
Intrinsic and Extrinsic Factors Impacting Cancer Stemness and Tumor Progression

 , and
, and 
Abstract
:Simple Summary
Abstract
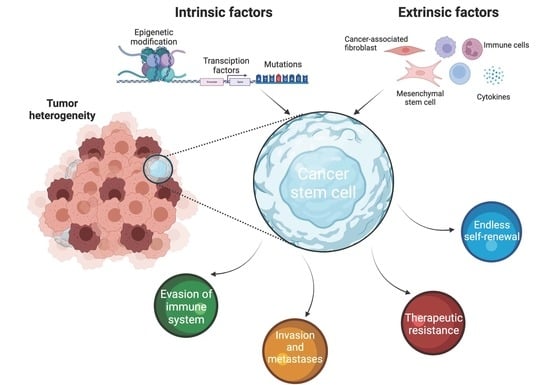
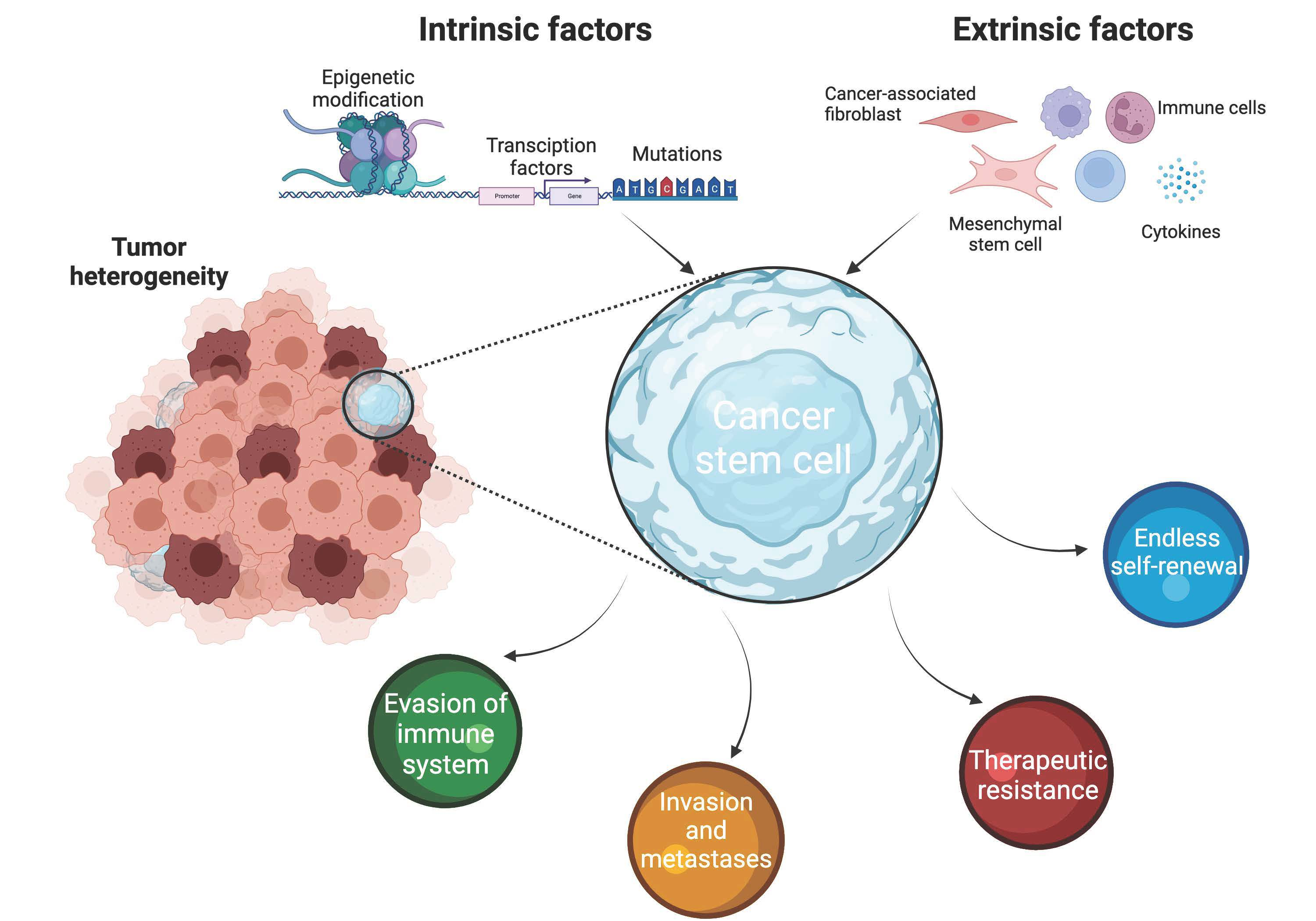
1. Introduction
2. Pluripotency Factors as Intrinsic Factors Regulating Cancer Stemness
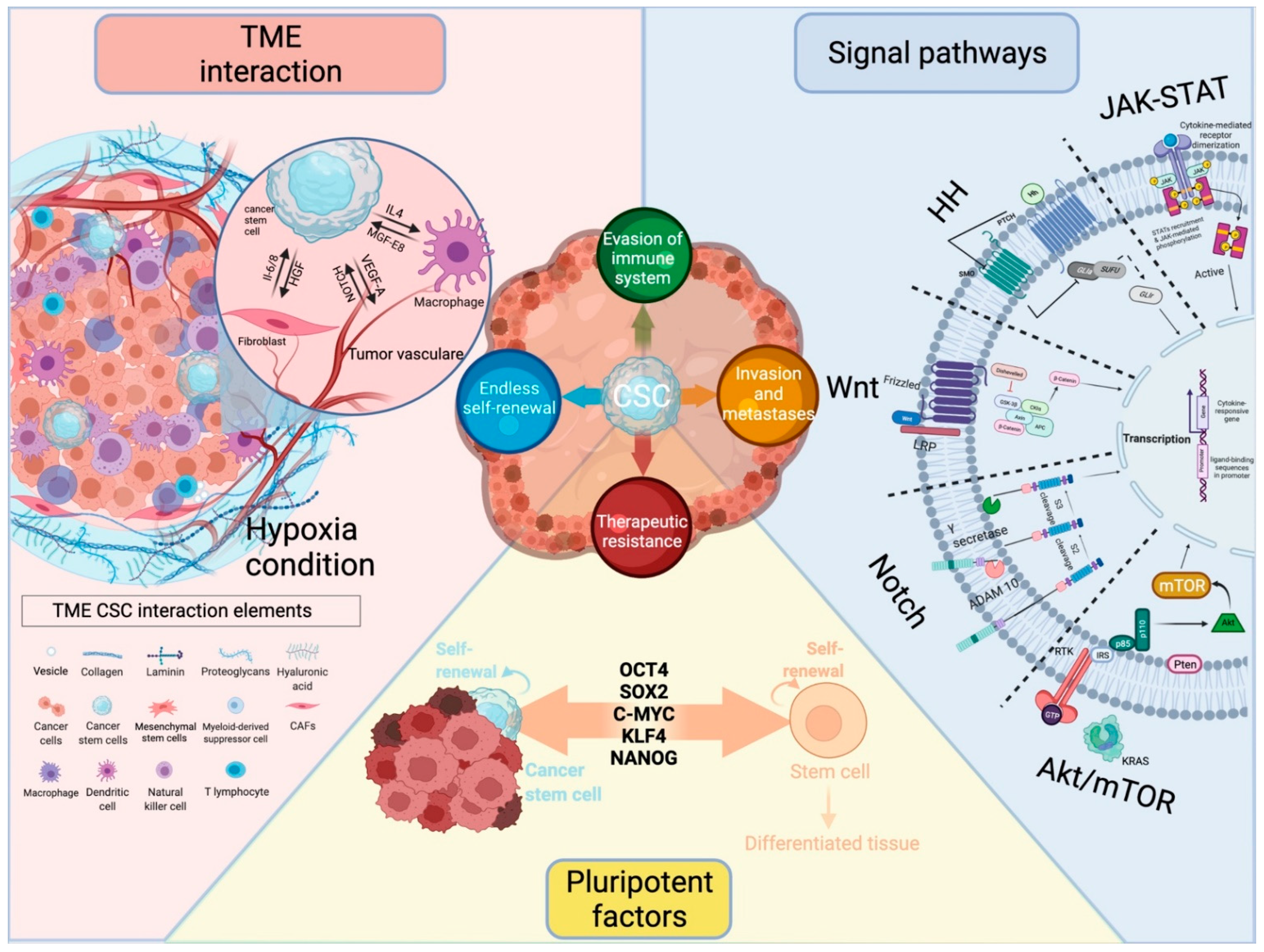
3. Signaling Pathways Modulate Cancer Stemness
3.1. Wnt Signaling
3.2. Notch Signaling
3.3. Hedgehog Signaling
3.4. JAK/STAT Signaling
3.5. AKT/mTOR Signaling
4. Influence of the Microenvironment on CSC
5. Models of Cancer Stemness and TME Interaction
5.1. Spheroids
5.2. Organoids
5.3. TME Interaction
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Elkashty, O.A.; Hariharan, A.; Tran, S.D. Aging, stem cells and cancer updated. Aging 2021, 13, 20854–20855. [Google Scholar] [CrossRef] [PubMed]
- Sell, S. On the Stem Cell Origin of Cancer. Am. J. Pathol. 2010, 176, 2584–2594. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Bruno, A.; Gallo, C.; Pajardi, G.; Noonan, D.M.; Dallaglio, K. Cancer stem cells and the tumor microenvironment: Interplay in tumor heterogeneity. Connect Tissue Res. 2015, 56, 414–425. [Google Scholar] [CrossRef] [Green Version]
- Girouard, S.D.; Murphy, G.F. Melanoma stem cells: Not rare, but well done. Lab. Investig. 2011, 91, 647–664. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Capp, J.P. Cancer Stem Cells: From Historical Roots to a New Perspective. J. Oncol. 2019, 2019, 5189232 . [Google Scholar] [CrossRef]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Fendler, A.; Bauer, D.; Busch, J.; Jung, K.; Wulf-Goldenberg, A.; Kunz, S.; Song, K.; Myszczyszyn, A.; Elezkurtaj, S.; Erguen, B.; et al. Inhibiting WNT and NOTCH in renal cancer stem cells and the implications for human patients. Nat. Commun. 2020, 11, 929. [Google Scholar] [CrossRef] [Green Version]
- Dou, J.; Gu, N. Emerging strategies for the identification and targeting of cancer stem cells. Tumor Biol. 2010, 31, 243–253. [Google Scholar] [CrossRef]
- Duan, J.J.; Qiu, W.; Xu, S.L.; Wang, B.; Ye, X.Z.; Ping, Y.F.; Zhang, X.; Bian, X.W.; Yu, S.C. Strategies for isolating and enriching cancer stem cells: Well begun is half done. Stem Cells Dev. 2013, 22, 2221–2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goossens, S.; Vandamme, N.; Van Vlierberghe, P.; Berx, G. EMT transcription factors in cancer development re-evaluated: Beyond EMT and MET. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, A.M. VEGF/Neuropilin Signaling in Cancer Stem Cells. Int. J. Mol. Sci. 2019, 20, 490. [Google Scholar] [CrossRef] [Green Version]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [Green Version]
- Farace, C.; Pisano, A.; Grinan-Lison, C.; Solinas, G.; Jimenez, G.; Serra, M.; Carrillo, E.; Scognamillo, F.; Attene, F.; Montella, A.; et al. Deregulation of cancer-stem-cell-associated miRNAs in tissues and sera of colorectal cancer patients. Oncotarget 2020, 11, 116–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.J. The Role of the Extracellular Matrix in Cancer Stemness. Front. Cell Dev. Biol. 2019, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Jijiwa, M.; Demir, H.; Gupta, S.; Leung, C.; Joshi, K.; Orozco, N.; Huang, T.; Yildiz, V.O.; Shibahara, I.; de Jesus, J.A.; et al. CD44v6 Regulates Growth of Brain Tumor Stem Cells Partially through the AKT-Mediated Pathway. PLoS ONE 2011, 6, e24217. [Google Scholar] [CrossRef]
- Matsui, W.; Huff, C.A.; Wang, Q.J.; Malehorn, M.T.; Barber, J.; Tanhehco, Y.; Smith, B.D.; Civin, C.I.; Jones, R.J. Characterization of clonogenic multiple myeloma cells. Blood 2004, 103, 2332–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.M.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szotek, P.P.; Pieretti-Vanmarcke, R.; Masiakos, P.T.; Dinulescut, D.M.; Connolly, D.; Foster, R.; Dombkowski, D.; Preffer, F.; MacLaughlin, D.T.; Donahoe, P.K. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc. Natl. Acad. Sci. USA 2006, 103, 11154–11159. [Google Scholar] [CrossRef] [Green Version]
- Sims-Mourtada, J.; Izzo, J.G.; Ajani, J.; Chao, K.S.C. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene 2007, 26, 5674–5679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, L.; Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Comparative genomics on PROM1 gene encoding stem cell marker CD133. Int. J. Mol. Med. 2007, 19, 967–970. [Google Scholar] [CrossRef]
- Van Der Flier, L.G.; Sabates-Bellver, J.; Oving, I.; Haegebarth, A.; De Palo, M.; Anti, M.; Van Gijn, M.E.; Suijkerbuijk, S.; Van De Wetering, M.; Marra, G.; et al. The intestinal Wnt/TCF signature. Gastroenterology 2007, 132, 628–632. [Google Scholar] [CrossRef]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.H.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, E.; Hermann, P.C.; Mueller, M.T.; Huber, S.; Balic, A.; Miranda-Lorenzo, I.; Zagorac, S.; Alcala, S.; Roderiguez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/Activin Signaling Drives Self-Renewal and Tumorigenicity of Pancreatic Cancer Stem Cells and Provides a Target for Combined Drug Therapy. Cell Stem Cell 2011, 9, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.W.; Lee, C.J.; Simeone, D.M. Identification of Human Pancreatic Cancer Stem Cells. In Cancer Stem Cells: Methods Protoc; Yu, J.S., Ed.; Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2009; Volume 568, pp. 161–173. [Google Scholar]
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.K.; Lam, C.T.; Poon, R.T.P.; Fan, S.T. Significance of CD90(+) cancer stem cells in human liver cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Cai, D.Y.; Dai, X.F. Modulating cancer stemness provides luminal a breast cancer cells with HER2 positive-like features. J. Cancer 2020, 11, 1162–1169. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.L.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.L.; Ginestier, C.; Ou, S.J.; Clouthier, S.G.; Patel, S.H.; Monville, F.; Korkaya, H.; Heath, A.; Dutcher, J.; Kleer, C.G.; et al. Breast Cancer Stem Cells Are Regulated by Mesenchymal Stem Cells through Cytokine Networks. Cancer Res. 2011, 71, 614–624. [Google Scholar] [CrossRef] [Green Version]
- Bocci, F.; Gearhart-Serna, L.; Boareto, M.; Ribeiro, M.; Ben-Jacob, E.; Devi, G.R.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2019, 116, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Viatour, P. Bridges between Cell Cycle Regulation and Self-Renewal Maintenance. Genes Cancer 2012, 3, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Simons, B.D.; Clevers, H. Strategies for homeostatic stem cell self-renewal in adult tissues. Cell 2011, 145, 851–862. [Google Scholar] [CrossRef] [Green Version]
- Brooks, M.D.; Burness, M.L.; Wicha, M.S. Therapeutic Implications of Cellular Heterogeneity and Plasticity in Breast Cancer. Cell Stem Cell 2015, 17, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, M.C.; Hollingsworth, R.E.; Hurt, E.M. Cancer stem cell plasticity and tumor hierarchy. World J. Stem Cells 2015, 7, 27–36. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [Green Version]
- Borovski, T.; Melo, F.D.E.; Vermeulen, L.; Medema, J.P. Cancer Stem Cell Niche: The Place to Be. Cancer Res. 2011, 71, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.; Han, Z.; Zhang, S.; Liu, Y.; Wei, L. Epithelial-Mesenchymal Transition in tumor microenvironment. Cell Biosci. 2011, 1, 29. [Google Scholar] [CrossRef] [Green Version]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Abell, A.N.; Johnson, G.L. Implications of Mesenchymal Cells in Cancer Stem Cell Populations: Relevance to EMT. Curr. Pathobiol. Rep. 2014, 2, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Mego, M.; Reuben, J.; Mani, S. Epithelial-Mesenchymal Transition (EMT) and Cancer Stem Cells (CSCs): The Traveling Metastasis; Cancer Drug Discovery and Development; Humana Press: Totowa, NJ, USA, 2017; p. 14. [Google Scholar]
- Tanabe, S.; Quader, S.; Cabral, H.; Ono, R. Interplay of EMT and CSC in Cancer and the Potential Therapeutic Strategies. Front. Pharm. 2020, 11, 904. [Google Scholar] [CrossRef]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating tumor cells: Biology and clinical significance. Signal Transduct Target 2021, 6, 404. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Laplane, L.; Solary, E. Towards a classification of stem cells. Elife 2019, 8, e46563. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Muller, M.; Hermann, P.C.; Liebau, S.; Weidgang, C.; Seufferlein, T.; Kleger, A.; Perkhofer, L. The role of pluripotency factors to drive stemness in gastrointestinal cancer. Stem Cell Res. 2016, 16, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Huang, S.; Chen, S.; Chen, J.; Wang, Z.; Wang, Y.; Zheng, H. SOX2 promotes chemoresistance, cancer stem cells properties, and epithelial-mesenchymal transition by beta-catenin and Beclin1/autophagy signaling in colorectal cancer. Cell Death Dis. 2021, 12, 449. [Google Scholar] [CrossRef]
- Yasumizu, Y.; Rajabi, H.; Jin, C.; Hata, T.; Pitroda, S.; Long, M.D.; Hagiwara, M.; Li, W.; Hu, Q.; Liu, S.; et al. MUC1-C regulates lineage plasticity driving progression to neuroendocrine prostate cancer. Nat. Commun. 2020, 11, 338. [Google Scholar] [CrossRef] [Green Version]
- Hepburn, A.C.; Steele, R.E.; Veeratterapillay, R.; Wilson, L.; Kounatidou, E.E.; Barnard, A.; Berry, P.; Cassidy, J.R.; Moad, M.; El-Sherif, A.; et al. The induction of core pluripotency master regulators in cancers defines poor clinical outcomes and treatment resistance. Oncogene 2019, 38, 4412–4424. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct Target 2021, 6, 153. [Google Scholar] [CrossRef]
- Wang, D.; Lu, P.; Zhang, H.; Luo, M.; Zhang, X.; Wei, X.; Gao, J.; Zhao, Z.; Liu, C. Oct-4 and Nanog promote the epithelial-mesenchymal transition of breast cancer stem cells and are associated with poor prognosis in breast cancer patients. Oncotarget 2014, 5, 10803–10815. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, K.; Semi, K.; Yamamoto, T.; Shimizu, M.; Tanaka, A.; Mitsunaga, K.; Okita, K.; Osafune, K.; Arioka, Y.; Maeda, T.; et al. Premature Termination of Reprogramming In Vivo Leads to Cancer Development through Altered Epigenetic Regulation. Cell 2014, 156, 663–677. [Google Scholar] [CrossRef] [Green Version]
- Hochedlinger, K.; Yamada, Y.; Beard, C.; Jaenisch, R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 2005, 121, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S.; Ninomiya, W.; Sakamoto, E.; Shibata, T.; Akiyama, H.; Tashiro, F. SRY and OCT4 Are Required for the Acquisition of Cancer Stem Cell-Like Properties and Are Potential Differentiation Therapy Targets. Stem Cells 2015, 33, 2652–2663. [Google Scholar] [CrossRef]
- Du, Z.; Jia, D.; Liu, S.; Wang, F.; Li, G.; Zhang, Y.; Cao, X.; Ling, E.A.; Hao, A. Oct4 is expressed in human gliomas and promotes colony formation in glioma cells. Glia 2009, 57, 724–733. [Google Scholar] [CrossRef]
- Wang, X.Q.; Ongkeko, W.M.; Chen, L.; Yang, Z.F.; Lu, P.; Chen, K.K.; Lopez, J.P.; Poon, R.T.P.; Fan, S.T. Octamer 4 (Oct4) Mediates Chemotherapeutic Drug Resistance in Liver Cancer Cells Through a Potential Oct4-AKT-ATP-Binding Cassette G2 Pathway. Hepatology 2010, 52, 528–539. [Google Scholar] [CrossRef]
- Fujino, S.; Miyoshi, N. Oct4 Gene Expression in Primary Colorectal Cancer Promotes Liver Metastasis. Stem Cells Int. 2019, 2019, 7896524. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.S.; Shiau, A.L.; Su, B.H.; Hsu, T.S.; Wang, C.T.; Su, Y.C.; Tsai, M.S.; Feng, Y.H.; Tseng, Y.L.; Yen, Y.T.; et al. Oct4 promotes M2 macrophage polarization through upregulation of macrophage colony-stimulating factor in lung cancer. J. Hematol. Oncol. 2020, 13, 62. [Google Scholar] [CrossRef]
- Hagey, D.W.; Klum, S.; Kurtsdotter, I.; Zaouter, C.; Topcic, D.; Andersson, O.; Bergsland, M.; Muhr, J. SOX2 regulates common and specific stem cell features in the CNS and endoderm derived organs. PLoS Genet. 2018, 14, e1007224. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, T.; Lengerke, C. SOX2 protein biochemistry in stemness, reprogramming, and cancer: The PI3K/AKT/SOX2 axis and beyond. Oncogene 2020, 39, 278–292. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Tang, H.; Song, C.; Wang, J.; Chen, B.; Huang, X.; Pei, X.; Liu, L. SOX2 Promotes Cell Proliferation and Metastasis in Triple Negative Breast Cancer. Front. Pharm. 2018, 9, 942. [Google Scholar] [CrossRef] [Green Version]
- Maurizi, G.; Verma, N.; Gadi, A.; Mansukhani, A.; Basilico, C. Sox2 is required for tumor development and cancer cell proliferation in osteosarcoma. Oncogene 2018, 37, 4626–4632. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Huang, T.; Wu, X.; Wang, X.; Liu, S.; Yang, W.; Shi, Q.; Li, H.; Hou, F. Prognostic Value of CD133 and SOX2 in Advanced Cancer. J. Oncol. 2019, 2019, 3905817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heurtier, V.; Owens, N.; Gonzalez, I.; Mueller, F.; Proux, C.; Mornico, D.; Clerc, P.; Dubois, A.; Navarro, P. The molecular logic of Nanog-induced self-renewal in mouse embryonic stem cells. Nat. Commun. 2019, 10, 1109. [Google Scholar] [CrossRef] [Green Version]
- Chiou, S.H.; Wang, M.L.; Chou, Y.T.; Chen, C.J.; Hong, C.F.; Hsieh, W.J.; Chang, H.T.; Chen, Y.S.; Lin, T.W.; Hsu, H.S.; et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010, 70, 10433–10444. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.; Ding, Y.Q.; Li, J.M. Overexpression of Nanog protein is associated with poor prognosis in gastric adenocarcinoma. Med. Oncol. 2012, 29, 878–885. [Google Scholar] [CrossRef]
- Chiou, S.H.; Yu, C.C.; Huang, C.Y.; Lin, S.C.; Liu, C.J.; Tsai, T.H.; Chou, S.H.; Chien, C.S.; Ku, H.H.; Lo, J.F. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin. Cancer Res. 2008, 14, 4085–4095. [Google Scholar] [CrossRef] [Green Version]
- de Vicente, J.C.; Rodriguez-Santamarta, T.; Rodrigo, J.P.; Allonca, E.; Vallina, A.; Singhania, A.; Donate-Perez Del Molino, P.; Garcia-Pedrero, J.M. The Emerging Role of NANOG as an Early Cancer Risk Biomarker in Patients with Oral Potentially Malignant Disorders. J. Clin. Med. 2019, 8, 1376. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jin, J.; Wan, F.; Zhao, L.; Chu, H.; Chen, C.; Liao, G.; Liu, J.; Yu, Y.; Teng, H.; et al. AMPK Promotes SPOP-Mediated NANOG Degradation to Regulate Prostate Cancer Cell Stemness. Dev. Cell 2019, 48, 345–360.e7. [Google Scholar] [CrossRef] [Green Version]
- Dehghan Harati, M.; Rodemann, H.P.; Toulany, M. Nanog Signaling Mediates Radioresistance in ALDH-Positive Breast Cancer Cells. Int J Mol Sci 2019, 20, 1151. [Google Scholar] [CrossRef] [Green Version]
- Ghaleb, A.M.; Yang, V.W. Kruppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef]
- Hsieh, M.H.; Chen, Y.T.; Chen, Y.T.; Lee, Y.H.; Lu, J.; Chien, C.L.; Chen, H.F.; Ho, H.N.; Yu, C.J.; Wang, Z.Q.; et al. PARP1 controls KLF4-mediated telomerase expression in stem cells and cancer cells. Nucleic Acids Res. 2017, 45, 10492–10503. [Google Scholar] [CrossRef] [PubMed]
- Riverso, M.; Montagnani, V.; Stecca, B. KLF4 is regulated by RAS/RAF/MEK/ERK signaling through E2F1 and promotes melanoma cell growth. Oncogene 2017, 36, 3322–3333. [Google Scholar] [CrossRef]
- Qi, X.T.; Li, Y.L.; Zhang, Y.Q.; Xu, T.; Lu, B.; Fang, L.; Gao, J.Q.; Yu, L.S.; Zhu, D.F.; Yang, B.; et al. KLF4 functions as an oncogene in promoting cancer stem cell-like characteristics in osteosarcoma cells. Acta Pharm. Sin. 2019, 40, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Zhong, T.; Jiang, L.; Huang, J.; Xia, Y.; Hu, R. miR-25 enhances cell migration and invasion in non-small-cell lung cancer cells via ERK signaling pathway by inhibiting KLF4. Mol. Med. Rep. 2018, 17, 7005–7016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Shen, A.; Ouyang, X.; Zhao, G.; Du, Z.; Huo, W.; Zhang, T.; Wang, Y.; Yang, C.; Dong, P.; et al. KLF4 expression enhances the efficacy of chemotherapy drugs in ovarian cancer cells. Biochem. Biophys. Res. Commun. 2017, 484, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Yoshida, G.J. Emerging roles of Myc in stem cell biology and novel tumor therapies. J. Exp. Clin. Cancer Res. 2018, 37, 1–20. [Google Scholar]
- Galardi, S.; Savino, M.; Scagnoli, F.; Pellegatta, S.; Pisati, F.; Zambelli, F.; Illi, B.; Annibali, D.; Beji, S.; Orecchini, E.; et al. Resetting cancer stem cell regulatory nodes upon MYC inhibition. EMBO Rep. 2016, 17, 1872–1889. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, I.; Malfettone, A.; Soukupova, J. New Insights into the Crossroads between EMT and Stemness in the Context of Cancer. J. Clin. Med. 2016, 5, 37. [Google Scholar] [CrossRef] [Green Version]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [Green Version]
- Harrington, B.S.; Annunziata, C.M. NF-kappaB Signaling in Ovarian Cancer. Cancers 2019, 11, 1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.S.; Kim, J.H.; Kim, S.L.; Lee, D.S. Disruption of the NF-kappaB/IL-8 Signaling Axis by Sulconazole Inhibits Human Breast Cancer Stem Cell Formation. Cells 2019, 8, 1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinkenbaugh, A.L.; Baldwin, A.S. The NF-kappaB Pathway and Cancer Stem Cells. Cells 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Networking of WNT, FGF, Notch, BMP, and Hedgehog signaling pathways during carcinogenesis. Stem Cell Rev. 2007, 3, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Clements, W.M.; Wang, J.; Sarnaik, A.; Kim, O.J.; MacDonald, J.; Fenoglio-Preiser, C.; Groden, J.; Lowy, A.M. beta-Catenin mutation is a frequent cause of Wnt pathway activation in gastric cancer. Cancer Res 2002, 62, 3503–3506. [Google Scholar]
- Abd El-Rehim, D.; Ali, M.M. Aberrant expression of beta-catenin in invasive ductal breast carcinomas. J. Egypt Natl. Canc. Inst. 2009, 21, 185–195. [Google Scholar]
- Kudo, J.; Nishiwaki, T.; Haruki, N.; Ishiguro, H.; Shibata, Y.; Terashita, Y.; Sugiura, H.; Shinoda, N.; Kimura, M.; Kuwabara, Y.; et al. Aberrant nuclear localization of beta-catenin without genetic alterations in beta-catenin or Axin genes in esophageal cancer. World J. Surg. Oncol. 2007, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Jiang, C.; Xu, R.; Liu, Q.; Liu, G.; Zhang, Y. TRIP6 enhances stemness property of breast cancer cells through activation of Wnt/beta-catenin. Cancer Cell Int. 2020, 20, 51. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Pan, R.; Zhou, D.; Ye, G.; Tan, W. BCL11A enhances stemness and promotes progression by activating Wnt/beta-catenin signaling in breast cancer. Cancer Manag. Res. 2019, 11, 2997–3007. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Pong, U.K.; Zhang, J.T.; Tsang, L.L.; Huang, J.W.; Tu, S.P.; Jiang, X.H. R-spodin2 enhances canonical Wnt signaling to maintain the stemness of glioblastoma cells. Cancer Cell Int. 2018, 18, 156. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, S.; Clarke, R.B.; Brennan, K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006, 66, 1517–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, E.V.; Kim, E.J.; Wu, J.J.; Hynes, M.; Bednar, F.; Proctor, E.; Wang, L.D.; Dziubinski, M.L.; Simeone, D.M. The Notch Pathway Is Important in Maintaining the Cancer Stem Cell Population in Pancreatic Cancer. PLoS ONE 2014, 9, e91983. [Google Scholar] [CrossRef] [PubMed]
- Koury, J.; Zhong, L.; Hao, J.J. Targeting Signaling Pathways in Cancer Stem Cells for Cancer Treatment. Stem Cells Int. 2017, 2017, 2925869 . [Google Scholar] [CrossRef]
- Yan, Y.Y.; Liu, F.X.; Han, L.; Zhao, L.; Chen, J.J.; Olopade, O.I.; He, M.; Wei, M.J. HIF-2 alpha promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J. Exp. Clin. Cancer Res. 2018, 37, 256. [Google Scholar] [CrossRef] [Green Version]
- Kang, L.; Mao, J.; Tao, Y.; Song, B.; Ma, W.; Lu, Y.; Zhao, L.; Li, J.; Yang, B.; Li, L. MicroRNA-34a suppresses the breast cancer stem cell-like characteristics by downregulating Notch1 pathway. Cancer Sci. 2015, 106, 700–708. [Google Scholar] [CrossRef]
- Zeng, F.; Chen, H.; Zhang, Z.H.; Yao, T.; Wang, G.; Zeng, Q.X.; Duan, S.H.; Zhan, Y.Q. Regulating glioma stem cells by hypoxia through the Notch1 and Oct3/4 signaling pathway. Oncol. Lett. 2018, 16, 6315–6322. [Google Scholar] [CrossRef]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [Green Version]
- Amantini, C.; Morelli, M.B.; Nabissi, M.; Cardinali, C.; Santoni, M.; Gismondi, A.; Santoni, G. Capsaicin triggers autophagic cell survival which drives epithelial mesenchymal transition and chemoresistance in bladder cancer cells in an Hedgehog-dependent manner. Oncotarget 2016, 7, 50180–50194. [Google Scholar] [CrossRef] [Green Version]
- Villegas, V.E.; Rondon-Lagos, M.; Annaratone, L.; Castellano, I.; Grismaldo, A.; Sapino, A.; Zaphiropoulos, P.G. Tamoxifen Treatment of Breast Cancer Cells: Impact on Hedgehog/GLI1 Signaling. Int. J. Mol. Sci. 2016, 17, 308. [Google Scholar] [CrossRef] [Green Version]
- Po, A.; Silvano, M.; Miele, E.; Capalbo, C.; Eramo, A.; Salvati, V.; Todaro, M.; Besharat, Z.M.; Catanzaro, G.; Cucchi, D.; et al. Noncanonical GLI1 signaling promotes stemness features and in vivo growth in lung adenocarcinoma. Oncogene 2017, 36, 4641–4652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borah, A.; Raveendran, S.; Rochani, A.; Maekawa, T.; Kumar, D.S. Targeting self-renewal pathways in cancer stem cells: Clinical implications for cancer therapy. Oncogenesis 2015, 4, e177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, R.; Gires, O.; Zhu, L.; Liu, J.; Li, J.; Yang, H.; Ju, G.; Huang, J.; Ge, W.; Chen, Y.; et al. TSPAN8 promotes cancer cell stemness via activation of sonic Hedgehog signaling. Nat. Commun. 2019, 10, 2863. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Altaba, A.R.I. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Mondal, S.; Butti, R.; Prasanna Gunasekaran, V.; Chatterjee, U.; Halder, A.; Kundu, G.C.; Mandal, C. Desialylation of Sonic-Hedgehog by Neu2 Inhibits Its Association with Patched1 Reducing Stemness-Like Properties in Pancreatic Cancer Sphere-forming Cells. Cells 2020, 9, 1512. [Google Scholar] [CrossRef] [PubMed]
- Chambers, I. The molecular basis of pluripotency in mouse embryonic stem cells. Cloning Stem Cells 2004, 6, 386–391. [Google Scholar] [CrossRef]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E., 3rd; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef] [Green Version]
- Kaowinn, S.; Kaewpiboon, C.; Koh, S.S.; Kramer, O.H.; Chung, Y.H. STAT1HDAC4 signaling induces epithelialmesenchymal transition and sphere formation of cancer cells overexpressing the oncogene, CUG2. Oncol. Rep. 2018, 40, 2619–2627. [Google Scholar] [CrossRef]
- Dey, N.; De, P.; Leyland-Jones, B. PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharm. 2017, 175, 91–106. [Google Scholar] [CrossRef]
- Tan, A.C. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, T.L.; Lertpiriyapong, K.; Cocco, L.; Martelli, A.M.; Libra, M.; Candido, S.; Montalto, G.; Cervello, M.; Steelman, L.; Abrams, S.L.; et al. Roles of EGFR and KRAS and their downstream signaling pathways in pancreatic cancer and pancreatic cancer stem cells. Adv. Biol Regul 2015, 59, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Madsen, R.R. PI3K in stemness regulation: From development to cancer. Biochem. Soc. Trans. 2020, 48, 301–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouspenskaia, T.; Matos, I.; Mertz, A.F.; Fiore, V.F.; Fuchs, E. WNT-SHH Antagonism Specifies and Expands Stem Cells prior to Niche Formation. Cell 2016, 164, 156–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlster, L.; Daley, G.Q. Progress towards generation of human haematopoietic stem cells. Nat. Cell Biol. 2016, 18, 1111–1117. [Google Scholar] [CrossRef]
- Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Cell Culture Based in vitro Test Systems for Anticancer Drug Screening. Front. Bioeng. Biotechnol. 2020, 8, 322. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Lambert, A.W.; Weinberg, R.A. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat. Rev. Cancer 2021, 21, 325–338. [Google Scholar] [CrossRef]
- Liu, Y.; Nenutil, R.; Appleyard, M.V.; Murray, K.; Boylan, M.; Thompson, A.M.; Coates, P.J. Lack of correlation of stem cell markers in breast cancer stem cells. Br. J. Cancer 2014, 110, 2063–2071. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Clevers, H. Reparative inflammation takes charge of tissue regeneration. Nature 2016, 529, 307–315. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, C.; Wang, H.Z.; Peng, Y.N.; Li, H.O.; Zhou, Y.J.; Liu, S.; Wang, F.; Liu, L.; Chang, Y.; et al. Soft fibrin matrix downregulates DAB2IP to promote Nanog-dependent growth of colon tumor-repopulating cells. Cell Death Dis 2019, 10, 151. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, L.; Pappan, L.; Galliher-Beckley, A.; Shi, J. IL-1beta promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol. Cancer 2012, 11, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, H.S.; Akhavan, J.; Lee, S.H.; Kim, R.H.; Kang, M.K.; Park, N.H.; Shin, K.H. Proinflammatory cytokine TNFalpha promotes HPV-associated oral carcinogenesis by increasing cancer stemness. Int. J. Oral Sci. 2020, 12, 3. [Google Scholar] [CrossRef] [Green Version]
- Ciardiello, C.; Leone, A.; Budillon, A. The Crosstalk between Cancer Stem Cells and Microenvironment Is Critical for Solid Tumor Progression: The Significant Contribution of Extracellular Vesicles. Stem Cells Int. 2018, 2018, 6392198. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1 alpha and Beyond. Front. Oncol. 2020, 10, 486. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Brosicke, N.; Faissner, A. Role of tenascins in the ECM of gliomas. Cell Adhes. Migr. 2015, 9, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Winer, J.P.; Janmey, P.A.; McCormick, M.E.; Funaki, M. Bone marrow-derived human mesenchymal stem cells become quiescent on soft substrates but remain responsive to chemical or mechanical stimuli. Tissue Eng. Part. A 2009, 15, 147–154. [Google Scholar] [CrossRef]
- You, Y.; Zheng, Q.; Dong, Y.; Xie, X.; Wang, Y.; Wu, S.; Zhang, L.; Wang, Y.; Xue, T.; Wang, Z.; et al. Matrix stiffness-mediated effects on stemness characteristics occurring in HCC cells. Oncotarget 2016, 7, 32221–32231. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Chulpanova, D.S.; Kitaeva, K.V.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Therapeutic Prospects of extracellular vesicles in Cancer Treatment. Front. Immunol. 2018, 9, 1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaimardanova, A.A.; Solovyeva, V.V.; Chulpanova, D.S.; James, V.; Kitaeva, K.V.; Rizvanov, A.A. Extracellular vesicles in the diagnosis and treatment of central nervous system diseases. Neural Regen. Res. 2020, 15, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Bronisz, A.; Wang, Y.; Nowicki, M.O.; Peruzzi, P.; Ansari, K.I.; Ogawa, D.; Balaj, L.; De Rienzo, G.; Mineo, M.; Nakano, I.; et al. Extracellular Vesicles Modulate the Glioblastoma Microenvironment via a Tumor Suppression Signaling Network Directed by miR-1. Cancer Res. 2014, 74, 738–750. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.B.; Yan, C.; Mu, L.; Huang, K.Y.; Li, X.L.; Tao, D.D.; Wu, Y.Q.; Qin, J.C. Fibroblast-Derived Exosomes Contribute to Chemoresistance through Priming Cancer Stem Cells in Colorectal Cancer. PLoS ONE 2015, 10, e0125625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, E.; Piva, M.; Rodriguez-Suarez, E.; Gil, D.; Royo, F.; Elortza, F.; Falcon-Perez, J.M.; Vivanco, M. Human mammospheres secrete hormone-regulated active extracellular vesicles. PLoS ONE 2014, 9, e83955. [Google Scholar] [CrossRef]
- Gilazieva, Z.; Ponomarev, A.; Rutland, C.; Rizvanov, A.; Solovyeva, V. Promising Applications of Tumor Spheroids and Organoids for Personalized Medicine. Cancers 2020, 12, 2727. [Google Scholar] [CrossRef]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Djordjevic, B.; Lange, C.S. Clonogenicity of mammalian cells in hybrid spheroids: A new assay method. Radiat. Env. Biophys. 1990, 29, 31–46. [Google Scholar] [CrossRef]
- Mikhail, A.S.; Eetezadi, S.; Allen, C. Multicellular tumor spheroids for evaluation of cytotoxicity and tumor growth inhibitory effects of nanomedicines in vitro: A comparison of docetaxel-loaded block copolymer micelles and Taxotere(R). PLoS ONE 2013, 8, e62630. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Yang, Z.T.; Dong, D.L.; Jang, T.S.; Knowles, J.C.; Kim, H.W.; Jin, G.Z.; Xuan, Y.H. 3D culture technologies of cancer stem cells: Promising ex vivo tumor models. J. Tissue Eng. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P. Patient-derived induced pluripotent stem cells in cancer research and precision oncology. Nat. Med. 2016, 22, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Spradling, A.C. Stem cells and niches: Mechanisms that promote stem cell maintenance throughout life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschhaeuser, F.; Menne, H.; Dittfeld, C.; West, J.; Mueller-Klieser, W.; Kunz-Schughart, L.A. Multicellular tumor spheroids: An underestimated tool is catching up again. J. Biotechnol. 2010, 148, 3–15. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Mooney, S.M.; Natsumeda, M.; Steiger, H.J.; Maciaczyk, J. Targeting cancer stem-like cells in glioblastoma and colorectal cancer through metabolic pathways. Int. J. Cancer 2017, 140, 10–22. [Google Scholar] [CrossRef]
- Puls, T.J.; Tan, X.H.; Whittington, C.F.; Voytik-Harbin, S.L. 3D collagen fibrillar microstructure guides pancreatic cancer cell phenotype and serves as a critical design parameter for phenotypic models of EMT. PLoS ONE 2017, 12, e0188870. [Google Scholar] [CrossRef] [Green Version]
- Pal, A.; Ashworth, J.C.; Collier, P.; Probert, C.; Jones, S.; Leza, E.P.; Meakin, M.L.; Ritchie, A.A.; Onion, D.; Clarke, P.A.; et al. A 3D Heterotypic Breast Cancer Model Demonstrates a Role for Mesenchymal Stem Cells in Driving a Proliferative and Invasive Phenotype. Cancers 2020, 12, 2290. [Google Scholar] [CrossRef]
- Xue, J.G.; Zhu, Y.; Sun, Z.X.; Ji, R.B.; Zhang, X.; Xu, W.R.; Yuan, X.; Zhang, B.; Yan, Y.M.; Yin, L.; et al. Tumorigenic hybrids between mesenchymal stem cells and gastric cancer cells enhanced cancer proliferation, migration and stemness. BMC Cancer 2015, 15, 793. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Devi, G.R. Three-dimensional culture systems in cancer research: Focus on tumor spheroid model. Pharmacol. Ther. 2016, 163, 94–108. [Google Scholar] [CrossRef] [Green Version]
- Maliszewska-Olejniczak, K.; Brodaczewska, K.K.; Bielecka, Z.F.; Solarek, W.; Kornakiewicz, A.; Szczylik, C.; Porta, C.; Czarnecka, A.M. Development of extracellular matrix supported 3D culture of renal cancer cells and renal cancer stem cells. Cytotechnology 2019, 71, 149–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.B.; Onder, T.T.; Jiang, G.Z.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of Selective Inhibitors of Cancer Stem Cells by High-Throughput Screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namiki, K.; Wongsirisin, P.; Yokoyama, S.; Sato, M.; Rawangkan, A.; Sakai, R.; Iida, K.; Suganuma, M. (-)-Epigallocatechin gallate inhibits stemness and tumourigenicity stimulated by AXL receptor tyrosine kinase in human lung cancer cells. Sci Rep. 2020, 10, 2444. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Rarnachandran, S.; Gupta, N.; Kaushik, I.; Srivastava, S.K. Cancer cells stemness: A doorstep to targeted therapy. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866. [Google Scholar] [CrossRef]
- Konishi, J.; Kawaguchi, K.S.; Vo, H.; Haruki, N.; Gonzalez, A.; Carbone, D.P.; Dang, T.P. gamma-secretase inhibitor prevents Notch3 activation and reduces proliferation in human lung cancers. Cancer Res. 2007, 67, 8051–8057. [Google Scholar] [CrossRef] [Green Version]
- Moore, G.; Annett, S.; McClements, L.; Robson, T. Top Notch Targeting Strategies in Cancer: A Detailed Overview of Recent Insights and Current Perspectives. Cells 2020, 9, 1503. [Google Scholar] [CrossRef]
- Cook, N.; Basu, B.; Smith, D.M.; Gopinathan, A.; Evans, J.; Steward, W.P.; Palmer, D.; Propper, D.; Venugopal, B.; Hategan, M.; et al. A phase I trial of the gamma-secretase inhibitor MK-0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. Br. J. Cancer 2018, 118, 793–801. [Google Scholar] [CrossRef]
- Herman, S.E.M.; Gordon, A.L.; Wagner, A.J.; Heerema, N.A.; Zhao, W.Q.; Flynn, J.M.; Jones, J.; Andritsos, L.; Puri, K.D.; Lannutti, B.J.; et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010, 116, 2078–2088. [Google Scholar] [CrossRef] [Green Version]
- Foster, P.; Yamaguchi, K.; Hsu, P.P.; Qian, F.; Du, X.N.; Wu, J.M.; Won, K.A.; Yu, P.W.; Jaeger, C.T.; Zhang, W.T.; et al. The Selective PI3K Inhibitor XL147 (SAR245408) Inhibits Tumor Growth and Survival and Potentiates the Activity of Chemotherapeutic Agents in Preclinical Tumor Models. Mol. Cancer Ther. 2015, 14, 931–940. [Google Scholar] [CrossRef] [Green Version]
- Eyler, C.E.; Foo, W.C.; Lafiura, K.M.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Brain Cancer Stem Cells Display Preferential Sensitivity to Akt Inhibition. Stem Cells 2008, 26, 3027–3036. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.K.; Reckamp, K.; Yu, H.; Figlin, R.A. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin. Investig. Drugs 2010, 19, 1355–1366. [Google Scholar] [CrossRef] [Green Version]
- Tagawa, S.T.; Chatta, G.S.; Mazhari, R.; Benaim, E. Archexin, a novel AKT-1-specific inhibitor for the treatment of metastatic renal cancer: Preliminary phase I data. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Lindemann, R.K. Stroma-initiated hedgehog signaling takes center stage in B-cell lymphoma. Cancer Res. 2008, 68, 961–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimeno, A.; Gordon, M.; Chugh, R.; Messersmith, W.; Mendelson, D.; Dupont, J.; Stagg, R.; Kapoun, A.M.; Xu, L.; Uttamsingh, S.; et al. A First-in-Human Phase I Study of the Anticancer Stem Cell Agent Ipafricept (OMP-54F28), a Decoy Receptor for Wnt Ligands, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 7490–7497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, A.H.; Chiorean, E.G.; Kwak, E.L.; Lenz, H.J.; Nadler, P.I.; Wood, D.L.; Fujimori, M.; Inada, T.; Kouji, H.; McWilliams, R.R. Final results of a phase Ib dose-escalation study of PRI-724, a CBP/beta-catenin modulator, plus gemcitabine (GEM) in patients with advanced pancreatic adenocarcinoma (APC) as second-line therapy after FOLFIRINOX or FOLFOX. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Cortes, J.E.; Faderl, S.; Pagel, J.; Jung, C.W.; Yoon, S.S.; Koh, Y.; Pardanani, A.D.; Hauptschein, R.S.; Lee, K.J.; Lee, J.H. Phase 1 study of CWP232291 in relapsed/refractory acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). J. Clin. Oncol. 2015, 33. [Google Scholar] [CrossRef]
- Jonker, D.J.; Stephenson, J.; Edenfield, W.J.; Supko, J.G.; Li, Y.Z.; Li, W.; Hitron, M.; Leggett, D.; Kerstein, D.; Li, C. A phase I extension study of BBI608, a first-in-class cancer stein cell (CSC) inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32. [Google Scholar] [CrossRef]
- Laurie, S.A.; Jonker, D.J.; Edenfield, W.J.; Stephenson, J.; Keller, D.; Hitron, M.; Li, W.; Li, Y.Z.; Gada, K.; Gao, Y.; et al. A phase 1 dose-escalation study of BBI503, a first-in-class cancer stemness kinase inhibitor in adult patients with advanced solid tumors. J. Clin. Oncol. 2014, 32. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Cancer Type | CSCs Markers | Reference |
|---|---|---|
| Blood tumors | CD34+ CD38− phenotype | [19] |
| Brain tumors | CD133+, CD49f+, CD90+, epidermal growth factor receptor (EGFR)+, c-series ganglioside-specific antigen A2B5+, L1 cell adhesion molecule (L1CAM)+ | [20,21,22] |
| Ovary tumors | CD24+, aldehyde dehydrogenase (ALDH)+, CD44+/CD117+, epithelial cell adhesion molecule (EpCAM)+, CD133+ | [23,24] |
| Prostate tumors | EpCAM+, CD117+, α2β1 integrin+, ALDH+, CD44+, enhancer of zeste homolog (EZH)+, CXC chemokine receptor type 4 (CXCR4)+, E-cadherin+, CD133+ | [13,25,26] |
| Colon tumors | CD133+, CD44+, CD166+, CD24+, EpCAM+), | [27,28,29,30] |
| Pancreatic tumors | CD133+, CD44+, CD24+, EpCAM+, tyrosine-protein kinase Met (cMet)+ | [31,32] |
| Liver tumors | CD44+, CD90+, CD206+, oval cell antigen 6 (OV-6)+), skin (CD20+, CD271+, ALDH+, CD133+ | [33] |
| Lung tumors | CD133+, ATP-binding cassette super-family G member 2 (ABCG2)high, CD166+, CD90+, CD87+, ALDH+, CD44+ | [34] |
| Breast tumors | ALDH1+, CD24+, CD44+, CD90+, CD133+, α6-integrin+ | [35,36,37,38] |
| Agent | Notes | Clinical Trials | Reference |
|---|---|---|---|
| MRK-003, MK-0752, R4733 | Notch inhibitors | NCT00106145 NCT01154452 | [167,168,169] |
| CAL-101, XL-147 | PI3K inhibitors | NCT01629615 NCT01613950 NCT00907205 | [170,171] |
| KRX-0401, RX-0201 | AKT inhibitors | NCT00590954 NCT01028495 | [172,173,174] |
| BMS-863923, IPI-926 | Hedgehog inhibitors | NCT01546038 NCT01130142 NCT01700049 | [175] |
| OMP-54F28, PRI-724, CWP232291 | Wnt inhibitors | NCT01606579 NCT01351103 NCT02092363 | [176,177,178] |
| BBI503, BBI608 | NANOG inhibitors | NCT02232633 NCT02315534 | [179,180] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponomarev, A.; Gilazieva, Z.; Solovyeva, V.; Allegrucci, C.; Rizvanov, A. Intrinsic and Extrinsic Factors Impacting Cancer Stemness and Tumor Progression. Cancers 2022, 14, 970. https://doi.org/10.3390/cancers14040970
Ponomarev A, Gilazieva Z, Solovyeva V, Allegrucci C, Rizvanov A. Intrinsic and Extrinsic Factors Impacting Cancer Stemness and Tumor Progression. Cancers. 2022; 14(4):970. https://doi.org/10.3390/cancers14040970
Chicago/Turabian StylePonomarev, Alexey, Zarema Gilazieva, Valeriya Solovyeva, Cinzia Allegrucci, and Albert Rizvanov. 2022. "Intrinsic and Extrinsic Factors Impacting Cancer Stemness and Tumor Progression" Cancers 14, no. 4: 970. https://doi.org/10.3390/cancers14040970
APA StylePonomarev, A., Gilazieva, Z., Solovyeva, V., Allegrucci, C., & Rizvanov, A. (2022). Intrinsic and Extrinsic Factors Impacting Cancer Stemness and Tumor Progression. Cancers, 14(4), 970. https://doi.org/10.3390/cancers14040970