou, T. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int. J. Cancer 2009, 124, 81–87. [Google Scholar] [CrossRef]
- Hu, X.; Estecio, M.R.; Chen, R.; Reuben, A.; Wang, L.; Fujimoto, J.; Carrot-Zhang, J.; McGranahan, N.; Ying, L.; Fukuoka, J.; et al. Evolution of DNA methylome from precancerous lesions to invasive lung adenocarcinomas. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Johnstone, S.E.; Reyes, A.; Qi, Y.; Adriaens, C.; Hegazi, E.; Pelka, K.; Chen, J.H.; Zou, L.S.; Drier, Y.; Hecht, V.; et al. Large-Scale Topological Changes Restrain Malignant Progression in Colorectal Cancer. Cell 2020, 182, 1474–1489.e23. [Google Scholar] [CrossRef]
- Siegmund, K.D.; Laird, P.W. Analysis of complex methylation data. Methods 2002, 27, 170–178. [Google Scholar] [CrossRef]
- Kalari, S.; Jung, M.; Kernstine, K.H.; Takahashi, T.; Pfeifer, G.P. The DNA methylation landscape of small cell lung cancer suggests a differentiation defect of neuroendocrine cells. Oncogene 2013, 32, 3559–3568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauch, T.; Wang, Z.; Zhang, X.; Zhong, X.; Wu, X.; Lau, S.K.; Kernstine, K.H.; Riggs, A.D.; Pfeifer, G.P. Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc. Natl. Acad. Sci. USA 2007, 104, 5527–5532. [Google Scholar] [CrossRef] [Green Version]
- Rauch, T.A.; Wang, Z.; Wu, X.; Kernstine, K.H.; Riggs, A.; Pfeifer, G.P. DNA methylation biomarkers for lung cancer. Tumor Biol. 2011, 33, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Khuri, F.R.; Lee, J.J.; Kemp, B.L.; Liu, D.; Hong, W.K.; Mao, L. Hypermethylation of the death-associated protein (DAP) kinase promoter and aggressiveness in stage I non-small-cell lung cancer. JNCI J. Natl. Cancer Inst. 2000, 92, 1511–1516. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Nelson, H.H.; Wiencke, J.K.; Christiani, D.C.; Wain, J.C.; Mark, E.J.; Kelsey, K.T. Promoter methylation of DAP-kinase: Association with advanced stage in non-small cell lung cancer. Oncogene 2001, 20, 1765–1770. [Google Scholar] [CrossRef]
- Hopkins-Donaldson, S.; Ziegler, A.; Kurtz, S.; Bigosch, C.; Kandioler, D.; Ludwig, C.; Zangemeister-Wittke, U.; Stahel, R. Silencing of death receptor and caspase-8 expression in small cell lung carcinoma cell lines and tumors by DNA methylation. Cell Death Differ. 2003, 10, 356–364. [Google Scholar] [CrossRef] [Green Version]
- Shivapurkar, N.; Toyooka, S.; Eby, M.T.; Huang, C.X.; Sathyanarayana, U.G.; Cunningham, H.T.; Reddy, J.L.; Brambilla, E.; Takahashi, T.; Minna, J.D.; et al. Differential inactivation of caspase-8 in lung cancers. Cancer Biol. Ther. 2002, 1, 65–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zöchbauer-Müller, S.; Fong, K.; Virmani, A.K.; Geradts, J.; Gazdar, A.F.; Minna, J.D. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001, 61, 249–255. [Google Scholar] [PubMed]
- Merlo, A.; Herman, J.G.; Mao, L.; Lee, D.J.; Gabrielson, E.; Burger, P.C.; Baylin, S.B.; Sidransky, D. 5’ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat. Med. 1995, 1, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Nelson, H.H.; Wiencke, J.K.; Zheng, S.; Christiani, D.C.; Wain, J.C.; Mark, E.J.; Kelsey, K.T. p16(INK4a) and histology-specific methylation of CpG islands by exposure to tobacco smoke in non-small cell lung cancer. Cancer Res. 2001, 61, 3419–3424. [Google Scholar]
- Belinsky, S.A.; Nikula, K.J.; Palmisano, W.A.; Michels, R.; Saccomanno, G.; Gabrielson, E.; Baylin, S.B.; Herman, J.G. Aberrant methylation of p16INK4a is an early event in lung cancer and a potential biomarker for early diagnosis. Proc. Natl. Acad. Sci. USA 1998, 95, 11891–11896. [Google Scholar] [CrossRef] [Green Version]
- Yanagawa, N.; Tamura, G.; Oizumi, H.; Kanauchi, N.; Endoh, M.; Sadahiro, M.; Motoyama, T. Promoter hypermethylation of RASSF1A and RUNX3 genes as an independent prognostic prediction marker in surgically resected non-small cell lung cancers. Lung Cancer 2007, 58, 131–138. [Google Scholar] [CrossRef]
- Brabender, J.; Usadel, H.; Metzger, R.; Schneider, P.M.; Park, J.; Salonga, D.; Tsao-Wei, D.D.; Groshen, S.; Lord, R.V.; Takebe, N.; et al. Quantitative O(6)-methylguanine DNA methyltransferase methylation analysis in curatively resected non-small cell lung cancer: Associations with clinical outcome. Clin. Cancer Res. 2003, 9, 223–227. [Google Scholar]
- Gomes, A.; Reis-Silva, M.; Alarcão, A.; Couceiro, P.; Sousa, V.; Carvalho, L. Promoter hypermethylation of DNA repair genes MLH1 and MSH2 in adenocarcinomas and squamous cell carcinomas of the lung. Rev. Port. Pneumol. 2014, 20, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Virmani, A.K.; Rathi, A.; Zöchbauer-Müller, S.; Sacchi, N.; Fukuyama, Y.; Bryant, D.; Maitra, A.; Heda, S.; Fong, K.M.; Thunnissen, T.; et al. Promoter methylation and silencing of the retinoic acid receptor-beta gene in lung carcinomas. J. Natl. Cancer Inst. 2000, 92, 1303–1307. [Google Scholar] [CrossRef]
- Sato, K.; Tomizawa, Y.; Iijima, H.; Saito, R.; Ishizuka, T.; Nakajima, T.; Mori, M. Epigenetic inactivation of the RUNX3 gene in lung cancer. Oncol. Rep. 2006, 15, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Grote, H.J.; Schmiemann, V.; Kiel, S.; Böcking, A.; Kappes, R.; Gabbert, H.E.; Sarbia, M. Aberrant methylation of the adenomatous polyposis coli promoter 1A in bronchial aspirates from patients with suspected lung cancer. Int. J. Cancer 2004, 110, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.; Liebenberg, V.; Dietrich, D.; Schlegel, T.; Kneip, C.; Seegebarth, A.; Flemming, N.; Seemann, S.; Distler, J.; Lewin, J.; et al. SHOX2 DNA Methylation is a Biomarker for the diagnosis of lung cancer based on bronchial aspirates. BMC Cancer 2010, 10, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.S.; Kim, M.J.; Lee, J.Y.; Kim, Y.Z.; Kim, E.J.; Park, J.Y. Aberrant methylation of E-cadherin and H-cadherin genes in nonsmall cell lung cancer and its relation to clinicopathologic features. Cancer 2007, 110, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.; Fong, K.; Girard, L.; Seidl, S.; End-Pfützenreuter, A.; Lang, G.; Gazdar, A.F.; Minna, J.D.; Zielinski, C.C.; Zöchbauer-Müller, S. Expression and methylation pattern of TSLC1 cascade genes in lung carcinomas. Oncogene 2005, 25, 959–968. [Google Scholar] [CrossRef] [Green Version]
- Tsou, J.A.; Hagen, J.A.; Carpenter, C.; Laird-Offringa, I.A. DNA methylation analysis: A powerful new tool for lung cancer diagnosis. Oncogene 2002, 21, 5450–5461. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.D.; Leão, R.; Komosa, M.; Gallo, M.; Zhang, C.H.; Lipman, T.; Remak, M.; Heidari, A.; Nunes, N.M.; Aplonio, J.D.; et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J. Clin. Investig. 2019, 129, 1801. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2015, 529, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Flavahan, W.A.; Drier, Y.; Johnstone, S.E.; Hemming, M.L.; Tarjan, D.R.; Hegazi, E.; Shareef, S.J.; Javed, N.M.; Raut, C.P.; Eschle, B.K.; et al. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature 2019, 575, 229–233. [Google Scholar] [CrossRef]
- Brabender, J.; Usadel, H.; Danenberg, K.D.; Metzger, R.; Schneider, P.M.; Lord, R.V.; Wickramasinghe, K.; Lum, C.E.; Park, J.; Salonga, D.; et al. Adenomatous polyposis coli gene promoter hypermethylation in non-small cell lung cancer is associated with survival. Oncogene 2001, 20, 3528–3532. [Google Scholar] [CrossRef] [Green Version]
- Virmani, A.K.; Rathi, A.; Sathyanarayana, U.G.; Padar, A.; Huang, C.X.; Cunnigham, H.T.; Farinas, A.J.; Milchgrub, S.; Euhus, D.M.; Gilcrease, M.; et al. Aberrant methylation of the adenomatous polyposis coli (APC) gene promoter 1A in breast and lung carcinomas. Clin. Cancer Res. 2001, 7, 1998–2004. [Google Scholar]
- Chen, L.; Guo, Q.; Liu, S.; Yu, Q. Clinicopathological significance and potential drug targeting of CDH1 in lung cancer: A meta-analysis and literature review. Drug Des. Dev. Ther. 2015, 9, 2171–2178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krohn, A.; Ahrens, T.; Yalcin, A.; Plönes, T.; Wehrle, J.; Taromi, S.; Wollner, S.; Follo, M.; Brabletz, T.; Mani, S.A.; et al. Tumor Cell Heterogeneity in Small Cell Lung Cancer (SCLC): Phenotypical and Functional Differences Associated with Epithelial-Mesenchymal Transition (EMT) and DNA Methylation Changes. PLoS ONE 2014, 9, e100249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyooka, K.O.; Toyooka, S.; Virmani, A.K.; Sathyanarayana, U.G.; Euhus, D.M.; Gilcrease, M.; Minna, J.D.; Gazdar, A.F. Loss of expression and aberrant methylation of the CDH13 (H-cadherin) gene in breast and lung carcinomas. Cancer Res. 2001, 61, 4556–4560. [Google Scholar]
- Nakata, S.; Sugio, K.; Uramoto, H.; Oyama, T.; Hanagiri, T.; Morita, M.; Yasumoto, K. The methylation status and protein expression of CDH1, p16(INK4A), and fragile histidine triad in nonsmall cell lung carcinoma: Epigenetic silencing, clinical features, and prognostic significance. Cancer 2006, 106, 2190–2199. [Google Scholar] [CrossRef]
- Zöchbauer-Müller, S.; Fong, K.; Maitra, A.; Lam, S.; Geradts, J.; Ashfaq, R.; Virmani, A.K.; Milchgrub, S.; Gazdar, A.F.; Minna, J.D. 5’ CpG island methylation of the FHIT gene is correlated with loss of gene expression in lung and breast cancer. Cancer Res. 2001, 61, 3581–3585. [Google Scholar] [PubMed]
- Esteller, M.; Sanchez-Cespedes, M.; Rosell, R.; Sidransky, D.; Baylin, S.B.; Herman, J.G. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res. 1999, 59, 67–70. [Google Scholar]
- Toyooka, S.; Toyooka, K.O.; Maruyama, R.; Virmani, A.K.; Girard, L.; Miyajima, K.; Harada, K.; Ariyoshi, Y.; Takahashi, T.; Sugio, K.; et al. DNA methylation profiles of lung tumors. Mol. Cancer Ther. 2001, 1, 61–67. [Google Scholar]
- Liu, L.; Broaddus, R.R.; Yao, J.C.; Xie, S.; White, J.A.; Wu, T.-T.; Hamilton, S.R.; Rashid, A. Epigenetic alterations in neuroendocrine tumors: Methylation of RAS-association domain family 1, isoform A and p16 genes are associated with metastasis. Mod. Pathol. 2005, 18, 1632–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, J.-C.; Lee, H.-Y.; Lee, J.I.; Wang, L.; Issa, J.-P.; Kemp, B.L.; Liu, D.D.; Kurie, J.M.; Mao, L.; Khuri, F.R. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin. Cancer Res. 2002, 8, 1178–1184. [Google Scholar]
- Dammann, R.; Takahashi, T.; Pfeifer, G.P. The CpG island of the novel tumor suppressor gene RASSF1A is intensely methylated in primary small cell lung carcinomas. Oncogene 2001, 20, 3563–3567. [Google Scholar] [CrossRef] [Green Version]
- Endoh, H.; Yatabe, Y.; Shimizu, S.; Tajima, K.; Kuwano, H.; Takahashi, T.; Mitsudomi, T. RASSF1A gene inactivation in non-small cell lung cancer and its clinical implication. Int. J. Cancer 2003, 106, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, T.; Trapasso, F.; Yendamuri, S.; Matsuyama, A.; Alder, H.; Williams, N.N.; Kaiser, L.R.; Croce, C.M. Allelic loss on chromosome 3p21.3 and promoter hypermethylation of semaphorin 3B in non-small cell lung cancer. Cancer Res. 2003, 63, 3352–3355. [Google Scholar]
- Loginov, V.I.; Dmitriev, A.A.; Senchenko, V.N.; Pronina, I.V.; Khodyrev, D.S.; Kudryavtseva, A.V.; Krasnov, G.S.; Gerashchenko, G.V.; Chashchina, L.I.; Kazubskaya, T.P.; et al. Tumor Suppressor Function of the SEMA3B Gene in Human Lung and Renal Cancers. PLoS ONE 2015, 10, e0123369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.T.; Chen, X.F.; Wang, M.H.; Wang, J.C.; Qi, Q.Y.; Zhang, R.M.; Xu, W.-Q.; Fei, Q.-Y.; Wang, F.; Cheng, Q.-Q.; et al. Defective expression of transforming growth factor beta receptor type II is associated with CpG methylated promoter in primary non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 2359–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukami, T.; Fukuhara, H.; Kuramochi, M.; Maruyama, T.; Isogai, K.; Sakamoto, M.; Takamoto, S.; Murakami, Y. Promoter methylation of theTSLC1 gene in advanced lung tumors and various cancer cell lines. Int. J. Cancer 2003, 107, 53–59. [Google Scholar] [CrossRef]
- Poirier, J.; Gardner, E.; Connis, N.; Moreira, A.L.; De Stanchina, E.; Hann, C.L.; Rudin, C.M. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene 2015, 34, 5869–5878. [Google Scholar] [CrossRef] [Green Version]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.M.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef]
- Sun, X.; Yi, J.; Yang, J.; Han, Y.; Qian, X.; Liu, Y.; Li, J.; Lu, B.; Zhang, J.; Pan, X.; et al. An integrated epigenomic-transcriptomic landscape of lung cancer reveals novel methylation driver genes of diagnostic and therapeutic relevance. Theranostics 2021, 11, 5346–5364. [Google Scholar] [CrossRef]
- Smith, L.T.; Lin, M.; Brena, R.M.; Lang, J.C.; Schuller, D.E.; Otterson, G.A.; Morrison, C.D.; Smiraglia, D.J.; Plass, C. Epigenetic regulation of the tumor suppressor gene TCF21 on 6q23-q24 in lung and head and neck cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 982–987. [Google Scholar] [CrossRef] [Green Version]
- Walter, R.F.H.; Rozynek, P.; Casjens, S.; Werner, R.; Mairinger, F.; Speel, E.J.M.; Hausen, A.Z.; Meier, S.; Wohlschlaeger, J.; Theegarten, D.; et al. Methylation of L1RE1, RARB, and RASSF1 function as possible biomarkers for the differential diagnosis of lung cancer. PLoS ONE 2018, 13, e0195716. [Google Scholar] [CrossRef]
- de Fraipont, F.; Levallet, G.; Creveuil, C.; Bergot, E.; Beau-Faller, M.; Mounawar, M.; Richard, N.; Antoine, M.; Rouquette, I.; Favrot, M.-C.; et al. An apoptosis methylation prognostic signature for early lung cancer in the IFCT-0002 trial. Clin. Cancer Res. 2012, 18, 2976–2986. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Nagae, G.; Motoi, N.; Miyauchi, E.; Ninomiya, H.; Uehara, H.; Mun, M.; Okumura, S.; Ohyanagi, F.; Nishio, M.; et al. Prognostic significance of CpG island methylator phenotype in surgically resected small cell lung carcinoma. Cancer Sci. 2016, 107, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Shinjo, K.; Okamoto, Y.; An, B.; Yokoyama, T.; Takeuchi, I.; Fujii, M.; Osada, H.; Usami, N.; Hasegawa, Y.; Ito, H.; et al. Integrated analysis of genetic and epigenetic alterations reveals CpG island methylator phenotype associated with distinct clinical characters of lung adenocarcinoma. Carcinogenesis 2012, 33, 1277–1285. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Yang, X.; Shi, W. Identifying differentially expressed genes and pathways in two types of non-small cell lung cancer: Adenocarcinoma and squamous cell carcinoma. Genet. Mol. Res. 2014, 13, 95–102. [Google Scholar] [CrossRef]
- Gandara, D.R.; Hammerman, P.S.; Sos, M.L.; Lara, P.N.; Hirsch, F.R. Squamous Cell Lung Cancer: From Tumor Genomics to Cancer Therapeutics. Clin. Cancer Res. 2015, 21, 2236–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.; Kawakami, K.; Fukui, Y.; Tsukioka, S.; Oda, M.; Watanabe, G.; Takechi, T.; Oka, T.; Minamoto, T. Different histological types of non-small cell lung cancer have distinct folate and DNA methylation levels. Cancer Sci. 2009, 100, 2325–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessema, M.; Yingling, C.M.; Picchi, M.A.; Wu, G.; Ryba, T.; Lin, Y.; Bungum, A.O.; Edell, E.S.; Spira, A.; Belinsky, S.A. ANK1 Methylation regulates expression of MicroRNA-486-5p and discriminates lung tumors by histology and smoking status. Cancer Lett. 2017, 410, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Hawes, S.E.; Stern, J.E.; Feng, Q.; Wiens, L.W.; Rasey, J.S.; Lu, H.; Kiviat, N.B.; Vesselle, H. DNA hypermethylation of tumors from non-small cell lung cancer (NSCLC) patients is associated with gender and histologic type. Lung Cancer 2010, 69, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Castro, M.; Grau, L.; Puerta, P.; Gimenez, L.; Venditti, J.; Quadrelli, S.; Sánchez-Carbayo, M. Multiplexed methylation profiles of tumor suppressor genes and clinical outcome in lung cancer. J. Transl. Med. 2010, 8, 86. [Google Scholar] [CrossRef] [Green Version]
- Niklinska, W.; Naumnik, W.; Sulewska, A.; Kozłowski, M.; Pankiewicz, W.; Milewski, R. Prognostic significance of DAPK and RASSF1A promoter hypermethylation in non-small cell lung cancer (NSCLC). Folia Histochem. Cytobiol. 2009, 47, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Hong, Q.; Jiang, J.; Chen, X.; Jiang, Z.; Wang, J.; Liu, S.; Duan, S.; Shi, S. AGTR1 promoter hypermethylation in lung squamous cell carcinoma but not in lung adenocarcinoma. Oncol. Lett. 2017, 14, 4989–4994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Jen, J.; Peikert, T.; Edell, E.; Tian, S.; Yang, P.; Huang, Y.; Zhpu, H. Selection of sensitive methylation markers for the detection of non-small cell lung cancer. J. Mol. Biomark. Diagn. 2015, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, M.; Liu, B. Exploring and comparing of the gene expression and methylation differences between lung adenocarcinoma and squamous cell carcinoma. J. Cell. Physiol. 2019, 234, 4454–4459. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Zhu, T.; Breeze, C.E.; Beck, S. EPISCORE: Cell type deconvolution of bulk tissue DNA methylomes from single-cell RNA-Seq data. Genome Biol. 2020, 21, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Terry, J.; Leung, S.; Laskin, J.; Leslie, K.O.; Gown, A.M.; Ionescu, D.N. Optimal Immunohistochemical Markers for Distinguishing Lung Adenocarcinomas from Squamous Cell Carcinomas in Small Tumor Samples. Am. J. Surg. Pathol. 2010, 34, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, R.H.; Hou, J.; Haberle, V.; Aerts, J.; Grosveld, F.; Lenhard, B.; Philipsen, S. Genomewide DNA Methylation Analysis Identifies Novel Methylated Genes in Non–Small-Cell Lung Carcinomas. J. Thorac. Oncol. 2013, 8, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Li, J.; Zhang, C.; Hong, Q.; Jiang, D.; Ye, M.; Duan, S. Distinguishing Lung Adenocarcinoma from Lung Squamous Cell Carcinoma by Two Hypomethylated and Three Hypermethylated Genes: A Meta-Analysis. PLoS ONE 2016, 11, e0149088. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.-X.; Wang, Y.; Li, X.; Zhang, W.; Zhou, H.-H.; Yin, J.-Y.; Liu, Z.-Q. Genome-wide DNA methylation profiling reveals novel epigenetic signatures in squamous cell lung cancer. BMC Genom. 2017, 18, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Jin, Z.; Cheng, L.; Zhang, B. Integrative Analysis of Methylation and Gene Expression in Lung Adenocarcinoma and Squamous Cell Lung Carcinoma. Front. Bioeng. Biotechnol. 2020, 8, 3. [Google Scholar] [CrossRef]
- Yauch, R.L.; Januario, T.; Eberhard, D.A.; Cavet, G.; Zhu, W.; Fu, L.; Pham, T.Q.; Soriano, R.; Stinson, J.; Seshagiri, S.; et al. Epithelial versus Mesenchymal Phenotype Determines In vitro Sensitivity and Predicts Clinical Activity of Erlotinib in Lung Cancer Patients. Clin. Cancer Res. 2005, 11 Pt 1, 8686–8698. [Google Scholar] [CrossRef] [Green Version]
- Walter, K.; Holcomb, T.; Januario, T.; Du, P.; Evangelista, M.; Kartha, N.; Iniguez, L.; Soriano, R.; Huw, L.; Stern, H.; et al. DNA Methylation Profiling Defines Clinically Relevant Biological Subsets of Non–Small Cell Lung Cancer. Clin. Cancer Res. 2012, 18, 2360–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Hou, M.; Pei, T. FAM83A Is a Prognosis Signature and Potential Oncogene of Lung Adenocarcinoma. DNA Cell Biol. 2020, 39, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Wu, D.; Chen, J.; Wang, P.; Lv, X.; Huang, J. Hypermethylation of the G protein-coupled receptor kinase 6 (GRK6) promoter inhibits binding of C/EBPα, and GRK6 knockdown promotes cell migration and invasion in lung adenocarcinoma cells. FEBS Open Bio 2019, 9, 605–617. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.-H.I.; Ziogas, A.; Zell, J.A. Prognostic Factors for Survival in Extensive Stage Small Cell Lung Cancer (ED-SCLC): The Importance of Smoking History, Socioeconomic and Marital Statuses, and Ethnicity. J. Thorac. Oncol. 2009, 4, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Smolle, E.; Pichler, M. Non-smoking-associated lung cancer: A distinct entity in terms of tumor biology, patient characteristics and impact of hereditary cancer predisposition. Cancers 2019, 11, 204. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Zhang, Y.; Breitling, L.P.; Brenner, H. Tobacco smoking and methylation of genes related to lung cancer development. Oncotarget 2016, 7, 59017–59028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Elgizouli, M.; Schöttker, B.; Holleczek, B.; Nieters, A.; Brenner, H. Smoking-associated DNA methylation markers predict lung cancer incidence. Clin. Epigenet. 2016, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Baglietto, L.; Ponzi, E.; Haycock, P.; Hodge, A.; Assumma, M.B.; Jung, C.-H.; Chung, J.; Fasanelli, F.; Guida, F.; Campanella, G.; et al. DNA methylation changes measured in pre-diagnostic peripheral blood samples are associated with smoking and lung cancer risk. Int. J. Cancer 2017, 140, 50–61. [Google Scholar] [CrossRef]
- Liu, Y.; Lan, Q.; Siegfried, J.M.; Luketich, J.D.; Keohavong, P. Aberrant Promoter Methylation of p16 and MGMT Genes in Lung Tumors from Smoking and Never-Smoking Lung Cancer Patients. Neoplasia 2006, 8, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Vaissière, T.; Hung, R.J.; Zaridze, D.; Moukeria, A.; Cuenin, C.; Fasolo, V.; Ferro, G.; Paliwal, A.; Hainaut, P.; Brennan, P.; et al. Quantitative Analysis of DNA Methylation Profiles in Lung Cancer Identifies Aberrant DNA Methylation of Specific Genes and Its Association with Gender and Cancer Risk Factors. Cancer Res. 2009, 69, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Pulling, L.C.; Divine, K.K.; Klinge, D.M.; Gilliland, F.D.; Kang, T.; Schwartz, A.G.; Bocklage, T.J.; Belinsky, S.A. Promoter hypermethylation of the O6-methylguanine-DNA methyltransferase gene: More common in lung adenocarcinomas from never-smokers than smokers and associated with tumor progression. Cancer Res. 2003, 63, 4842–4848. [Google Scholar] [PubMed]
- Wu, J.-Y.; Wang, J.; Lai, J.-C.; Cheng, Y.-W.; Yeh, K.-T.; Wu, T.-C.; Chen, C.-Y.; Lee, H. Association of O6-Methylguanine-DNA Methyltransferase (MGMT) Promoter Methylation with p53 Mutation Occurrence in Non-Small Cell Lung Cancer with Different Histology, Gender, and Smoking Status. Ann. Surg. Oncol. 2008, 15, 3272–3277. [Google Scholar] [CrossRef]
- Damiani, L.A.; Yingling, C.M.; Leng, S.; Romo, P.E.; Nakamura, J.; Belinsky, S.A. Carcinogen-Induced Gene Promoter Hypermethylation Is Mediated by DNMT1 and Causal for Transformation of Immortalized Bronchial Epithelial Cells. Cancer Res. 2008, 68, 9005–9014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.-K.; Hsieh, Y.-S.; Lin, P.; Hsu, H.-S.; Chen, C.-Y.; Tang, Y.-A.; Lee, C.-F.; Wang, Y.-C. The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J. Clin. Investig. 2010, 120, 521–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hagan, H.M.; Wang, W.; Sen, S.; Lee, S.S.; Clements, E.G.; Cai, Y.; Van Neste, L.; Easwaran, H.; Baylin, S.B. Abstract LB-185: Oxidative damage targets complexes containing DNA methyltransferases, SIRT1 and polycomb members to promoter CpG islands. Cell. Mol. Biol. 2011, 71, LB-185. [Google Scholar] [CrossRef]
- Vaz, M.; Hwang, S.Y.; Kagiampakis, I.; Phallen, J.; Patil, A.; O’Hagan, H.M.; Murphy, L.; Zahnow, C.A.; Gabrielson, E.; Velculescu, V.E.; et al. Chronic Cigarette Smoke-Induced Epigenomic Changes Precede Sensitization of Bronchial Epithelial Cells to Single-Step Transformation by KRAS Mutations. Cancer Cell 2017, 32, 360–376.e6. [Google Scholar] [CrossRef] [Green Version]
- Pesek, M.; Kopeckova, M.; Benesova, L.; Meszarosova, A.; Mukensnabl, P.; Bruha, F.; Minarik, M. Clinical significance of hypermethylation status in NSCLC: Evaluation of a 30-gene panel in patients with advanced disease. Anticancer Res. 2011, 31, 4647–4652. [Google Scholar]
- Satelli, A.; Rao, P.S.; Thirumala, S.; Rao, U.S. Galectin-4 functions as a tumor suppressor of human colorectal cancer. Int. J. Cancer 2011, 129, 799–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakulski, K.M.; Dou, J.; Lin, N.; London, S.J.; Colacino, J.A. DNA methylation signature of smoking in lung cancer is enriched for exposure signatures in newborn and adult blood. Sci. Rep. 2019, 9, 4576. [Google Scholar] [CrossRef] [Green Version]
- Adderley, H.; Blackhall, F.H.; Lindsay, C.R. KRAS-mutant non-small cell lung cancer: Converging small molecules and immune checkpoint inhibition. EBioMedicine 2019, 41, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyooka, S.; Tokumo, M.; Shigematsu, H.; Matsuo, K.; Asano, H.; Tomii, K.; Ichihara, S.; Suzuki, M.; Aoe, M.; Date, H.; et al. Mutational and Epigenetic Evidence for Independent Pathways for Lung Adenocarcinomas Arising in Smokers and Never Smokers. Cancer Res. 2006, 66, 1371–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinoue, T.; Weisenberger, D.J.; Lange, C.P.; Shen, H.; Byun, H.-M.; Van Den Berg, D.; Malik, S.; Pan, F.; Noushmehr, H.; van Dijk, C.M.; et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012, 22, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, Z.; Dai, Z.; Popkie, A.P.; Plass, C.; Morrison, C.; Wang, Y.; You, M. RASSF1A Promoter Methylation and Kras2 Mutations in Non Small Cell Lung Cancer. Neoplasia 2003, 5, 362–366. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gao, W.; Siegfried, J.M.; Weissfeld, J.L.; Luketich, J.D.; Keohavong, P. Promoter methylation of RASSF1A and DAPK and mutations of K-ras, p53, and EGFR in lung tumors from smokers and never-smokers. BMC Cancer 2007, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Tew, B.Y.; Durand, J.K.; Bryant, K.L.; Hayes, T.K.; Peng, S.; Tran, N.L.; Gooden, G.C.; Buckley, D.N.; Der, C.J.; Baldwin, A.S.; et al. Genome-wide DNA methylation analysis of KRAS mutant cell lines. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Yuan, J.-Q.; Wang, K.-F.; Fu, X.-H.; Han, X.-R.; Threapleton, D.; Yang, Z.-Y.; Mao, C.; Tang, J.-L. The prevalence of EGFR mutation in patients with non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget 2016, 7, 78985–78993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Qin, F.; Yuan, L.; Wei, J.; Sun, Y.; Qin, J.; Deng, K.; Zheng, T.; Li, S. EGFR DNA Methylation Correlates with EGFR Expression, Immune Cell Infiltration, and Overall Survival in Lung Adenocarcinoma. Front. Oncol. 2021, 11, 691915. [Google Scholar] [CrossRef]
- Li, X.-Y.; Zhong, Y.-J.; Wu, J.-Z.; Cao, H.-X.; Ma, R.; Feng, J.-F. Blockade of DNA methylation enhances the therapeutic effect of gefitinib in non-small cell lung cancer cells. Oncol. Rep. 2013, 29, 1975–1982. [Google Scholar] [CrossRef] [Green Version]
- Rønneberg, J.A.; Fleischer, T.; Solvang, H.K.; Nordgard, S.H.; Edvardsen, H.; Potapenko, I.; Nebdal, D.; Daviaud, C.; Gut, I.; Bukholm, I.; et al. Methylation profiling with a panel of cancer related genes: Association with estrogen receptor, TP53 mutation status and expression subtypes in sporadic breast cancer. Mol. Oncol. 2010, 5, 61–76. [Google Scholar] [CrossRef]
- Network, C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyota, M.; Ohe-Toyota, M.; Ahuja, N.; Issa, J.-P. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc. Natl. Acad. Sci. USA 2000, 97, 710–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haiman, C.A.; Stram, D.O.; Wilkens, L.R.; Pike, M.C.; Kolonel, L.N.; Henderson, B.E.; Le Marchand, L. Ethnic and Racial Differences in the Smoking-Related Risk of Lung Cancer. N. Engl. J. Med. 2006, 354, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Rivera, M.; Stover, D.E. Gender and lung cancer. Clin. Chest Med. 2004, 25, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Stram, D.O.; Park, S.L.; Haiman, C.A.; Murphy, S.E.; Patel, Y.; Hecht, S.S.; Le Marchand, L. Racial/Ethnic Differences in Lung Cancer Incidence in the Multiethnic Cohort Study: An Update. JNCI J. Natl. Cancer Inst. 2019, 111, 811–819. [Google Scholar] [CrossRef]
- Zang, E.A.; Wynder, E.L. Differences in Lung Cancer Risk Between Men and Women: Examination of the Evidence. JNCI J. Natl. Cancer Inst. 1996, 88, 183–192. [Google Scholar] [CrossRef]
- Adkins, R.M.; Krushkal, J.; Tylavsky, F.A.; Thomas, F. Racial differences in gene-specific DNA methylation levels are present at birth. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 728–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.V.; Smith, A.K.; Conneely, K.N.; Chang, Q.; Li, W.; Lazarus, A.; Smith, J.; Almli, L.; Binder, E.B.; Klengel, T.; et al. Epigenomic association analysis identifies smoking-related DNA methylation sites in African Americans. Qual. Life Res. 2013, 132, 1027–1037. [Google Scholar] [CrossRef] [Green Version]
- Campesi, I.; Carru, C.; Zinellu, A.; Occhioni, S.; Sanna, M.; Palermo, M.; Tonolo, G.; Mercuro, G.; Franconi, F. Regular cigarette smoking influences the transsulfuration pathway, endothelial function, and inflammation biomarkers in a sex-gender specific manner in healthy young humans. Am. J. Transl. Res. 2013, 5, 497–509. [Google Scholar]
- Sarter, B.; Long, T.I.; Tsong, W.H.; Koh, W.-P.; Yu, M.C.; Laird, P.W. Sex differential in methylation patterns of selected genes in Singapore Chinese. Qual. Life Res. 2005, 117, 402–403. [Google Scholar] [CrossRef]
- McCarthy, N.S.; Melton, P.E.; Cadby, G.; Yazar, S.; Franchina, M.; Moses, E.K.; Mackey, D.A.; Hewitt, A.W. Meta-analysis of human methylation data for evidence of sex-specific autosomal patterns. BMC Genom. 2014, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis-Owen, S.A.G.; Domingo-Sabugo, C.; Starren, E.; Liang, L.; Freidin, M.B.; Arseneault, M.; Zhang, Y.; Lu, S.K.; Popat, S.; Lim, E.; et al. Y disruption, autosomal hypomethylation and poor male lung cancer survival. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Lai, J.-C.; Cheng, Y.-W.; Chiou, H.-L.; Wu, M.-F.; Chen, C.-Y.; Lee, H. Gender difference in estrogen receptor alpha promoter hypermethylation and its prognostic value in non-small cell lung cancer. Int. J. Cancer 2005, 117, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.Z.; Lin, R.; Carrillo, J.; Bhutani, M.; Pathak, A.; Ren, H.; Li, Y.; Song, J.; Mao, L. ∆ DNMT3B4-del Contributes to Aberrant DNA Methylation Patterns in Lung Tumorigenesis. EBioMedicine 2015, 2, 1340–1350. [Google Scholar] [CrossRef] [Green Version]
- Parry, A.; Rulands, S.; Reik, W. Active turnover of DNA methylation during cell fate decisions. Nat. Rev. Genet. 2021, 22, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Easwaran, H.; Johnstone, S.E.; Van Neste, L.; Ohm, J.; Mosbruger, T.; Wang, Q.; Aryee, M.J.; Joyce, P.; Ahuja, N.; Weisenberger, D.; et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 2012, 22, 837–849. [Google Scholar] [CrossRef] [Green Version]
- Cheung, W.K.; Nguyen, D.X. Lineage factors and differentiation states in lung cancer progression. Oncogene 2015, 34, 5771–5780. [Google Scholar] [CrossRef]
- Smiraglia, D.J.; Rush, L.J.; Frühwald, M.C.; Dai, Z.; Held, W.A.; Costello, J.F.; Lang, J.C.; Eng, C.; Li, B.; Wright, F.A.; et al. Excessive CpG island hypermethylation in cancer cell lines versus primary human malignancies. Hum. Mol. Genet. 2001, 10, 1413–1419. [Google Scholar] [CrossRef] [Green Version]
- Licchesi, J.D.; Westra, W.H.; Hooker, C.M.; Herman, J.G. Promoter Hypermethylation of Hallmark Cancer Genes in Atypical Adenomatous Hyperplasia of the Lung. Clin. Cancer Res. 2008, 14, 2570–2578. [Google Scholar] [CrossRef] [Green Version]
- Landi, M.T.; Synnott, N.C.; Rosenbaum, J.; Zhang, T.; Zhu, B.; Shi, J.; Zhao, W.; Kebede, M.; Sang, J.; Choi, J.; et al. Tracing Lung Cancer Risk Factors Through Mutational Signatures in Never-Smokers. Am. J. Epidemiol. 2021, 190, 962–976. [Google Scholar] [CrossRef]
- Hua, X.; Zhao, W.; Pesatori, A.C.; Consonni, D.; Caporaso, N.E.; Zhang, T.; Zhu, B.; Wang, M.; Jones, K.; Hicks, B.; et al. Genetic and epigenetic intratumor heterogeneity impacts prognosis of lung adenocarcinoma. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mazor, T.; Pankov, A.; Johnson, B.E.; Hong, C.; Hamilton, E.; Bell, R.J.; Smirnov, I.V.; Reis, G.F.; Phillips, J.J.; Barnes, M.J.; et al. DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell 2015, 28, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brocks, D.; Assenov, Y.; Minner, S.; Bogatyrova, O.; Simon, R.; Koop, C.; Oakes, C.; Zucknick, M.; Lipka, D.B.; Weischenfeldt, J.; et al. Intratumor DNA Methylation Heterogeneity Reflects Clonal Evolution in Aggressive Prostate Cancer. Cell Rep. 2014, 8, 798–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karemaker, I.; Vermeulen, M. Single-Cell DNA Methylation Profiling: Technologies and Biological Applications. Trends Biotechnol. 2018, 36, 952–965. [Google Scholar] [CrossRef]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016, 26, 304–319. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoang, P.H.; Landi, M.T. DNA Methylation in Lung Cancer: Mechanisms and Associations with Histological Subtypes, Molecular Alterations, and Major Epidemiological Factors. Cancers 2022, 14, 961. https://doi.org/10.3390/cancers14040961
Hoang PH, Landi MT. DNA Methylation in Lung Cancer: Mechanisms and Associations with Histological Subtypes, Molecular Alterations, and Major Epidemiological Factors. Cancers. 2022; 14(4):961. https://doi.org/10.3390/cancers14040961
Chicago/Turabian StyleHoang, Phuc H., and Maria Teresa Landi. 2022. "DNA Methylation in Lung Cancer: Mechanisms and Associations with Histological Subtypes, Molecular Alterations, and Major Epidemiological Factors" Cancers 14, no. 4: 961. https://doi.org/10.3390/cancers14040961
APA StyleHoang, P. H., & Landi, M. T. (2022). DNA Methylation in Lung Cancer: Mechanisms and Associations with Histological Subtypes, Molecular Alterations, and Major Epidemiological Factors. Cancers, 14(4), 961. https://doi.org/10.3390/cancers14040961