
Comprehensive Metabolic Profiling of MYC-Amplified Medulloblastoma Tumors Reveals Key Dependencies on Amino Acid, Tricarboxylic Acid and Hexosamine Pathways

 ,
,
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.1.1. Cell Culture
2.1.2. Animal Studies
2.2. In Vivo Stable Isotope Labeling and Metabolite Extraction and Analyses
2.3. Mass Spectrometry Analysis
2.3.1. Metabolite Analysis
2.3.2. Sample Normalization and Statistical Analysis
2.4. Human RNAseq Data
2.5. Antibodies and Reagents
Western Blots
3. Results
3.1. Orthotopic D425MED and MED211 Xenografts Showed Upregulation of Nucleotide Metabolism, Amino Acid, and Glutathione Synthesis
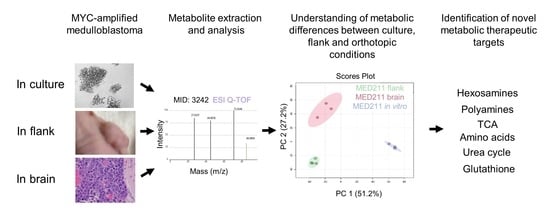
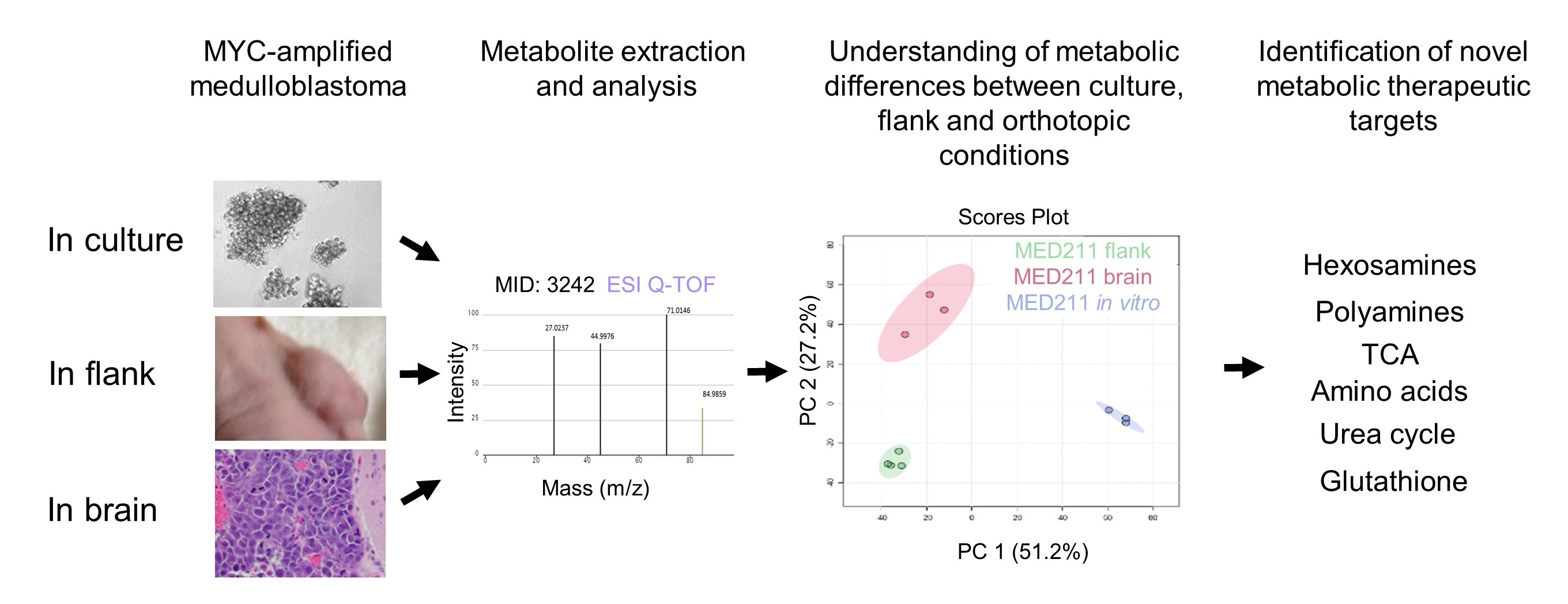
3.2. The Metabolic Profile of MYC-Amplified Medulloblastoma In Vitro Models Was Distinct from In Vivo Flank and Orthotopic Xenograft Tumor Models
3.3. Glucose Is the Main Carbon Source Fueling the TCA Cycle in Normal Brain and Orthotopic D425MED High MYC Medulloblastoma
3.4. Glucose-Derived Glutamate Was Used Differently in the In Vitro vs. In Vivo Setting
3.5. Gluconeogenesis Contributed to the Hexosamine Biosynthetic Pathway and Was Upregulated in Orthotopic D425MED High-MYC Medulloblastoma Tumor Compared to Normal Brain
3.6. The Glutaminase II Pathway Was the Main Pathway Metabolizing Glutamine In Vivo
3.7. Glutamine Derived Glutathione Synthesis Was Mainly through GTK, and It Was Upregulated in Orthotopic D425MED High MYC Xenograft Tumors Compared to Normal Brain
3.8. Overall Model of the Metabolomics of Orthotopic MYC-Amplified Medulloblastoma Reveals Key Dependencies That May Be Therapeutically Targetable
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Land, H.; Parada, L.F.; Weinberg, R.A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983, 304, 596–602. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.D.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Smits, A.M.; Bos, J.L. Genetic Alterations during Colorectal-Tumor Development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stine, Z.E.; Walton, Z.E.; Altman, B.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor Suppression in the Absence of p53-Mediated Cell-Cycle Arrest, Apoptosis, and Senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [Green Version]
- Cantor, J.R.; Sabatini, D.M. Cancer Cell Metabolism: One Hallmark, Many Faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 Is Required for Tumor Initiation and Maintenance and Its Systemic Deletion Is Therapeutic in Mouse Models of Cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavell, L.A.; Gelber, R.D.; Cohen, H.J.; Hitchcock-Bryan, S.; Cassady, J.R.; Tarbell, N.J.; Blattner, S.R.; Tantravahi, R.; Leavitt, P.; Sallan, S.E. Four-Agent Induction and Intensive Asparaginase Therapy for Treatment of Childhood Acute Lymphoblastic Leukemia. N. Engl. J. Med. 1986, 315, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Ochocki, J.D.; Simon, M.C. Nutrient-sensing pathways and metabolic regulation in stem cells. J. Cell Biol. 2013, 203, 23–33. [Google Scholar] [CrossRef]
- Yuan, H.-X.; Xiong, Y.; Guan, K.-L. Nutrient Sensing, Metabolism, and Cell Growth Control. Mol. Cell 2013, 49, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Metallo, C.M.; Heiden, M.G.V. Understanding Metabolic Regulation and Its Influence on Cell Physiology. Mol. Cell 2013, 49, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Ding, Y.; Zhou, M.; Rini, B.I.; Petillo, D.; Qian, C.-N.; Kahnoski, R.; Futreal, P.A.; Furge, K.A.; Teh, B.T. Interleukin-8 Mediates Resistance to Antiangiogenic Agent Sunitinib in Renal Cell Carcinoma. Cancer Res. 2010, 70, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Rini, B.I.; Atkins, M.B. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009, 10, 992–1000. [Google Scholar] [CrossRef]
- Smit, E.F.; de Vries, E.G.E.; Timmer-Bosscha, H.; de Leij, L.F.H.M.; Oosterhuis, J.W.; Scheper, R.J.; Weening, J.J.; Postmus, P.E.; Mulder, N.H. In vitro response of human small-cell lung-cancer cell lines to chemotherapeutic drugs; no correlation with clinical data. Int. J. Cancer 1992, 51, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Gaglio, D.; Valtorta, S.; Ripamonti, M.; Bonanomi, M.; Damiani, C.; Todde, S.; Negri, A.S.; Sanvito, F.; Mastroianni, F.; Di Campli, A.; et al. Divergent in vitro/in vivo responses to drug treatments of highly aggressive NIH-Ras cancer cells: A PET imaging and metabolomics-mass-spectrometry study. Oncotarget 2016, 7, 52017–52031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weil, A.G.; Wang, A.C.; Westwick, H.J.; Ibrahim, G.M.; Ariani, R.T.; Crevier, L.; Perreault, S.; Davidson, T.; Tseng, C.-H.; Fallah, A. Survival in pediatric medulloblastoma: A population-based observational study to improve prognostication. J. Neuro-Oncology 2016, 132, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, C.G.; Kratz, J.; Wang, Y.; Summers, K.; Stearns, D.; Cohen, K.; Dang, C.V.; Burger, P.C. Histopathological and Molecular Prognostic Markers in Medulloblastoma. J. Neuropathol. Exp. Neurol. 2004, 63, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.-J.; Tsherniak, A.; Tamayo, P.; Santagata, S.; Ligon, A.; Greulich, H.; Berhoukim, R.; Amani, V.; Goumnerova, L.; Eberhart, C.G.; et al. Integrative Genomic Analysis of Medulloblastoma Identifies a Molecular Subgroup That Drives Poor Clinical Outcome. J. Clin. Oncol. 2011, 29, 1424–1430. [Google Scholar] [CrossRef]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2011, 123, 465–472. [Google Scholar] [CrossRef] [Green Version]
- He, X.M.; Wikstrand, C.J.; Friedman, H.S.; Bigner, S.H.; Pleasure, S.; Trojanowski, J.Q.; Bigner, D.D. Differentiation characteristics of newly established medulloblastoma cell lines (D384 Med, D425 Med, and D458 Med) and their transplantable xenografts. Lab. Investig. 1991, 64, 833–843. [Google Scholar]
- Hanaford, A.R.; Alt, J.; Rais, R.; Wang, S.Z.; Kaur, H.; Thorek, D.L.; Eberhart, C.G.; Slusher, B.S.; Martin, A.M.; Raabe, E.H. Orally bioavailable glutamine antagonist prodrug JHU-083 penetrates mouse brain and suppresses the growth of MYC-driven medulloblastoma. Transl. Oncol. 2019, 12, 1314–1322. [Google Scholar] [CrossRef]
- Nedelcovych, M.T.; Tenora, L.; Kim, B.-H.; Kelschenbach, J.; Chao, W.; Hadas, E.; Jančařík, A.; Prchalová, E.; Zimmermann, S.C.; Dash, R.P.; et al. N-(Pivaloyloxy)alkoxy-carbonyl Prodrugs of the Glutamine Antagonist 6-Diazo-5-oxo-l-norleucine (DON) as a Potential Treatment for HIV Associated Neurocognitive Disorders. J. Med. Chem. 2017, 60, 7186–7198. [Google Scholar] [CrossRef]
- Brabetz, S.; Leary, S.E.S.; Gröbner, S.N.; Nakamoto, M.W.; Şeker-Cin, H.; Girard, E.; Cole, B.; Strand, A.D.; Bloom, K.L.; Hovestadt, V.; et al. A biobank of patient-derived pediatric brain tumor models. Nat. Med. 2018, 24, 1752–1761. [Google Scholar] [CrossRef]
- Stearns, D.; Chaudhry, A.; Abel, T.W.; Burger, P.C.; Dang, C.V.; Eberhart, C.G. c-myc overexpression causes anaplasia in medul-loblastoma. Cancer Res. 2006, 66, 673–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meister, A.; Tice, S.V. Transamination from glutamine to α-keto acids. J. Biol. Chem. 1950, 187, 173–187. [Google Scholar] [CrossRef]
- Agrawal, S.; Kumar, S.; Sehgal, R.; George, S.; Gupta, R.; Poddar, S.; Jha, A.; Pathak, S. El-MAVEN: A Fast, Robust, and User-Friendly Mass Spectrometry Data Processing Engine for Metabolomics. In Methods in Pharmacology and Toxicology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2019; Volume 1978, pp. 301–321. [Google Scholar]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]
- Mason, S. Lactate Shuttles in Neuroenergetics—Homeostasis, Allostasis and Beyond. Front. Neurosci. 2017, 11, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meldrum, B.S. Glutamate as a Neurotransmitter in the Brain: Review of Physiology and Pathology. J. Nutr. 2000, 130, 1007S–1015S. [Google Scholar] [CrossRef] [Green Version]
- Yelamanchi, S.D.; Jayaram, S.; Thomas, J.K.; Gundimeda, S.; Khan, A.A.; Singhal, A.; Prasad, T.S.K.; Pandey, A.; Somani, B.L.; Gowda, H. A pathway map of glutamate metabolism. J. Cell Commun. Signal. 2016, 10, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, M.; van der Donk, W.A. The many roles of glutamate in metabolism. J. Ind. Microbiol. Biotechnol. 2016, 43, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.J.L.; Jeitner, T.M. Central Role of Glutamate Metabolism in the Maintenance of Nitrogen Homeostasis in Normal and Hyperammonemic Brain. Biomol. 2016, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Maynard, R.E.; Poore, B.; Hanaford, A.R.; Pham, K.; James, M.; Alt, J.; Park, Y.; Slusher, B.S.; Tamayo, P.; Mesirov, J.; et al. TORC1/2 kinase inhibition depletes glutathione and synergizes with carboplatin to suppress the growth of MYC-driven medulloblastoma. Cancer Lett. 2021, 504, 137–145. [Google Scholar] [CrossRef]
- Akella, N.M.; Ciraku, L.; Reginato, M.J. Fueling the fire: Emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 2019, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nat. 2009, 458, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Errera, M.; Greenstein, J.P. Phosphate-activated glutaminase in kidney and other tissues. J. Biol. Chem. 1949, 178, 495–502. [Google Scholar] [CrossRef]
- Meister, A.; Sober, H.A.; Tice, S.V.; Fraser, P.E. Transamination and associated deamidation of asparagine and glutamine. J. Biol. Chem. 1952, 197, 319–330. [Google Scholar] [CrossRef]
- Copper, A.J.L.; Meister, A. Isolation and properties of highly purified glutamine transaminase. Biochemistry 1972, 11, 661–671. [Google Scholar] [CrossRef]
- Cooper, A.J.; Meister, A. Isolation and Properties of a New Glutamine Transaminase from Rat Kidney. J. Biol. Chem. 1974, 249, 2554–2561. [Google Scholar] [CrossRef]
- Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Narayanan, S.P. Arginase: A Multifaceted Enzyme Important in Health and Disease. Physiol. Rev. 2018, 98, 641–665. [Google Scholar] [CrossRef] [Green Version]
- Vardon, A.; Dandapani, M.; Cheng, D.; Cheng, P.; De Santo, C.; Mussai, F. Arginine auxotrophic gene signature in paediatric sarcomas and brain tumours provides a viable target for arginine depletion therapies. Oncotarget 2017, 8, 63506–63517. [Google Scholar] [CrossRef] [PubMed]
- Albaugh, V.L.; Mukherjee, K.; Barbul, A. Proline Precursors and Collagen Synthesis: Biochemical Challenges of Nutrient Supplementation and Wound Healing. J. Nutr. 2017, 147, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; MacKenzie, E.D.; Karim, S.A.; Hedley, A.; Blyth, K.; Kalna, G.; Watson, D.G.; Szlosarek, P.; Frezza, C.; Gottlieb, E. Reversed argininosuccinate lyase activity in fumarate hydratase-deficient cancer cells. Cancer Metab. 2013, 1, 12. [Google Scholar] [CrossRef] [Green Version]
- Towle, H.C. Metabolic Regulation of Gene Transcription in Mammals. J. Biol. Chem. 1995, 270, 23235–23238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Meng, Y.; Wu, X.; Sun, Y. Polyamines and related signaling pathways in cancer. Cancer Cell Int. 2020, 20, 1–16. [Google Scholar] [CrossRef]
- Kp, M.; Kumar, A.; Biswas, D.; Moiyadi, A.; Shetty, P.; Gupta, T.; Epari, S.; Shirsat, N.; Srivastava, S. The proteomic analysis shows enrichment of RNA surveillance pathways in adult SHH and extensive metabolic reprogramming in Group 3 medulloblastomas. Brain Tumor Pathol. 2021, 38, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.-L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of Tumor Metabolism Reveals Mitochondrial Glucose Oxidation in Genetically Diverse Human Glioblastomas in the Mouse Brain In Vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ast, T.; Mootha, V.K. Oxygen and mammalian cell culture: Are we repeating the experiment of Dr. Ox? Nat. Metab. 2019, 1, 858–860. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On Respiratory Impairment in Cancer Cells. Sci. 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Owen, O.E.; Kalhan, S.; Hanson, R.W. The Key Role of Anaplerosis and Cataplerosis for Citric Acid Cycle Function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [Green Version]
- Boukouris, A.E.; Zervopoulos, S.; Michelakis, E.D. Metabolic Enzymes Moonlighting in the Nucleus: Metabolic Regulation of Gene Transcription. Trends Biochem. Sci. 2016, 41, 712–730. [Google Scholar] [CrossRef]
- Dorai, T.; Pinto, J.T.; Denton, T.T.; Krasnikov, B.F.; Cooper, A.J. The metabolic importance of the glutaminase II pathway in normal and cancerous cells. Anal. Biochem. 2020, 114083. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-specific Expression by Genome-wide Integration of Transcriptomics and Antibody-based Proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagamani, S.C.; Erez, A. A metabolic link between the urea cycle and cancer cell proliferation. Mol. Cell. Oncol. 2016, 3, e1127314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, E.A.; LaFleur, B.; Gerner, E.W. Polyamines are oncometabolites that regulate the LIN28/let-7 pathway in colorectal cancer cells. Mol. Carcinog. 2014, 53, E96–E106. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.; Taurino, G.; Bianchi, M.G.; Kilberg, M.S.; Bussolati, O. Asparagine Synthetase in Cancer: Beyond Acute Lymphoblastic Leukemia. Front. Oncol. 2019, 9, 1480. [Google Scholar] [CrossRef]
- Dominguez, M.; Brüne, B.; Namgaladze, D. Exploring the Role of ATP-Citrate Lyase in the Immune System. Front. Immunol. 2021, 12, 632526. [Google Scholar] [CrossRef]
- Gamble, L.D.; Purgato, S.; Murray, J.; Xiao, L.; Yu, D.M.T.; Hanssen, K.M.; Giorgi, F.M.; Carter, D.R.; Gifford, A.J.; Valli, E.; et al. Inhibition of polyamine synthesis and uptake reduces tumor progression and prolongs survival in mouse models of neuroblastoma. Sci. Transl. Med. 2019, 11, eaau1099. [Google Scholar] [CrossRef] [Green Version]
- Evageliou, N.F.; Haber, M.; Vu, A.; Laetsch, T.W.; Murray, J.; Gamble, L.; Cheng, N.C.; Liu, K.; Reese, M.; Corrigan, K.A.; et al. Polyamine Antagonist Therapies Inhibit Neuroblastoma Initiation and Progression. Clin. Cancer Res. 2016, 22, 4391–4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogarty, M.D.; Norris, M.D.; Davis, K.; Liu, X.; Evageliou, N.F.; Hayes, C.S.; Pawel, B.; Guo, R.; Zhao, H.; Sekyere, E.; et al. ODC1Is a Critical Determinant ofMYCNOncogenesis and a Therapeutic Target in Neuroblastoma. Cancer Res. 2008, 68, 9735–9745. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Gamble, L.D.; Upton, D.H.; Ung, C.; Yu, D.M.T.; Ehteda, A.; Pandher, R.; Mayoh, C.; Hébert, S.; Jabado, N.; et al. Dual targeting of polyamine synthesis and uptake in diffuse intrinsic pontine gliomas. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- D’Amico, D.; Antonucci, L.; Di Magno, L.; Coni, S.; Sdruscia, G.; Macone, A.; Miele, E.; Infante, P.; Di Marcotullio, L.; De Smaele, E.; et al. Non-canonical Hedgehog/AMPK-Mediated Control of Polyamine Metabolism Supports Neuronal and Medulloblastoma Cell Growth. Dev. Cell 2015, 35, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Delage, B.; Fennell, D.A.; Nicholson, L.; McNeish, I.; Lemoine, N.R.; Crook, T.; Szlosarek, P.W. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int. J. Cancer 2010, 126, 2762–2772. [Google Scholar] [CrossRef] [PubMed]
- Helenius, I.T.; Madala, H.R.; Yeh, J.-R.J. An Asp to Strike Out Cancer? Therapeutic Possibilities Arising from Aspartate’s Emerging Roles in Cell Proliferation and Survival. Biomolecules 2021, 11, 1666. [Google Scholar] [CrossRef] [PubMed]
- Johnston, K.; Pachnis, P.; Tasdogan, A.; Faubert, B.; Zacharias, L.G.; Vu, H.S.; Rodgers-Augustyniak, L.; Johnson, A.; Huang, F.; Ricciardio, S.; et al. Isotope tracing reveals glycolysis and oxidative metabolism in childhood tumors of multiple histologies. Med 2021, 2, 395–410.e4. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, K.; Hanaford, A.R.; Poore, B.A.; Maxwell, M.J.; Sweeney, H.; Parthasarathy, A.; Alt, J.; Rais, R.; Slusher, B.S.; Eberhart, C.G.; et al. Comprehensive Metabolic Profiling of MYC-Amplified Medulloblastoma Tumors Reveals Key Dependencies on Amino Acid, Tricarboxylic Acid and Hexosamine Pathways. Cancers 2022, 14, 1311. https://doi.org/10.3390/cancers14051311
Pham K, Hanaford AR, Poore BA, Maxwell MJ, Sweeney H, Parthasarathy A, Alt J, Rais R, Slusher BS, Eberhart CG, et al. Comprehensive Metabolic Profiling of MYC-Amplified Medulloblastoma Tumors Reveals Key Dependencies on Amino Acid, Tricarboxylic Acid and Hexosamine Pathways. Cancers. 2022; 14(5):1311. https://doi.org/10.3390/cancers14051311
Chicago/Turabian StylePham, Khoa, Allison R. Hanaford, Brad A. Poore, Micah J. Maxwell, Heather Sweeney, Akhila Parthasarathy, Jesse Alt, Rana Rais, Barbara S. Slusher, Charles G. Eberhart, and et al. 2022. "Comprehensive Metabolic Profiling of MYC-Amplified Medulloblastoma Tumors Reveals Key Dependencies on Amino Acid, Tricarboxylic Acid and Hexosamine Pathways" Cancers 14, no. 5: 1311. https://doi.org/10.3390/cancers14051311
APA StylePham, K., Hanaford, A. R., Poore, B. A., Maxwell, M. J., Sweeney, H., Parthasarathy, A., Alt, J., Rais, R., Slusher, B. S., Eberhart, C. G., & Raabe, E. H. (2022). Comprehensive Metabolic Profiling of MYC-Amplified Medulloblastoma Tumors Reveals Key Dependencies on Amino Acid, Tricarboxylic Acid and Hexosamine Pathways. Cancers, 14(5), 1311. https://doi.org/10.3390/cancers14051311