
Epigenetic Regulation: A Link between Inflammation and Carcinogenesis

 ,
,  , ,
, ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction: The Complexity of Carcinogenesis
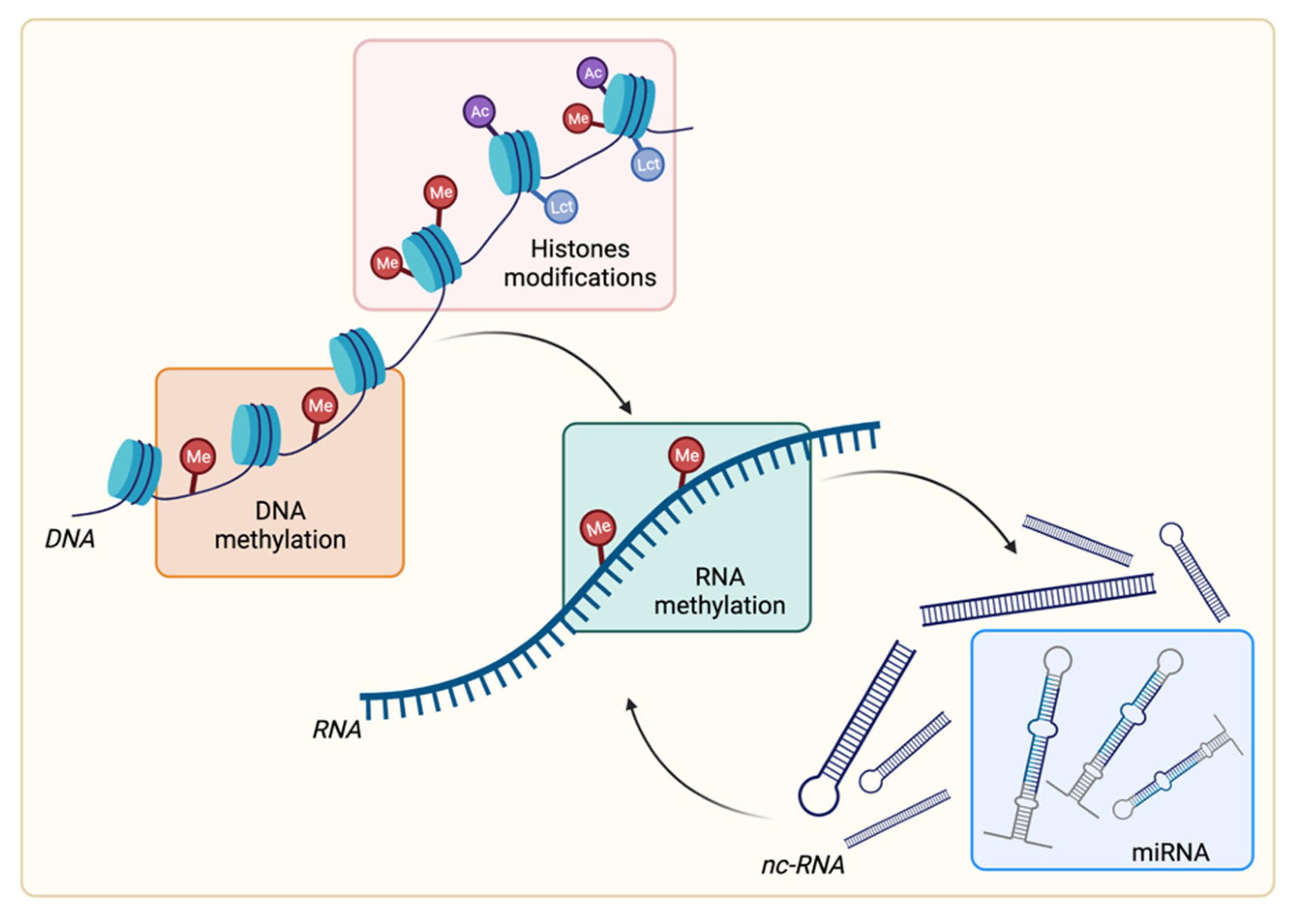
2. Overview of Epigenetic Modifications
2.1. DNA Methylation
2.2. RNA Methylation
2.3. Histone Posttranslational Modifications
2.4. Noncoding RNA: Focus on microRNA
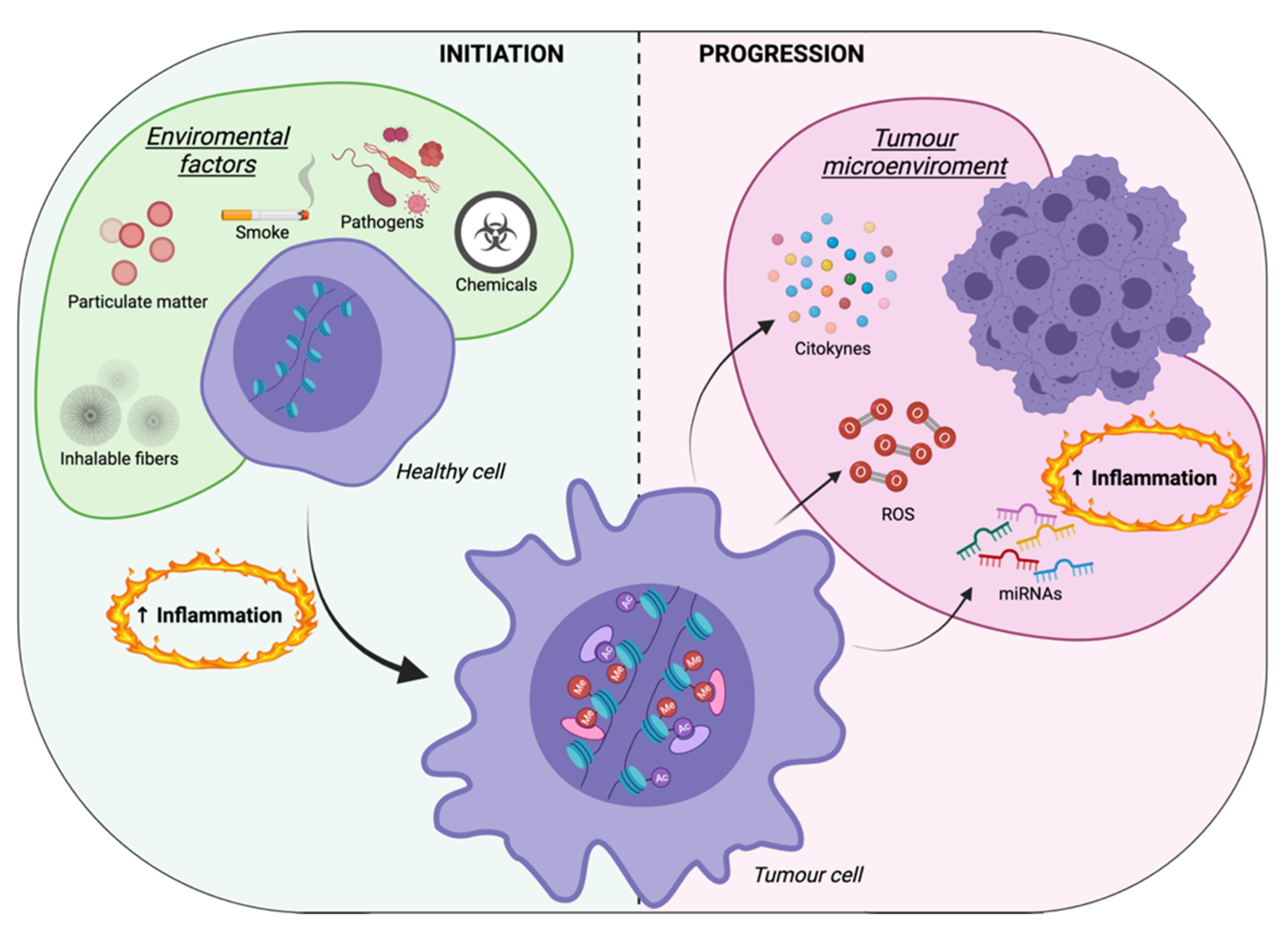
3. Inflammation-Mediated Epigenetic Modifications: A Focus on Cancer Cells
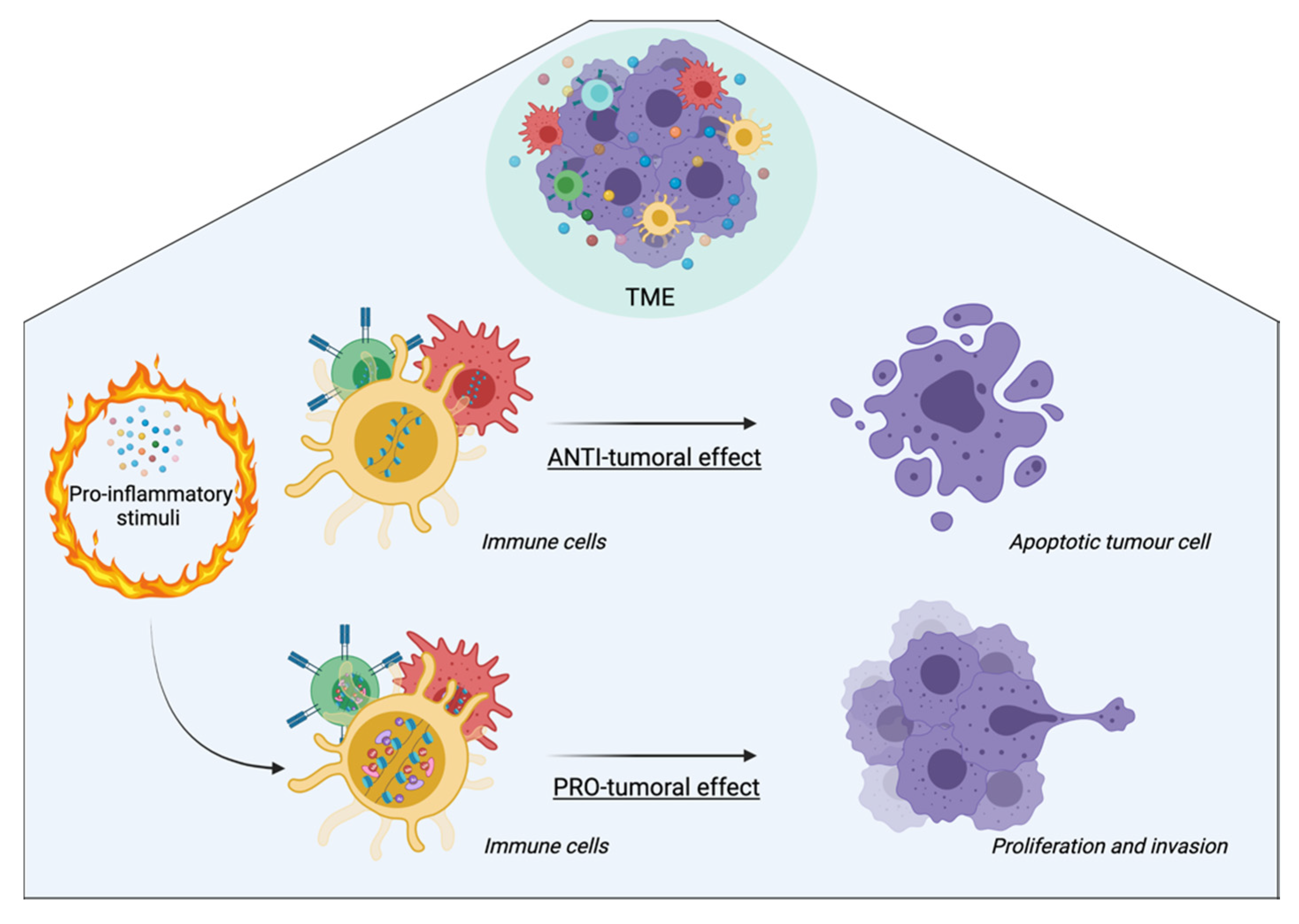
4. Inflammation-Mediated Epigenetic Modifications: A Focus on the Tumor Microenvironment
5. Anti-Inflammatory and Epidrug Applications in Cancer Therapy
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Easwaran, H.; Tsai, H.-C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; da Silva Santos, R.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.W.; Zhu, X.Y.; Li, Y.Y.; Meng, Z.Q. Epigenetic regulation and cancer (review). Oncol. Rep. 2014, 31, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Chan, Y.-T.; Tan, H.-Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef]
- Cedar, H.; Bergman, Y. Programming of DNA methylation patterns. Annu. Rev. Biochem. 2012, 81, 97–117. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Ziller, M.J.; Müller, F.; Liao, J.; Zhang, Y.; Gu, H.; Bock, C.; Boyle, P.; Epstein, C.B.; Bernstein, B.E.; Lengauer, T.; et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011, 7, e1002389. [Google Scholar] [CrossRef] [Green Version]
- Ramsahoye, B.H.; Biniszkiewicz, D.; Lyko, F.; Clark, V.; Bird, A.P.; Jaenisch, R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. USA 2000, 97, 5237–5242. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panneerdoss, S.; Eedunuri, V.K.; Yadav, P.; Timilsina, S.; Rajamanickam, S.; Viswanadhapalli, S.; Abdelfattah, N.; Onyeagucha, B.C.; Cui, X.; Lai, Z.; et al. Cross-talk among writers, readers, and erasers of m6A regulates cancer growth and progression. Sci. Adv. 2018, 4, eaar8263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klutstein, M.; Nejman, D.; Greenfield, R.; Cedar, H. DNA Methylation in Cancer and Aging. Cancer Res. 2016, 76, 3446–3450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welner, S.A.; Koty, Z.C.; Boksa, P. Chromaffin cell grafts to rat cerebral cortex reverse lesion-induced memory deficits. Brain Res. 1990, 527, 163–166. [Google Scholar] [CrossRef]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.L.; Morey, L. Emerging Roles for Polycomb-Group Proteins in Stem Cells and Cancer. Trends Biochem. Sci. 2019, 44, 688–700. [Google Scholar] [CrossRef]
- Issa, J.-P. Aging and epigenetic drift: A vicious cycle. J. Clin. Investig. 2014, 124, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Chen, Y.; Petersen, I. Expression and promoter DNA methylation of MLH1 in colorectal cancer and lung cancer. Pathol. Res. Pract. 2017, 213, 333–338. [Google Scholar] [CrossRef]
- Botezatu, A.; Iancu, I.V.; Plesa, A.; Manda, D.; Popa, O.; Bostan, M.; Mihaila, M.; Albulescu, A.; Fudulu, A.; Vladoiu, S.V.; et al. Methylation of tumour suppressor genes associated with thyroid cancer. Cancer Biomark. 2019, 25, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Takehara, K.; Sugimoto, N.; Kaneko, K.; Fujimoto, E.; Okazawa-Sakai, M.; Okame, S.; Shiroyama, Y.; Yokoyama, T.; Teramoto, N.; et al. Lynch syndrome-associated endometrial carcinoma with MLH1 germline mutation and MLH1 promoter hypermethylation: A case report and literature review. BMC Cancer 2018, 18, 576. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Missiroli, S.; Perrone, M.; Fiorica, F.; Pinton, P.; Giorgi, C. Mitochondrial Control of Genomic Instability in Cancer. Cancers 2021, 13, 1914. [Google Scholar] [CrossRef]
- Hughes, L.A.E.; Melotte, V.; de Schrijver, J.; de Maat, M.; Smit, V.T.H.B.M.; Bovée, J.V.M.G.; French, P.J.; van den Brandt, P.A.; Schouten, L.J.; de Meyer, T.; et al. The CpG island methylator phenotype: What’s in a name? Cancer Res. 2013, 73, 5858–5868. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Chen, C.; Ji, X.; Liu, J.; Zhou, Q.; Wang, G.; Yuan, W.; Kan, Q.; Sun, Z. The interplay between m6A RNA methylation and noncoding RNA in cancer. J. Hematol. Oncol. 2019, 12, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.-M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.-L.; Song, S.-H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef]
- Sun, T.; Wu, R.; Ming, L. The role of m6A RNA methylation in cancer. Biomed. Pharmacother. 2019, 112, 108613. [Google Scholar] [CrossRef] [PubMed]
- Sadakierska-Chudy, A.; Filip, M. A comprehensive view of the epigenetic landscape. Part II: Histone post-translational modification, nucleosome level, and chromatin regulation by ncRNAs. Neurotox. Res. 2015, 27, 172–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Voss, A.K.; Thomas, T. Histone Lysine and Genomic Targets of Histone Acetyltransferases in Mammals. Bioessays 2018, 40, e1800078. [Google Scholar] [CrossRef] [PubMed]
- Gayther, S.A.; Batley, S.J.; Linger, L.; Bannister, A.; Thorpe, K.; Chin, S.F.; Daigo, Y.; Russell, P.; Wilson, A.; Sowter, H.M.; et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 2000, 24, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Fei, H.-J.; Zu, L.-D.; Wu, J.; Jiang, X.-S.; Wang, J.-L.; Chin, Y.E.; Fu, G.-H. PCAF acts as a gastric cancer suppressor through a novel PCAF-p16-CDK4 axis. Am. J. Cancer Res. 2016, 6, 2772–2786. [Google Scholar] [PubMed]
- Wan, J.; Xu, W.; Zhan, J.; Ma, J.; Li, X.; Xie, Y.; Wang, J.; Zhu, W.-G.; Luo, J.; Zhang, H. PCAF-mediated acetylation of transcriptional factor HOXB9 suppresses lung adenocarcinoma progression by targeting oncogenic protein JMJD6. Nucleic Acids Res. 2016, 44, 10662–10675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, R.; Banerjee, S.; Amin, S.A.; Adhikari, N.; Jha, T. Histone deacetylase 3 (HDAC3) inhibitors as anticancer agents: A review. Eur. J. Med. Chem. 2020, 192, 112171. [Google Scholar] [CrossRef] [PubMed]
- Zee, B.M.; Levin, R.S.; Xu, B.; LeRoy, G.; Wingreen, N.S.; Garcia, B.A. In vivo residue-specific histone methylation dynamics. J. Biol. Chem. 2010, 285, 3341–3350. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Charvin, G.; Siggia, E.D.; Cross, F.R. Nucleosome-depleted regions in cell-cycle-regulated promoters ensure reliable gene expression in every cell cycle. Dev. Cell 2010, 18, 544–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collings, C.K.; Anderson, J.N. Links between DNA methylation and nucleosome occupancy in the human genome. Epigenet. Chromatin 2017, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Soufi, A.; Donahue, G.; Zaret, K.S. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 2012, 151, 994–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onder, T.T.; Kara, N.; Cherry, A.; Sinha, A.U.; Zhu, N.; Bernt, K.M.; Cahan, P.; Marcarci, B.O.; Unternaehrer, J.; Gupta, P.B.; et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature 2012, 483, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 2016, 16, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Béguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosen, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013, 23, 677–692. [Google Scholar] [CrossRef] [Green Version]
- Hodges, C.; Kirkland, J.G.; Crabtree, G.R. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026930. [Google Scholar] [CrossRef] [Green Version]
- Berns, K.; Sonnenblick, A.; Gennissen, A.; Brohée, S.; Hijmans, E.M.; Evers, B.; Fumagalli, D.; Desmedt, C.; Loibl, S.; Denkert, C.; et al. Loss of ARID1A Activates ANXA1, which Serves as a Predictive Biomarker for Trastuzumab Resistance. Clin. Cancer Res. 2016, 22, 5238–5248. [Google Scholar] [CrossRef] [Green Version]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184.e7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Latham, T.; Mackay, L.; Sproul, D.; Karim, M.; Culley, J.; Harrison, D.J.; Hayward, L.; Langridge-Smith, P.; Gilbert, N.; Ramsahoye, B.H. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. 2012, 40, 4794–4803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genders, A.J.; Martin, S.D.; McGee, S.L.; Bishop, D.J. A physiological drop in pH decreases mitochondrial respiration, and HDAC and Akt signaling, in L6 myocytes. Am. J. Physiol. Cell Physiol. 2019, 316, C404–C414. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovat, F.; Fassan, M.; Sacchi, D.; Ranganathan, P.; Palamarchuk, A.; Bill, M.; Karunasiri, M.; Gasparini, P.; Nigita, G.; Distefano, R.; et al. Knockout of both miR-15/16 loci induces acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2018, 115, 13069–13074. [Google Scholar] [CrossRef] [Green Version]
- Young, L.E.; Moore, A.E.; Sokol, L.; Meisner-Kober, N.; Dixon, D.A. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol. Cancer Res. 2012, 10, 167–180. [Google Scholar] [CrossRef] [Green Version]
- Bonci, D.; Coppola, V.; Musumeci, M.; Addario, A.; Giuffrida, R.; Memeo, L.; D’Urso, L.; Pagliuca, A.; Biffoni, M.; Labbaye, C.; et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat. Med. 2008, 14, 1271–1277. [Google Scholar] [CrossRef]
- Sun, C.-Y.; She, X.-M.; Qin, Y.; Chu, Z.-B.; Chen, L.; Ai, L.-S.; Zhang, L.; Hu, Y. miR-15a and miR-16 affect the angiogenesis of multiple myeloma by targeting VEGF. Carcinogenesis 2013, 34, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Huang, E.; Liu, R.; Chu, Y. miRNA-15a/16: As tumor suppressors and more. Future Oncol. 2015, 11, 2351–2363. [Google Scholar] [CrossRef]
- Hünten, S.; Siemens, H.; Kaller, M.; Hermeking, H. The p53/microRNA network in cancer: Experimental and bioinformatics approaches. Adv. Exp. Med. Biol. 2013, 774, 77–101. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Hao, X.; Meng, Y.; Zhang, M.; Desano, J.; Fan, D.; Xu, L. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer 2008, 8, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warne, M.S.; Connell, D.W.; Hawker, D.W. Comparison of the critical concentration and critical volume hypotheses to model non-specific toxicity of individual compounds. Toxicology 1991, 66, 187–195. [Google Scholar] [CrossRef]
- Xia, H.; Ng, S.S.; Jiang, S.; Cheung, W.K.C.; Sze, J.; Bian, X.-W.; Kung, H.-F.; Lin, M.C. miR-200a-mediated downregulation of ZEB2 and CTNNB1 differentially inhibits nasopharyngeal carcinoma cell growth, migration and invasion. Biochem. Biophys. Res. Commun. 2010, 391, 535–541. [Google Scholar] [CrossRef]
- Pecot, C.V.; Rupaimoole, R.; Yang, D.; Akbani, R.; Ivan, C.; Lu, C.; Wu, S.; Han, H.-D.; Shah, M.Y.; Rodriguez-Aguayo, C.; et al. Tumour angiogenesis regulation by the miR-200 family. Nat. Commun. 2013, 4, 2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, T.V.; Mendell, J.T. Myc: Maestro of MicroRNAs. Genes Cancer 2010, 1, 568–575. [Google Scholar] [CrossRef]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef] [Green Version]
- Kent, O.A.; Chivukula, R.R.; Mullendore, M.; Wentzel, E.A.; Feldmann, G.; Lee, K.H.; Liu, S.; Leach, S.D.; Maitra, A.; Mendell, J.T. Repression of the miR-143/145 cluster by oncogenic Ras initiates a tumor-promoting feed-forward pathway. Genes Dev. 2010, 24, 2754–2759. [Google Scholar] [CrossRef] [Green Version]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [Green Version]
- Lujambio, A.; Calin, G.A.; Villanueva, A.; Ropero, S.; Sánchez-Céspedes, M.; Blanco, D.; Montuenga, L.M.; Rossi, S.; Nicoloso, M.S.; Faller, W.J.; et al. A microRNA DNA methylation signature for human cancer metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 13556–13561. [Google Scholar] [CrossRef] [Green Version]
- Lujambio, A.; Ropero, S.; Ballestar, E.; Fraga, M.F.; Cerrato, C.; Setién, F.; Casado, S.; Suarez-Gauthier, A.; Sanchez-Cespedes, M.; Git, A.; et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007, 67, 1424–1429. [Google Scholar] [CrossRef] [Green Version]
- Neves, R.; Scheel, C.; Weinhold, S.; Honisch, E.; Iwaniuk, K.M.; Trompeter, H.-I.; Niederacher, D.; Wernet, P.; Santourlidis, S.; Uhrberg, M. Role of DNA methylation in miR-200c/141 cluster silencing in invasive breast cancer cells. BMC Res. Notes 2010, 3, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Rimessi, A.; Previati, M.; Nigro, F.; Wieckowski, M.R.; Pinton, P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 2016, 81, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. J. Clin. Med. 2020, 9, 740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Hiraku, Y.; Tabata, T.; Ma, N.; Murata, M.; Ding, X.; Kawanishi, S. Nitrative and oxidative DNA damage in cervical intraepithelial neoplasia associated with human papilloma virus infection. Cancer Sci. 2007, 98, 964–972. [Google Scholar] [CrossRef]
- Hemmat, N.; Bannazadeh Baghi, H. Association of human papillomavirus infection and inflammation in cervical cancer. Pathog. Dis. 2019, 77, ftz048. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Kawanishi, M.; Hiraku, Y.; Murata, M.; Huang, G.-W.; Huang, Y.; Luo, D.-Z.; Mo, W.-G.; Fukui, Y.; Kawanishi, S. Reactive nitrogen species-dependent DNA damage in EBV-associated nasopharyngeal carcinoma: The relation to STAT3 activation and EGFR expression. Int. J. Cancer 2008, 122, 2517–2525. [Google Scholar] [CrossRef]
- Wang, F.; Meng, W.; Wang, B.; Qiao, L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014, 345, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Adachi, Y.; Hiraku, Y.; Horiki, N.; Horiike, S.; Imoto, I.; Pinlaor, S.; Murata, M.; Semba, R.; Kawanishi, S. Accumulation of 8-nitroguanine in human gastric epithelium induced by Helicobacter pylori infection. Biochem. Biophys. Res. Commun. 2004, 319, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Tsukamoto, T.; Toyoda, T.; Mori, A.; Tanaka, H.; Maekita, T.; Ichinose, M.; Tatematsu, M.; Ushijima, T. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010, 70, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; Coffelt, S.B.; Duits, D.E.M.; van Miltenburg, M.H.; Slagter, M.; de Rink, I.; Henneman, L.; Kas, S.M.; Prekovic, S.; Hau, C.-S.; et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 2019, 572, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Shawki, S.; Ashburn, J.; Signs, S.A.; Huang, E. Colon Cancer: Inflammation-Associated Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 269–287. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.P.; Li, J.; Tewari, A.K. Inflammation and prostate cancer: The role of interleukin 6 (IL-6). BJU Int. 2014, 113, 986–992. [Google Scholar] [CrossRef]
- Yang, Y.M.; Kim, S.Y.; Seki, E. Inflammation and Liver Cancer: Molecular Mechanisms and Therapeutic Targets. Semin. Liver Dis. 2019, 39, 26–42. [Google Scholar] [CrossRef]
- Hillegass, J.M.; Shukla, A.; Lathrop, S.A.; MacPherson, M.B.; Beuschel, S.L.; Butnor, K.J.; Testa, J.R.; Pass, H.I.; Carbone, M.; Steele, C.; et al. Inflammation precedes the development of human malignant mesotheliomas in a SCID mouse xenograft model. Ann. N. Y. Acad. Sci. 2010, 1203, 7–14. [Google Scholar] [CrossRef]
- Seiler, C.L.; Song, J.U.M.; Kotandeniya, D.; Chen, J.; Kono, T.J.Y.; Han, Q.; Colwell, M.; Auch, B.; Sarver, A.L.; Upadhyaya, P.; et al. Inhalation exposure to cigarette smoke and inflammatory agents induces epigenetic changes in the lung. Sci. Rep. 2020, 10, 11290. [Google Scholar] [CrossRef]
- Li, R.; Ong, S.L.; Tran, L.M.; Jing, Z.; Liu, B.; Park, S.J.; Huang, Z.L.; Walser, T.C.; Heinrich, E.L.; Lee, G.; et al. Author Correction: Chronic IL-1β-induced inflammation regulates epithelial-to-mesenchymal transition memory phenotypes via epigenetic modifications in non-small cell lung cancer. Sci. Rep. 2020, 10, 4386. [Google Scholar] [CrossRef] [Green Version]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; DeStefano Shields, C.E.; Niknafs, N.; Yen, R.-W.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017, 171, 1284–1300.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, J.Y.; Skrypek, N.; Yang, H.H.; Merchant, A.S.; Nelson, G.W.; Chen, W.-D.; Ishii, H.; Chen, J.M.; Hu, G.; Achyut, B.R.; et al. Induction of DNMT3B by PGE2 and IL6 at Distant Metastatic Sites Promotes Epigenetic Modification and Breast Cancer Colonization. Cancer Res. 2020, 80, 2612–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.C.; Kang, W.; Xu, J.; Qian, Y.; Luk, S.T.Y.; Chen, H.; Li, W.; Zhao, L.; Zhang, X.; Chiu, P.W.; et al. Prostaglandin E2 induces DNA hypermethylation in gastric cancer in vitro and in vivo. Theranostics 2019, 9, 6256–6268. [Google Scholar] [CrossRef]
- Lai, S.-C.; Su, Y.-T.; Chi, C.-C.; Kuo, Y.-C.; Lee, K.-F.; Wu, Y.-C.; Lan, P.-C.; Yang, M.-H.; Chang, T.-S.; Huang, Y.-H. DNMT3b/OCT4 expression confers sorafenib resistance and poor prognosis of hepatocellular carcinoma through IL-6/STAT3 regulation. J. Exp. Clin. Cancer Res. 2019, 38, 474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsunaga, S.; Ikeda, M.; Shimizu, S.; Ohno, I.; Furuse, J.; Inagaki, M.; Higashi, S.; Kato, H.; Terao, K.; Ochiai, A. Serum levels of IL-6 and IL-1β can predict the efficacy of gemcitabine in patients with advanced pancreatic cancer. Br. J. Cancer 2013, 108, 2063–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.I.; Katsura, A.; Matsuyama, H.; Miyazono, K. MicroRNA regulons in tumor microenvironment. Oncogene 2015, 34, 3085–3094. [Google Scholar] [CrossRef] [Green Version]
- Hai Ping, P.; Feng Bo, T.; Li, L.; Nan Hui, Y.; Hong, Z. IL-1β/NF-kb signaling promotes colorectal cancer cell growth through miR-181a/PTEN axis. Arch. Biochem. Biophys. 2016, 604, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Liu, J.; Wang, Z.; Gu, X.; Fan, Y.; Zhang, W.; Xu, L.; Zhang, J.; Cai, D. NF-kappaB-dependent microRNA-425 upregulation promotes gastric cancer cell growth by targeting PTEN upon IL-1β induction. Mol. Cancer 2014, 13, 40. [Google Scholar] [CrossRef] [Green Version]
- Arts, N.; Cané, S.; Hennequart, M.; Lamy, J.; Bommer, G.; Van den Eynde, B.; De Plaen, E. microRNA-155, induced by interleukin-1ß, represses the expression of microphthalmia-associated transcription factor (MITF-M) in melanoma cells. PLoS ONE 2015, 10, e0122517. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.-M.; Zu, Y.-F.; Wang, J.; Bai, S.; Shi, Y.-F.; Shi, R.; Duan, J.; Cui, D.; Chen, J.; Xiang, Y.; et al. Interleukin-1β-mediated suppression of microRNA-101 and upregulation of enhancer of zeste homolog 2 is involved in particle-induced lung cancer. Med. Oncol. 2015, 32, 387. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.-F.; Wu, J.; Xu, S.-J.; Xu, Y.-Y.; Li, D.; Lou, J.-T.; Liu, M.-F. IL-1β-mediated repression of microRNA-101 is crucial for inflammation-promoted lung tumorigenesis. Cancer Res. 2014, 74, 4720–4730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rokavec, M.; Öner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J. Clin. Investig. 2014, 124, 1853–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, B.; Shi, Z.; Wang, J.; Wu, J.; Yang, Z.; Fang, K. IL-6 Inhibits the Targeted Modulation of PDCD4 by miR-21 in Prostate Cancer. PLoS ONE 2015, 10, e0134366. [Google Scholar] [CrossRef] [PubMed]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Savage, P.A.; Leventhal, D.S.; Malchow, S. Shaping the repertoire of tumor-infiltrating effector and regulatory T cells. Immunol. Rev. 2014, 259, 245–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, D.L.; Sjaastad, L.E.; Farrar, M.A. Regulatory T Cell Development in the Thymus. J. Immunol. 2019, 203, 2031–2041. [Google Scholar] [CrossRef]
- Ohkura, N.; Hamaguchi, M.; Morikawa, H.; Sugimura, K.; Tanaka, A.; Ito, Y.; Osaki, M.; Tanaka, Y.; Yamashita, R.; Nakano, N.; et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 2012, 37, 785–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floess, S.; Freyer, J.; Siewert, C.; Baron, U.; Olek, S.; Polansky, J.; Schlawe, K.; Chang, H.-D.; Bopp, T.; Schmitt, E.; et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007, 5, e38. [Google Scholar] [CrossRef]
- Selvaraj, R.K.; Geiger, T.L. A kinetic and dynamic analysis of Foxp3 induced in T cells by TGF-beta. J. Immunol. 2007, 179, 7667–7677. [Google Scholar] [CrossRef] [Green Version]
- Miyao, T.; Floess, S.; Setoguchi, R.; Luche, H.; Fehling, H.J.; Waldmann, H.; Huehn, J.; Hori, S. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity 2012, 36, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Zhang, N.; Yopp, A.C.; Chen, D.; Mao, M.; Chen, D.; Zhang, H.; Ding, Y.; Bromberg, J.S. TGF-beta induces Foxp3 + T-regulatory cells from CD4 + CD25—Precursors. Am. J. Transplant. 2004, 4, 1614–1627. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hong, S.; Li, H.; Park, J.; Hong, B.; Wang, L.; Zheng, Y.; Liu, Z.; Xu, J.; He, J.; et al. Th9 cells promote antitumor immune responses in vivo. J. Clin. Investig. 2012, 122, 4160–4171. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Wu, B.; Yang, T.; Zhang, L.; Jin, K. The outstanding antitumor capacity of CD4+ T helper lymphocytes. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188439. [Google Scholar] [CrossRef]
- Martin-Orozco, N.; Muranski, P.; Chung, Y.; Yang, X.O.; Yamazaki, T.; Lu, S.; Hwu, P.; Restifo, N.P.; Overwijk, W.W.; Dong, C. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity 2009, 31, 787–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9, a022236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seoane, J.; Gomis, R.R. TGF-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-β: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 2014, 106, djt369. [Google Scholar] [CrossRef]
- Scharer, C.D.; Barwick, B.G.; Youngblood, B.A.; Ahmed, R.; Boss, J.M. Global DNA methylation remodeling accompanies CD8 T cell effector function. J. Immunol. 2013, 191, 3419–3429. [Google Scholar] [CrossRef] [Green Version]
- Saleh, R.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Role of Epigenetic Modifications in Inhibitory Immune Checkpoints in Cancer Development and Progression. Front. Immunol. 2020, 11, 1469. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan Nair, V.; El Salhat, H.; Taha, R.Z.; John, A.; Ali, B.R.; Elkord, E. DNA methylation and repressive H3K9 and H3K27 trimethylation in the promoter regions of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, and PD-L1 genes in human primary breast cancer. Clin. Epigenet. 2018, 10, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasidharan Nair, V.; Toor, S.M.; Taha, R.Z.; Shaath, H.; Elkord, E. DNA methylation and repressive histones in the promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and galectin-9 genes in human colorectal cancer. Clin. Epigenet. 2018, 10, 104. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Barozzi, I.; Termanini, A.; Prosperini, E.; Recchiuti, A.; Dalli, J.; Mietton, F.; Matteoli, G.; Hiebert, S.; Natoli, G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E2865–E2874. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wang, X.; Liu, D.; Yu, L.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol. Endocrinol. 2014, 28, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Lee, D.U.; Agarwal, S.; Rao, A. Th2 lineage commitment and efficient IL-4 production involves extended demethylation of the IL-4 gene. Immunity 2002, 16, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Makar, K.W.; Pérez-Melgosa, M.; Shnyreva, M.; Weaver, W.M.; Fitzpatrick, D.R.; Wilson, C.B. Active recruitment of DNA methyltransferases regulates interleukin 4 in thymocytes and T cells. Nat. Immunol. 2003, 4, 1183–1190. [Google Scholar] [CrossRef]
- Valapour, M.; Guo, J.; Schroeder, J.T.; Keen, J.; Cianferoni, A.; Casolaro, V.; Georas, S.N. Histone deacetylation inhibits IL4 gene expression in T cells. J. Allergy Clin. Immunol. 2002, 109, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Young, H.A.; Ghosh, P.; Ye, J.; Lederer, J.; Lichtman, A.; Gerard, J.R.; Penix, L.; Wilson, C.B.; Melvin, A.J.; McGurn, M.E. Differentiation of the T helper phenotypes by analysis of the methylation state of the IFN-gamma gene. J. Immunol. 1994, 153, 3603–3610. [Google Scholar]
- Makar, K.W.; Wilson, C.B. DNA methylation is a nonredundant repressor of the Th2 effector program. J. Immunol. 2004, 173, 4402–4406. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Wang, Z.; Wu, H.; Xue, M.; Lin, P.; Wang, S.; Lin, N.; Huang, X.; Pan, W.; Liu, M.; et al. Epigenetic Regulation of CXCL12 Plays a Critical Role in Mediating Tumor Progression and the Immune Response In Osteosarcoma. Cancer Res. 2018, 78, 3938–3953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurj, A.; Zanoaga, O.; Braicu, C.; Lazar, V.; Tomuleasa, C.; Irimie, A.; Berindan-Neagoe, I. A Comprehensive Picture of Extracellular Vesicles and Their Contents. Molecular Transfer to Cancer Cells. Cancers 2020, 12, 298. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Dean, D.C.; Hornicek, F.J.; Shi, H.; Duan, Z. Exosomes promote pre-metastatic niche formation in ovarian cancer. Mol. Cancer 2019, 18, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Zhou, W.; Zhou, Y.; Chen, X.; Ning, T.; Chen, H.; Guo, Q.; Zhang, Y.; Liu, P.; Zhang, Y.; Li, C.; et al. Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials 2021, 268, 120546. [Google Scholar] [CrossRef]
- Li, I.; Nabet, B.Y. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol. Cancer 2019, 18, 32. [Google Scholar] [CrossRef]
- Rimessi, A.; Patergnani, S.; Ioannidi, E.; Pinton, P. Chemoresistance and Cancer-Related Inflammation: Two Hallmarks of Cancer Connected by an Atypical Link, PKCζ. Front. Oncol. 2013, 3, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretz, N.P.; Ridinger, J.; Rupp, A.-K.; Rimbach, K.; Keller, S.; Rupp, C.; Marmé, F.; Umansky, L.; Umansky, V.; Eigenbrod, T.; et al. Body fluid exosomes promote secretion of inflammatory cytokines in monocytic cells via Toll-like receptor signaling. J. Biol. Chem. 2013, 288, 36691–36702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Orjalo, A.V.; Rodier, F.; Lithgow, G.J.; Campisi, J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging 2009, 1, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Liu, Y.; Zhang, C.; Su, Z.-Y.; Li, W.; Huang, M.-T.; Kong, A.-N. The epigenetic effects of aspirin: The modification of histone H3 lysine 27 acetylation in the prevention of colon carcinogenesis in azoxymethane- and dextran sulfate sodium-treated CF-1 mice. Carcinogenesis 2016, 37, 616–624. [Google Scholar] [CrossRef]
- Son, D.-S.; Wilson, A.J.; Parl, A.K.; Khabele, D. The effects of the histone deacetylase inhibitor romidepsin (FK228) are enhanced by aspirin (ASA) in COX-1 positive ovarian cancer cells through augmentation of p21. Cancer Biol. Ther. 2010, 9, 928–935. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Zhang, X.; Du, R.; Gao, W.; Wang, J.; Bao, Y.; Yang, W.; Luo, N.; Li, J. Ibuprofen mediates histone modification to diminish cancer cell stemness properties via a COX2-dependent manner. Br. J. Cancer 2020, 123, 730–741. [Google Scholar] [CrossRef]
- Mota, M.; Porrini, V.; Parrella, E.; Benarese, M.; Bellucci, A.; Rhein, S.; Schwaninger, M.; Pizzi, M. Neuroprotective epi-drugs quench the inflammatory response and microglial/macrophage activation in a mouse model of permanent brain ischemia. J. Neuroinflamm. 2020, 17, 361. [Google Scholar] [CrossRef]
- Samanta, S.; Zhou, Z.; Rajasingh, S.; Panda, A.; Sampath, V.; Rajasingh, J. DNMT and HDAC inhibitors together abrogate endotoxemia mediated macrophage death by STAT3-JMJD3 signaling. Int. J. Biochem. Cell Biol. 2018, 102, 117–127. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Gallenkamp, D.; Gelato, K.A.; Haendler, B.; Weinmann, H. Bromodomains and their pharmacological inhibitors. ChemMedChem 2014, 9, 438–464. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Bui, M.H.; Lin, X.; Albert, D.H.; Li, L.; Lam, L.T.; Faivre, E.J.; Warder, S.E.; Huang, X.; Wilcox, D.; Donawho, C.K.; et al. Preclinical Characterization of BET Family Bromodomain Inhibitor ABBV-075 Suggests Combination Therapeutic Strategies. Cancer Res. 2017, 77, 2976–2989. [Google Scholar] [CrossRef] [Green Version]
- Klein, K.; Kabala, P.A.; Grabiec, A.M.; Gay, R.E.; Kolling, C.; Lin, L.-L.; Gay, S.; Tak, P.P.; Prinjha, R.K.; Ospelt, C.; et al. The bromodomain protein inhibitor I-BET151 suppresses expression of inflammatory genes and matrix degrading enzymes in rheumatoid arthritis synovial fibroblasts. Ann. Rheum. Dis. 2016, 75, 422–429. [Google Scholar] [CrossRef]
- Ullmann, T.; Luckhardt, S.; Wolf, M.; Parnham, M.J.; Resch, E. High-Throughput Screening for CEBPD-Modulating Compounds in THP-1-Derived Reporter Macrophages Identifies Anti-Inflammatory HDAC and BET Inhibitors. Int. J. Mol. Sci. 2021, 22, 3022. [Google Scholar] [CrossRef]
- Chan, A.O.-O.; Lam, S.-K.; Wong, B.C.-Y.; Wong, W.-M.; Yuen, M.-F.; Yeung, Y.-H.; Hui, W.-M.; Rashid, A.; Kwong, Y.-L. Promoter methylation of E-cadherin gene in gastric mucosa associated with Helicobacter pylori infection and in gastric cancer. Gut 2003, 52, 502–506. [Google Scholar] [CrossRef] [Green Version]
- Maekita, T.; Nakazawa, K.; Mihara, M.; Nakajima, T.; Yanaoka, K.; Iguchi, M.; Arii, K.; Kaneda, A.; Tsukamoto, T.; Tatematsu, M.; et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin. Cancer Res. 2006, 12, 989–995. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vezzani, B.; Carinci, M.; Previati, M.; Giacovazzi, S.; Della Sala, M.; Gafà, R.; Lanza, G.; Wieckowski, M.R.; Pinton, P.; Giorgi, C. Epigenetic Regulation: A Link between Inflammation and Carcinogenesis. Cancers 2022, 14, 1221. https://doi.org/10.3390/cancers14051221
Vezzani B, Carinci M, Previati M, Giacovazzi S, Della Sala M, Gafà R, Lanza G, Wieckowski MR, Pinton P, Giorgi C. Epigenetic Regulation: A Link between Inflammation and Carcinogenesis. Cancers. 2022; 14(5):1221. https://doi.org/10.3390/cancers14051221
Chicago/Turabian StyleVezzani, Bianca, Marianna Carinci, Maurizio Previati, Stefania Giacovazzi, Mario Della Sala, Roberta Gafà, Giovanni Lanza, Mariusz R. Wieckowski, Paolo Pinton, and Carlotta Giorgi. 2022. "Epigenetic Regulation: A Link between Inflammation and Carcinogenesis" Cancers 14, no. 5: 1221. https://doi.org/10.3390/cancers14051221
APA StyleVezzani, B., Carinci, M., Previati, M., Giacovazzi, S., Della Sala, M., Gafà, R., Lanza, G., Wieckowski, M. R., Pinton, P., & Giorgi, C. (2022). Epigenetic Regulation: A Link between Inflammation and Carcinogenesis. Cancers, 14(5), 1221. https://doi.org/10.3390/cancers14051221