
Effects of Metformin as Add-On Therapy against Glioblastoma: An Old Medicine for Novel Oncology Therapeutics

 , ,
, ,  , , , ,
, , , ,  and
and
Abstract
:Simple Summary
Abstract
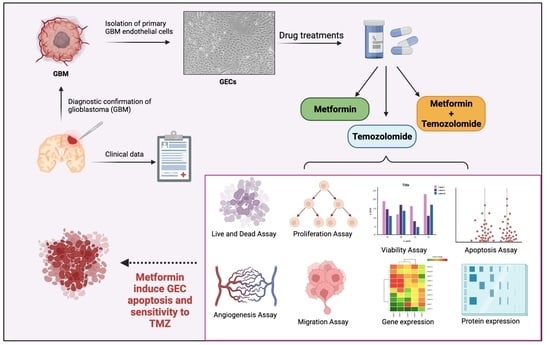
1. Introduction
2. Materials and Methods
2.1. Tumor Samples Processing and GEC Isolation
2.2. Pharmacological Treatment
2.3. MTT Assay
2.4. Estimation of Proliferation Rate
2.5. Live and Dead Assay
2.6. Annexin V Apoptosis
2.7. Caspase 3/7 Activity
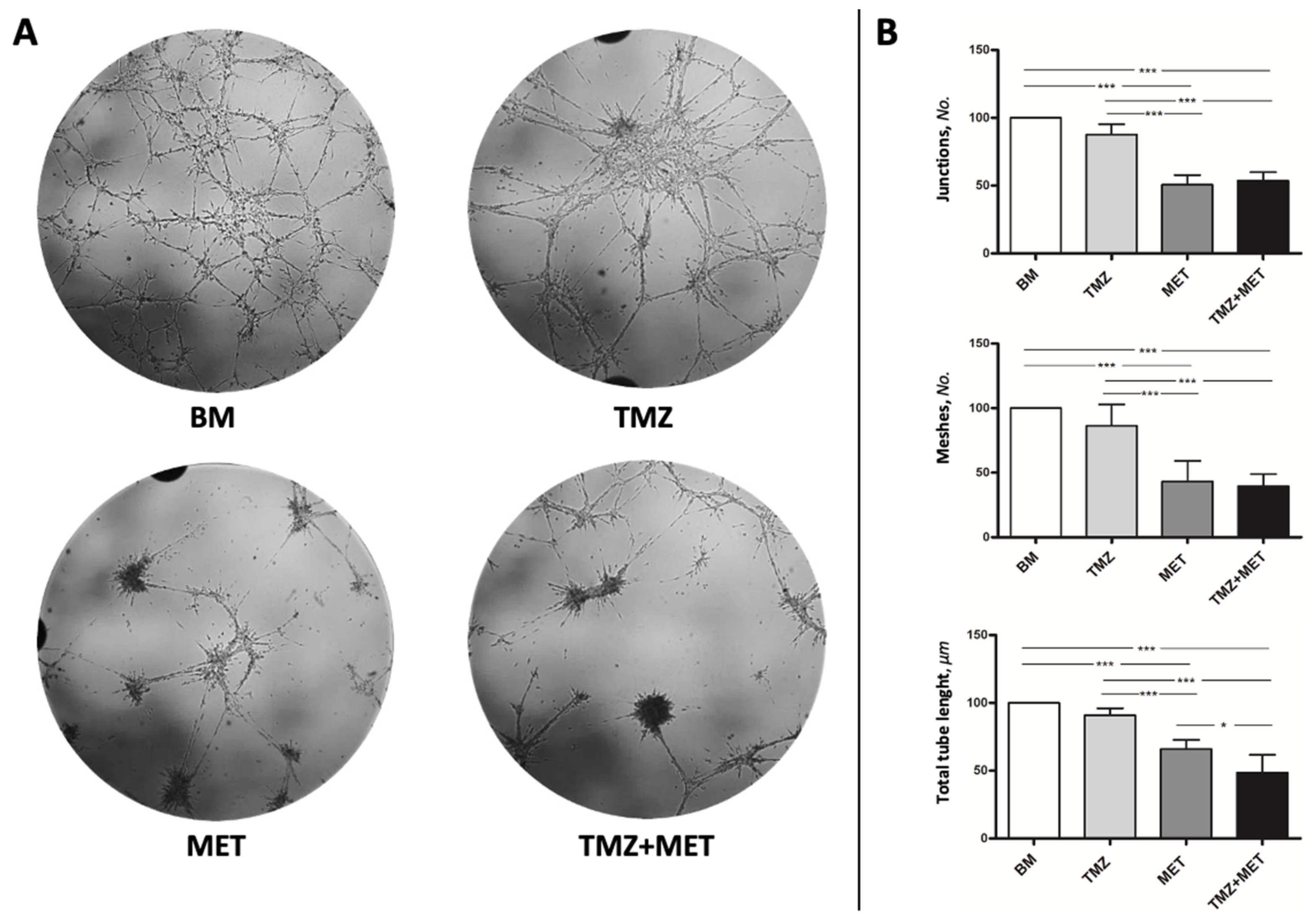
2.8. Tube-like Structure Formation Assay
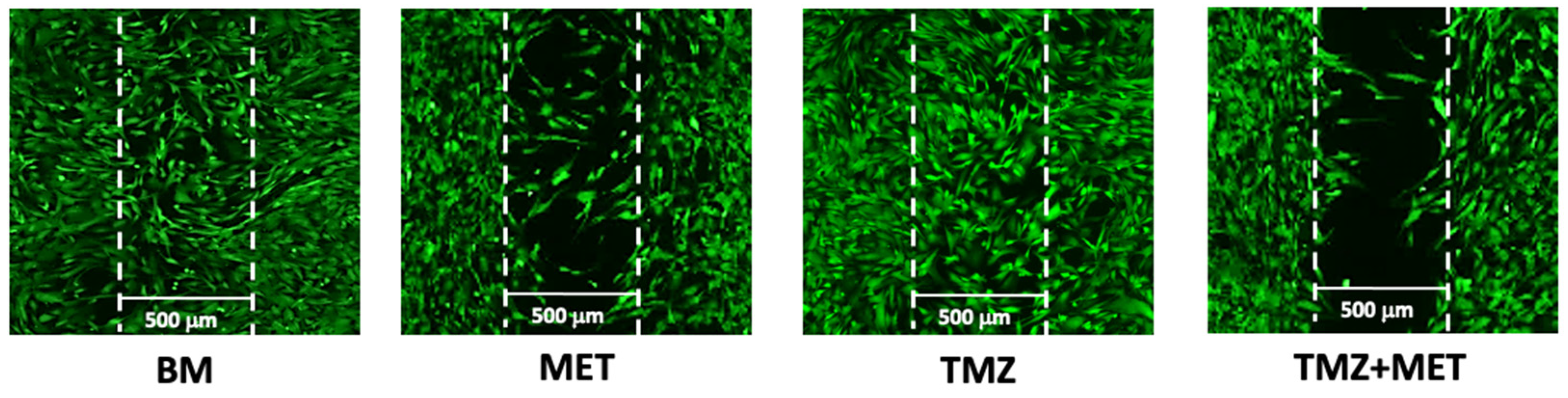
2.9. Migration Assay
2.10. Quantitative Real-Time PCR Analysis (qRT-PCR)
2.11. Western Blot Analyses
2.12. Statistical Analyses
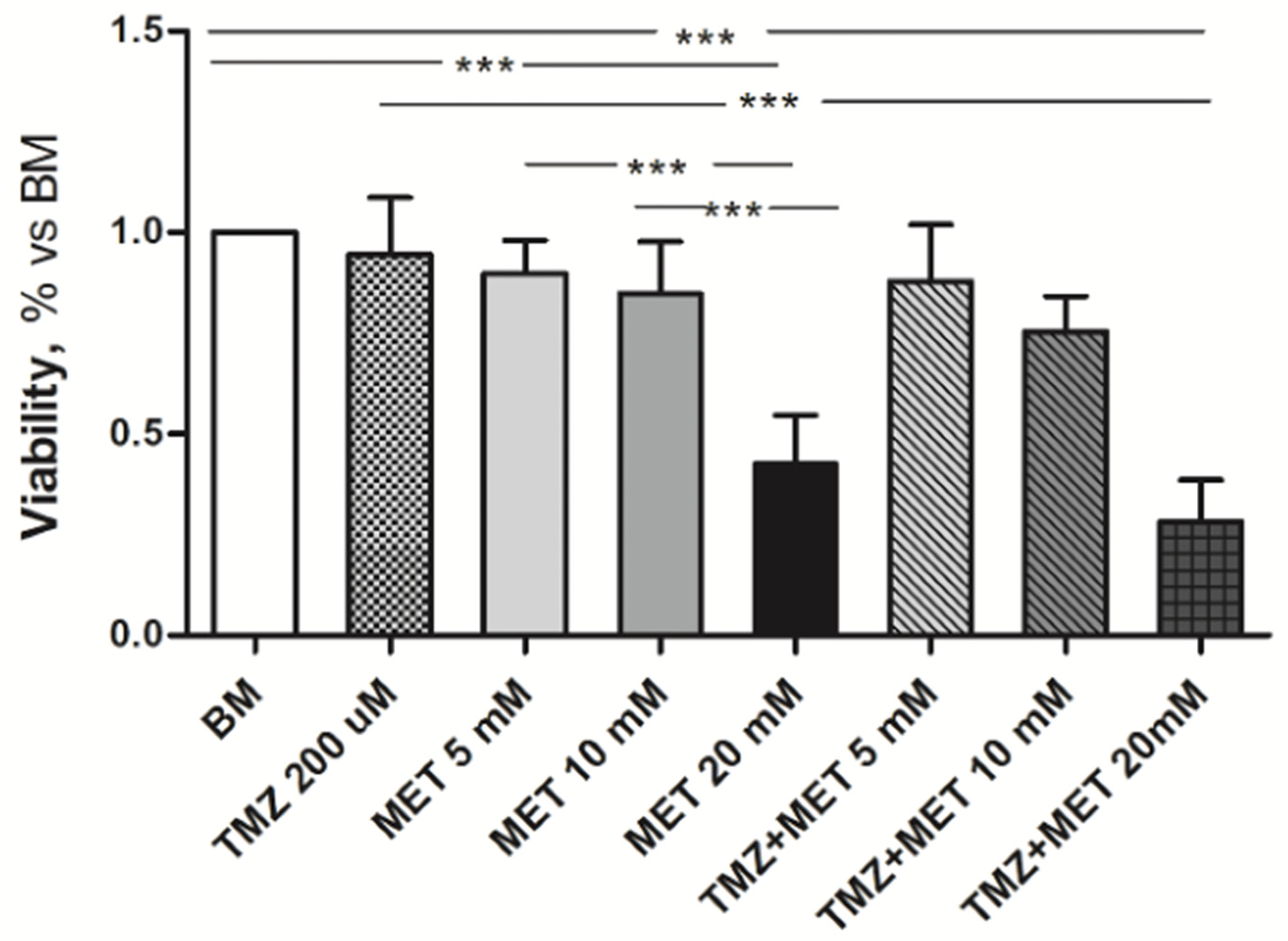
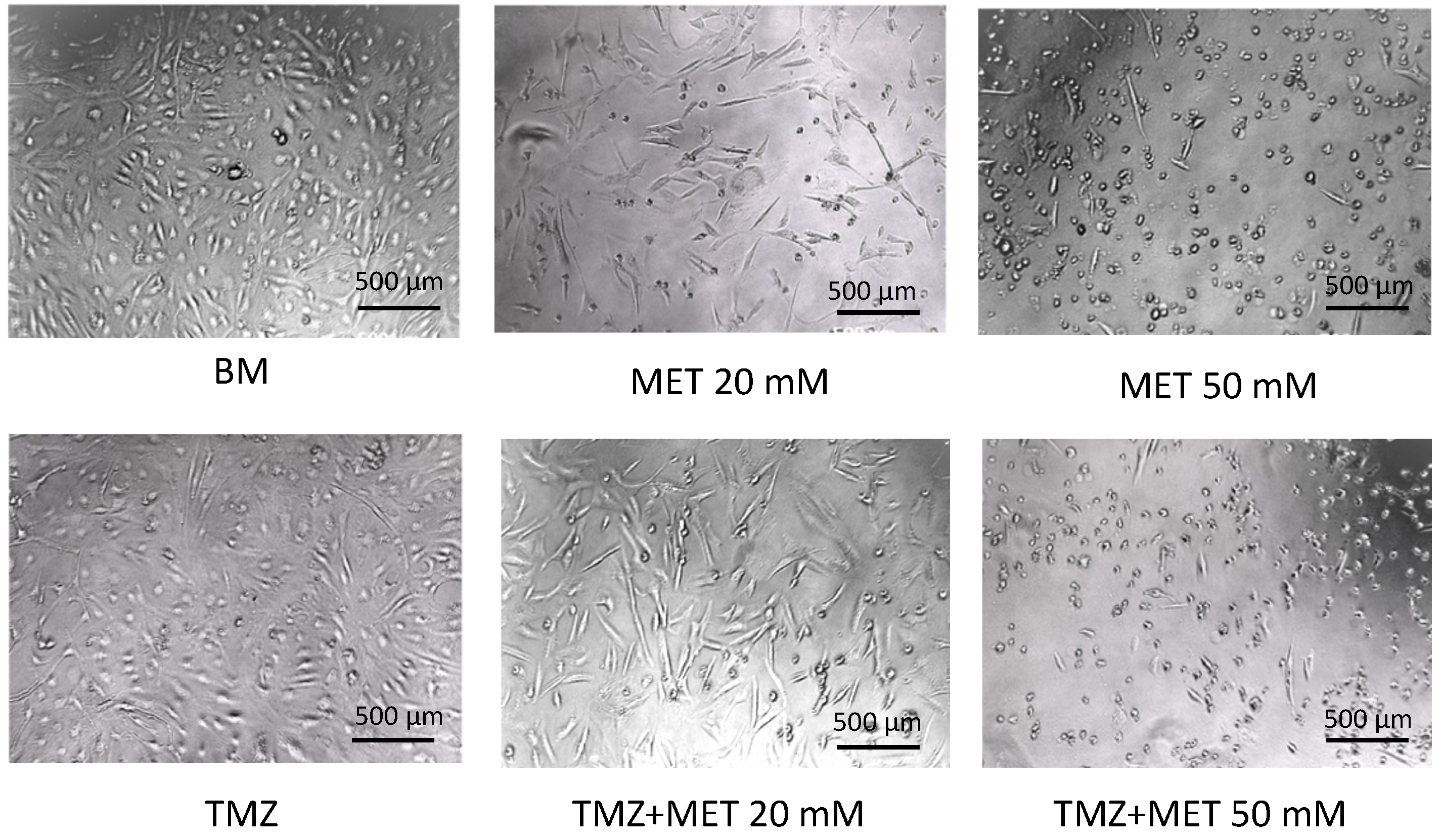
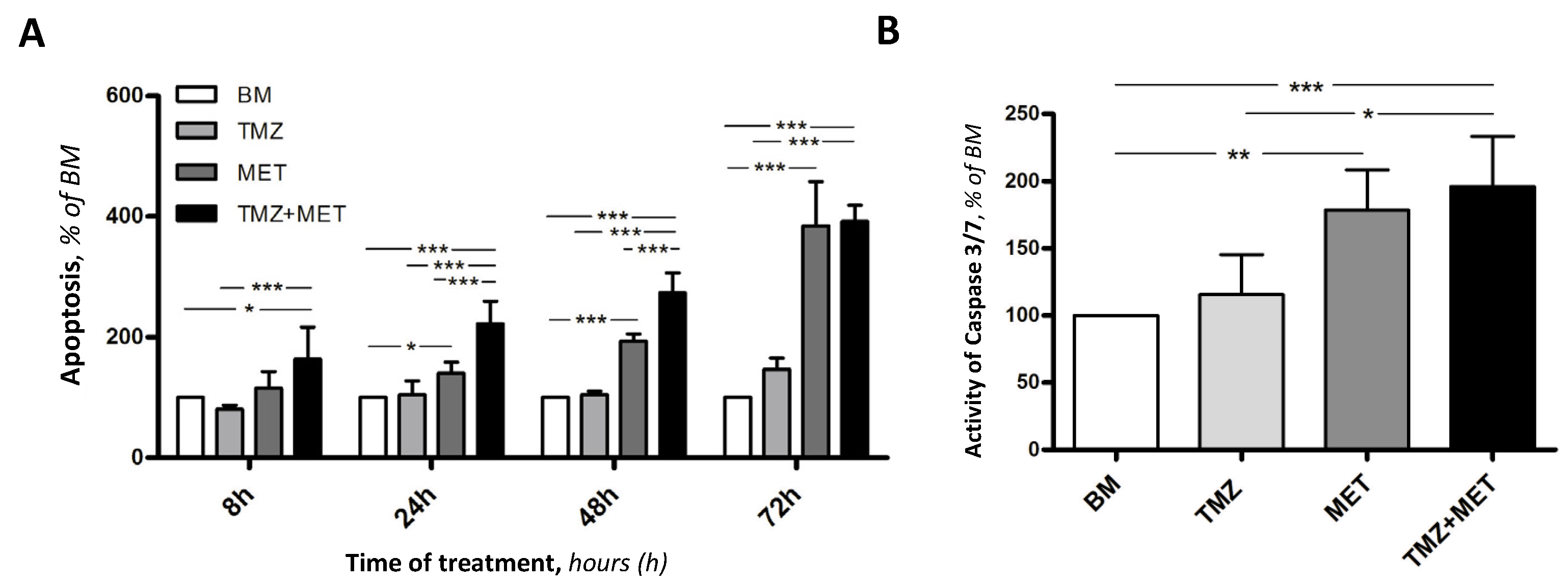
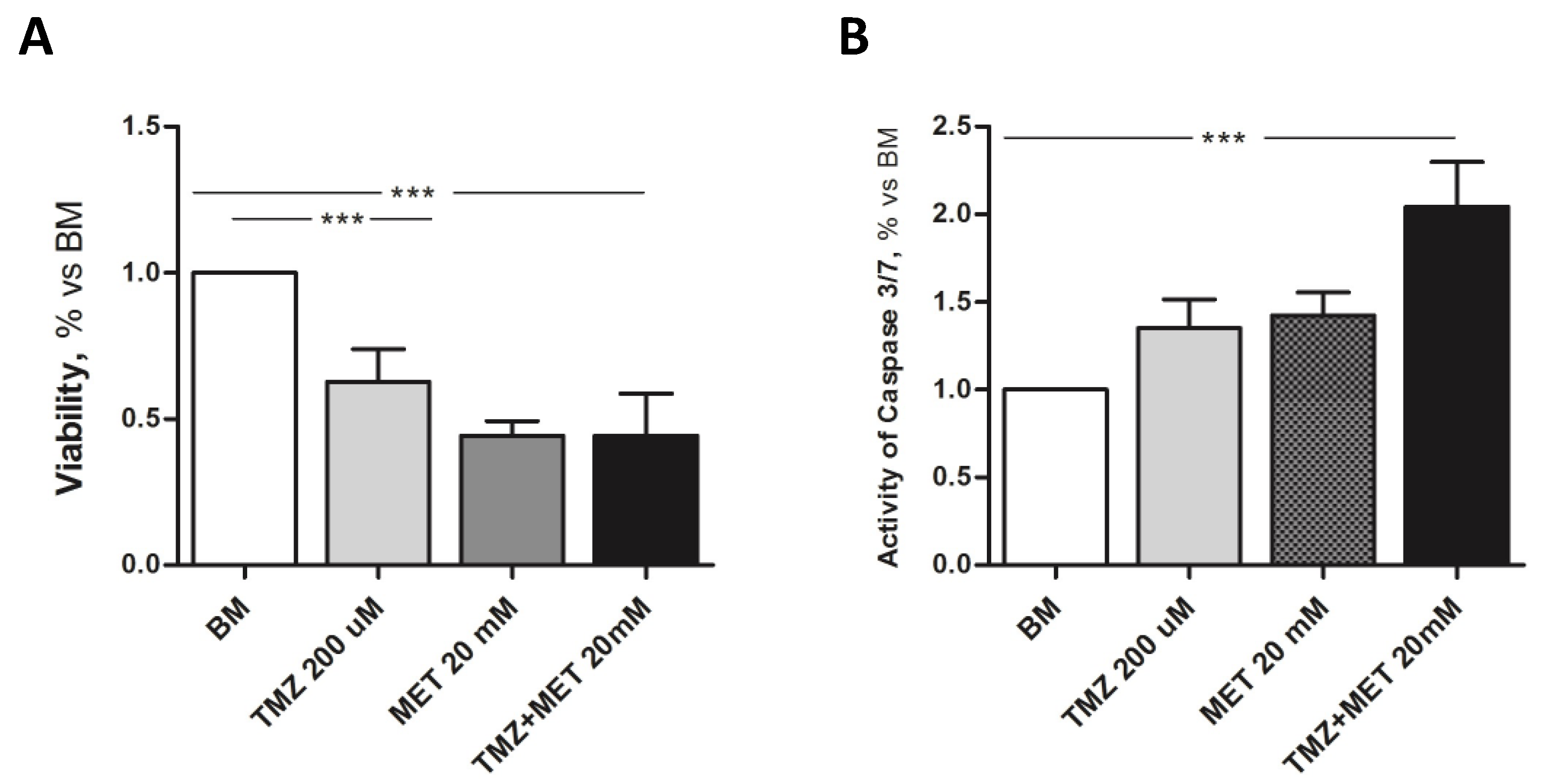
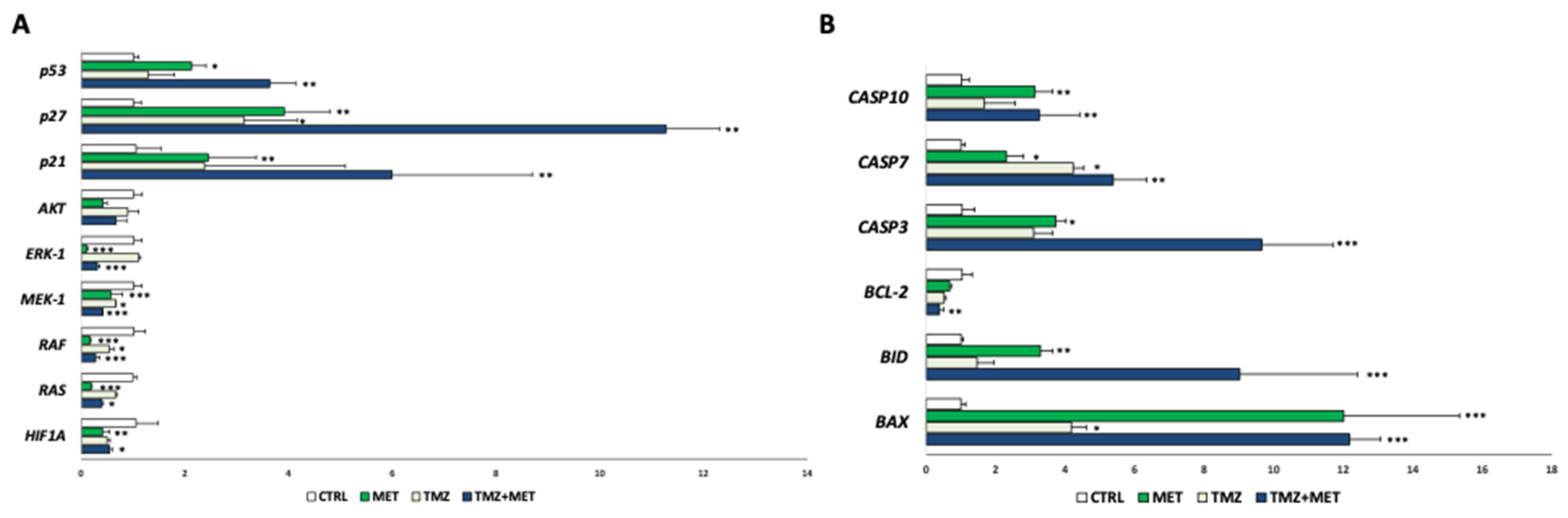
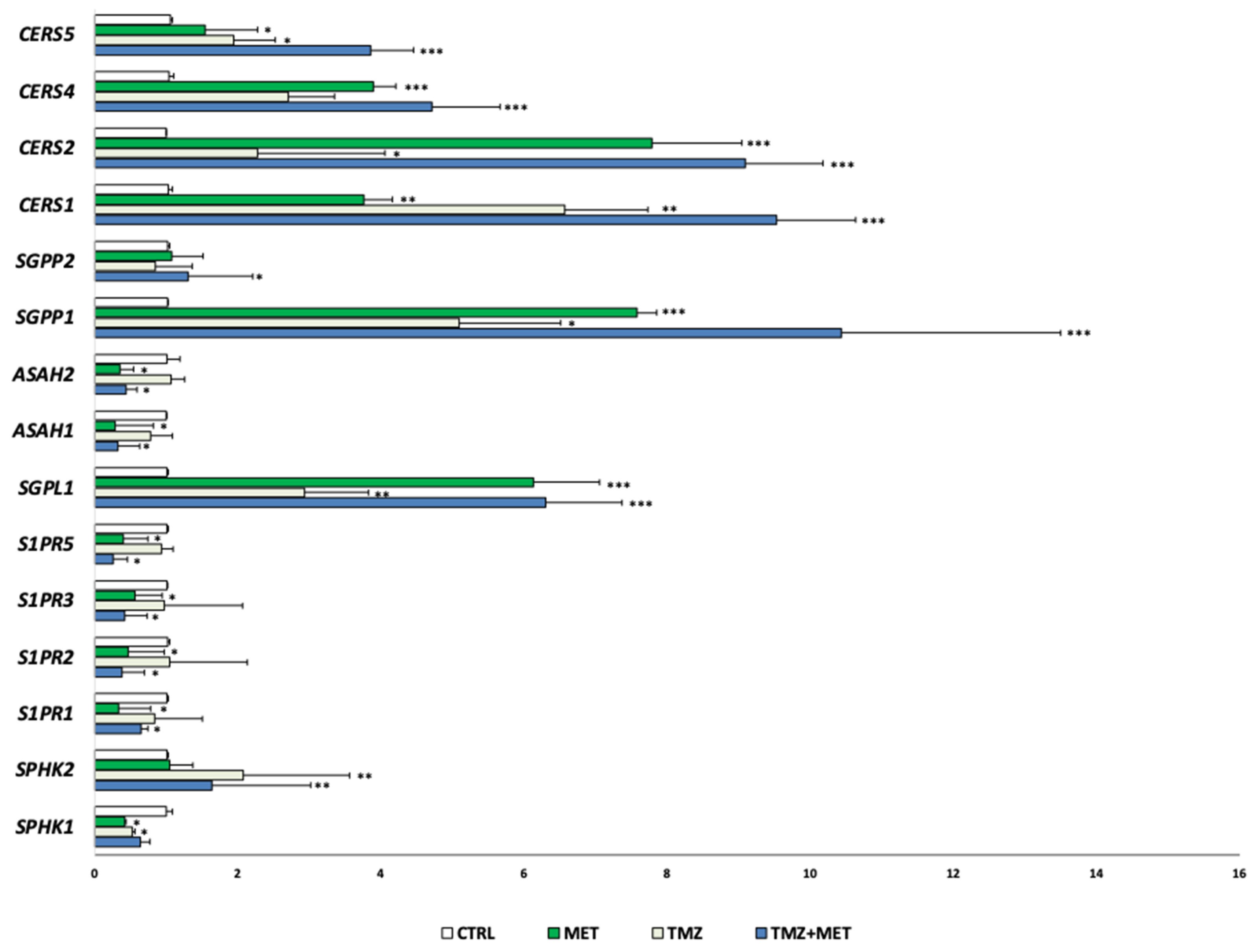
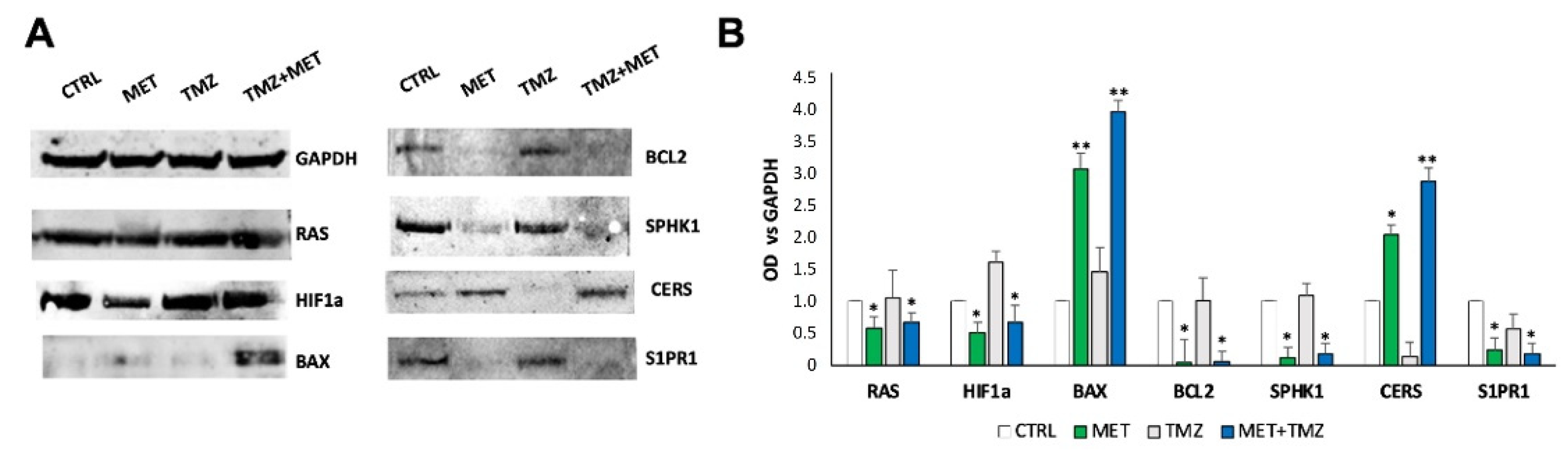
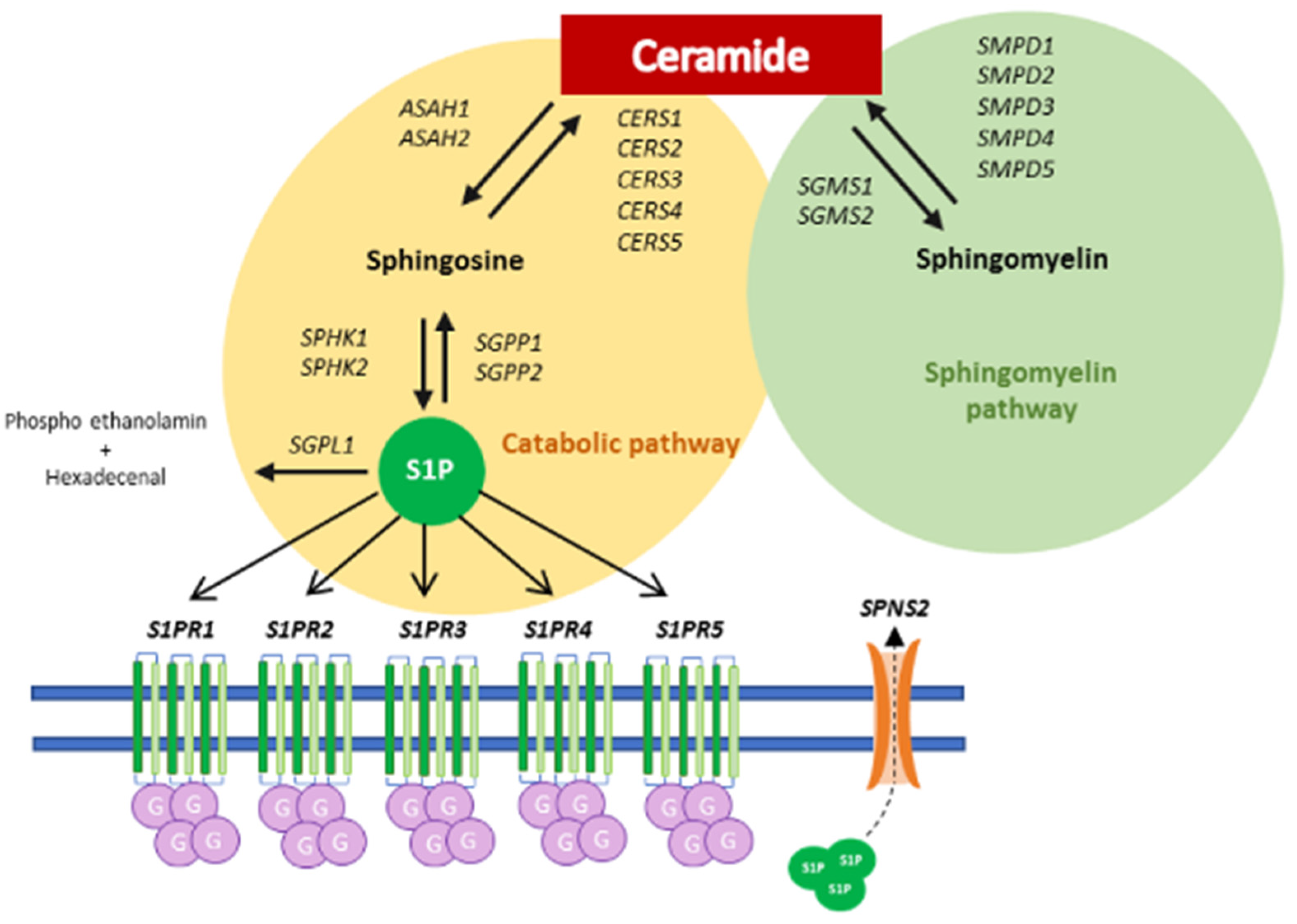
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro. Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Soomro, S.H.; Ting, L.R.; Qing, Y.Y.; Ren, M. Molecular biology of glioblastoma: Classification and mutational locations. J. Pak. Med. Assoc. 2017, 67, 1410–1414. [Google Scholar] [PubMed]
- Bastien, J.I.L.; McNeill, K.A.; Fine, H.A. Molecular characterizations of glioblastoma, targeted therapy, and clinical results to date. Cancer 2015, 121, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro. Oncol. 2015, 17 (Suppl. S4), iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Prim. 2015, 1, 15017. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gilbert, M.R.; Chakravarti, A. Chemoradiotherapy in malignant glioma: Standard of care and future directions. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 4127–4136. [Google Scholar] [CrossRef] [PubMed]
- Guarnaccia, L.; Navone, S.E.; Trombetta, E.; Cordiglieri, C.; Cherubini, A.; Crisà, F.M.; Rampini, P.; Miozzo, M.; Fontana, L.; Caroli, M.; et al. Angiogenesis in human brain tumors: Screening of drug response through a patient-specific cell platform for personalized therapy. Sci. Rep. 2018, 8, 8748. [Google Scholar] [CrossRef] [PubMed]
- Navone, S.E.; Guarnaccia, L.; Cordiglieri, C.; Crisà, F.M.; Caroli, M.; Locatelli, M.; Schisano, L.; Rampini, P.; Miozzo, M.; La Verde, N.; et al. Aspirin Affects Tumor Angiogenesis and Sensitizes Human Glioblastoma Endothelial Cells to Temozolomide, Bevacizumab, and Sunitinib, Impairing Vascular Endothelial Growth Factor-Related Signaling. World Neurosurg. 2018, 120, e380–e391. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.R.; Grommes, C. Retrospective analysis of the effects of steroid therapy and antidiabetic medication on survival in diabetic glioblastoma patients. CNS Oncol. 2013, 2, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Orgel, E.; Mittelman, S. The links between insulin resistance, diabetes, and cancer. Curr. Diab. Rep. 2013, 13, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelgadir, E.; Ali, R.; Rashid, F.; Bashier, A. Effect of Metformin on Different Non-Diabetes Related Conditions, a Special Focus on Malignant Conditions: Review of Literature. J. Clin. Med. Res. 2017, 9, 388–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, D.M.; Buse, J.B.; Davidson, M.B.; Ferrannini, E.; Holman, R.R.; Sherwin, R.; Zinman, B. Medical management of hyperglycemia in type 2 diabetes: A consensus algorithm for the initiation and adjustment of therapy. Diabetes Care 2009, 32, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The beneficial effects of metformin on cancer prevention and therapy: A comprehensive review of recent advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazurek, M.; Litak, J.; Kamieniak, P.; Kulesza, B.; Jonak, K.; Baj, J.; Grochowski, C. Metformin as Potential Therapy for High-Grade Glioma. Cancers 2020, 12, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattan, R.; Giri, S.; Hartmann, L.C.; Shridhar, V. Metformin attenuates ovarian cancer cell growth in an AMP-kinase dispensable manner. J. Cell. Mol. Med. 2011, 15, 166–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sesen, J.; Dahan, P.; Scotland, S.J.; Saland, E.; Dang, V.T.; Lemarié, A.; Tyler, B.M.; Brem, H.; Toulas, C.; Moyal, E.C.J.; et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PLoS ONE 2015, 10, e0123721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Zhao, G.; Li, P.; Li, Y.; Zhou, G.; Chen, Y.; Xie, G. Temozolomide in combination with metformin act synergistically to inhibit proliferation and expansion of glioma stem-like cells. Oncol. Lett. 2016, 11, 2792–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Dico, A.; Valtorta, S.; Ottobrini, L.; Moresco, R.M. Role of metformin and AKT axis modulation in the reversion of hypoxia induced TMZ-resistance in glioma cells. Front. Oncol. 2019, 9, 463. [Google Scholar] [CrossRef] [PubMed]
- Pradas, I.; Rovira-Llopis, S.; Naudí, A.; Bañuls, C.; Rocha, M.; Hernandez-Mijares, A.; Pamplona, R.; Victor, V.M.; Jové, M. Metformin induces lipid changes on sphingolipid species and oxidized lipids in polycystic ovary syndrome women. Sci. Rep. 2019, 9, 16033. [Google Scholar] [CrossRef] [PubMed]
- Campanella, R.; Guarnaccia, L.; Cordiglieri, C.; Trombetta, E.; Caroli, M.; Carrabba, G.; La Verde, N.; Rampini, P.; Gaudino, C.; Costa, A.; et al. Tumor-Educated Platelets and Angiogenesis in Glioblastoma: Another Brick in the Wall for Novel Prognostic and Targetable Biomarkers, Changing the Vision from a Localized Tumor to a Systemic Pathology. Cells 2020, 9, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marfia, G.; Campanella, R.; Navone, S.E.; Di Vito, C.; Riccitelli, E.; Hadi, L.A.; Bornati, A.; de Rezende, G.; Giussani, P.; Tringali, C.; et al. Autocrine/paracrine sphingosine-1-phosphate fuels proliferative and stemness qualities of glioblastoma stem cells. Glia 2014, 62, 1968–1981. [Google Scholar] [CrossRef] [PubMed]
- Di Vito, C.; Hadi, L.A.; Navone, S.E.; Marfia, G.; Campanella, R.; Mancuso, M.E.; Riboni, L. Platelet-derived sphingosine-1-phosphate and inflammation: From basic mechanisms to clinical implications. Platelets 2016, 27, 393–401. [Google Scholar] [CrossRef]
- Riccitelli, E.; Giussani, P.; Di Vito, C.; Condomitti, G.; Tringali, C.; Caroli, M.; Galli, R.; Viani, P.; Riboni, L. Extracellular sphingosine-1-phosphate: A novel actor in human glioblastoma stem cell survival. PLoS ONE 2013, 8, e68229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel Hadi, L.; Anelli, V.; Guarnaccia, L.; Navone, S.; Beretta, M.; Moccia, F.; Tringali, C.; Urechie, V.; Campanella, R.; Marfia, G.; et al. A bidirectional crosstalk between glioblastoma and brain endothelial cells potentiates the angiogenic and proliferative signaling of sphingosine-1-phosphate in the glioblastoma microenvironment. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1179–1192. [Google Scholar] [CrossRef]
- Lapointe, S.; Perry, A.; Butowski, N. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Fontana, L.; Tabano, S.; Bonaparte, E.; Marfia, G.; Pesenti, C.; Falcone, R.; Augello, C.; Carlessi, N.; Silipigni, R.; Guerneri, S.; et al. MGMT-Methylated Alleles Are Distributed Heterogeneously Within Glioma Samples Irrespective of IDH Status and Chromosome 10q Deletion. J. Neuropathol. Exp. Neurol. 2016, 75, 791–800. [Google Scholar] [CrossRef] [Green Version]
- Brentnall, M.; Rodriguez-Menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Ghatak, S.B.; Dhamecha, P.S.; Bhadada, S.V.; Panchal, S.J. Investigation of the potential effects of metformin on atherothrombotic risk factors in hyperlipidemic rats. Eur. J. Pharmacol. 2011, 659, 213–223. [Google Scholar] [CrossRef]
- Mazza, A.; Fruci, B.; Garinis, G.A.; Giuliano, S.; Malaguarnera, R.; Belfiore, A. The role of metformin in the management of NAFLD. Exp. Diabetes Res. 2012, 2012, 716404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gritti, M.; Würth, R.; Angelini, M.; Barbieri, F.; Peretti, M.; Pizzi, E.; Pattarozzi, A.; Carra, E.; Sirito, R.; Daga, A.; et al. Metformin repositioning as antitumoral agent: Selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget 2014, 5, 11252–11268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, S.-M.; Hay, N. The dark face of AMPK as an essential tumor promoter. Cell. Logist. 2012, 2, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-Initiating Cell Elimination by Metformin Activation of FOXO3 via AMPK. Stem Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Würth, R.; Pattarozzi, A.; Gatti, M.; Bajetto, A.; Corsaro, A.; Parodi, A.; Sirito, R.; Massollo, M.; Marini, C.; Zona, G.; et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle 2013, 12, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarnaccia, L.; Marfia, G.; Masseroli, M.M.; Navone, S.E.; Balsamo, M.; Caroli, M.; Valtorta, S.; Moresco, R.M.; Campanella, R.; Garzia, E.; et al. Frontiers in Anti-Cancer Drug Discovery: Challenges and Perspectives of Metformin as Anti-Angiogenic Add-On Therapy in Glioblastoma. Cancers 2021, 14, 112. [Google Scholar] [CrossRef] [PubMed]
- Valtorta, S.; Lo Dico, A.; Raccagni, I.; Gaglio, D.; Belloli, S.; Politi, L.S.; Martelli, C.; Diceglie, C.; Bonanomi, M.; Ercoli, G.; et al. Metformin and temozolomide, a synergic option to overcome resistance in glioblastoma multiforme models. Oncotarget 2017, 8, 113090–113104. [Google Scholar] [CrossRef] [Green Version]
- Wojnowski, L.; Stancato, L.F.; Larner, A.C.; Rapp, U.R.; Zimmer, A. Overlapping and specific functions of Braf and Craf-1 proto-oncogenes during mouse embryogenesis. Mech. Dev. 2000, 91, 97–104. [Google Scholar] [CrossRef]
- Baumann, B.; Weber, C.K.; Troppmair, J.; Whiteside, S.; Israel, A.; Rapp, U.R.; Wirth, T. Raf induces NF-kappaB by membrane shuttle kinase MEKK1, a signaling pathway critical for transformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4615–4620. [Google Scholar] [CrossRef] [Green Version]
- Colman, H.; Zhang, L.; Sulman, E.P.; McDonald, J.M.; Shooshtari, N.L.; Rivera, A.; Popoff, S.; Nutt, C.L.; Louis, D.N.; Cairncross, J.G.; et al. A multigene predictor of outcome in glioblastoma. Neuro Oncol. 2010, 12, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Ekerot, M.; Stavridis, M.P.; Delavaine, L.; Mitchell, M.P.; Staples, C.; Owens, D.M.; Keenan, I.D.; Dickinson, R.J.; Storey, K.G.; Keyse, S.M. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem. J. 2008, 412, 287–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vališ, K.; Novák, P. Targeting ERK-Hippo interplay in cancer therapy. Int. J. Mol. Sci. 2020, 21, 3236. [Google Scholar] [CrossRef] [PubMed]
- Unni, A.M.; Harbourne, B.; Oh, M.H.; Wild, S.; Ferrarone, J.R.; Lockwood, W.W.; Varmus, H. Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. Elife 2018, 7, e33718. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmer, J.C.; Salvesen, G.S. Caspase substrates. Cell Death Differ. 2007, 14, 66–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutelle, A.M.; Attardi, L.D. p53 and Tumor Suppression: It Takes a Network. Trends Cell Biol. 2021, 31, 298–310. [Google Scholar] [CrossRef] [PubMed]
- CChu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; He, M.; Wang, Y.; Zhao, X.; He, Y.; Shi, B. Sphingolipid metabolism in type 2 diabetes and associated cardiovascular complications. Exp. Ther. Med. 2019, 18, 3603–3614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riboni, L.; Hadi, L.A.; Navone, S.E.; Guarnaccia, L.; Campanella, R.; Marfia, G. Sphingosine-1-Phosphate in the Tumor Microenvironment: A Signaling Hub Regulating Cancer Hallmarks. Cells 2020, 9, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadi, L.A.; Di Vito, C.; Marfia, G.; Navone, S.E.; Campanella, R.; Riboni, L. The role and function of sphingolipids in glioblastoma multiforme. In Bioactive Sphingolipids in Cancer Biology and Therapy; Springer International Publisher: Berlin/Heidelberg, Germany, 2015; pp. 259–293. [Google Scholar]
- Liu, X.; Zhang, Q.-H.; Yi, G.-H. Regulation of metabolism and transport of sphingosine-1-phosphate in mammalian cells. Mol. Cell. Biochem. 2012, 363, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Urtz, N.; Gaertner, F.; Legate, K.R.; Petzold, T.; Lorenz, M.; Mazharian, A.; Watson, S.P.; Massberg, S. Sphingosine kinase 2 (Sphk2) regulates platelet biogenesis by providing intracellular sphingosine 1-phosphate (S1P). Blood 2013, 122, 791–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, M.G.; Wesseling, P.; Wurdinger, T. Tumor-Educated Platelets as a Noninvasive Biomarker Source for Cancer Detection and Progression Monitoring. Cancer Res. 2018, 78, 3407–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, P.C.; Chiyoda, T.; Liu, X.; Weigert, M.; Curtis, M.; Chiang, C.-Y.; Loth, R.; Lastra, R.; McGregor, S.M.; Locasale, J.W.; et al. SPHK1 Is a Novel Target of Metformin in Ovarian Cancer. Mol. Cancer Res. 2019, 17, 870–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenouda, G.; Souhami, L.; Petrecca, K.; Panet-Raymond, V.; Owen, S.; Diaz, R.; Hall, J.; Guiot, M.; Carvalho, T.; Abdulkarim, B. A Phase 2 Study of Neo-adjuvant Metformin and Temozolomide followed by Hypofractionated Accelerated RadioTherapy (HART) with Concomitant and Adjuvant Metformin and Temozolomide (TMZ) in Patients with Glioblastoma. Int. J. Radiat. Oncol. 2020, 108, S21. [Google Scholar] [CrossRef]
- Seliger, C.; Genbrugge, E.; Gorlia, T.; Chinot, O.; Stupp, R.; Nabors, B.; Weller, M.; Hau, P. Use of metformin and outcome of patients with newly diagnosed glioblastoma: Pooled analysis. Int. J. Cancer 2020, 146, 803–809. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Age | Sex | Tumor Location | KPS | IDH | MGMT | MIB-1 |
|---|---|---|---|---|---|---|---|
| Poli 09 | 81 | F | FP left | 60 | wt | 4% | 15% |
| Poli 182 | 41 | F | T left | 70 | wt | 49% | 55% |
| Poli 183 | 41 | M | T right | 80 | wt | 14% | 40% |
| Poli 187 | 45 | M | F right | 100 | wt | 37% | 20% |
| Poli 208 | 60 | M | P right | 80 | wt | 14% | 30% |
| Poli 210 | 52 | M | P left | 70 | wt | 15% | 55% |
| Poli 214 | 66 | M | T left | 80 | wt | 60% | 65% |
| Poli 230 | 82 | F | T left | 70 | wt | 3% | 27% |
| Poli 231 | 80 | M | T right | 80 | wt | 2% | 40% |
| Gene | Forward Primer (5′-3′) | Reverse Primer (3′-5′) | Tm (°C) |
|---|---|---|---|
| 18 S | ACTTTCGATGGTAGTCGCCGT | CCTTGGATGTGGTAGCCGTTT | 61 |
| AKT | TCT ATG GCG CTG AGA TTG TG | CTT AAT GTG CCC GTC CTT GT | 58 |
| BAX | AGC AAA CTG GTG CTC AAG G | TCT TGG ATC CAG CCC AAC | 57 |
| BCL-2 | AGT ACC TGA ACC GGC ACC T | GCC GTA CAG TTC CAC AAA GG | 60 |
| BID | ACC GTG GTC TTT CCA GCA CC | TCT GCG GAA GCT GTT GTC AG | 61 |
| SPHK1 | TGCAGTTGGTCAGGAGGTCT | GCTCTGGTGGTCATGTCTGG | 66 |
| SPHK2 | CCCCGGTTGCTTCTATTGGT | ATCCCACTCACTCAGGCTCA | 66 |
| S1PR1 | GGGAGCAATAACTTCCGCCT | AAGCAGAGTGAAGACCGTGG | 66 |
| S1PR2 | CCTGTACGTGCGCATCTACT | GCTTTGTAGAGGATCGGGCA | 65 |
| S1PR3 | CAACCACAACAACTCGGAGC | GCCAACACGATGAACCACTG | 64 |
| S1PR5 | CATCTACTGCCAGGTACGCG | GAGCAACAGCAGCAGGAAGA | 65 |
| SGPL1 | AAGCATATCGGGATCTGGCC | TAGCTCTTCTCATTGCCCGC | 65 |
| SGPP1 | CGTGGTCAAGTTGGAGGTCT | GGCAAACTAGAGAACACCAGC | 63 |
| SGPP2 | AGGATGTCTTGAAGTGGCCC | CCATCACCAGTCCCAACACA | 66 |
| ASAH1 | TTC TTT GCC TCT GCT GGA GTC | TGG AAC TGC ACC TCT GTA CG | 60 |
| ASAH2 | CAT GGC AGA ACC TGA TGG GT | GTC TGT TCA GGA CCT CCA GC | 61 |
| CERS1 | CCC TTC TTC CAT GAC CCA CC | CTC AGT GGC TTC TCG GCT TT | 61 |
| CERS2 | TTT GCC CCT CAC TCA GGA TG | CGT AGA CAC GTC CAT CTC GG | 61 |
| CERS4 | AGG AGC AGA GTC CGG CTG | CCT GCC AAA ACC ACT CGT TG | 60 |
| CERS5 | GCT CTT CGA GCG ATT TAT TGC C | ATT CAC CCG ATT GGC ACC AT | 60 |
| CASPASE-3 | ATG GTT TGA GCC TGA GCA GA | GGC AGC ATC ATC CAC ACA TAC | 60 |
| CASPASE-7 | GAG CAG GGG GTT GAG GAT TC | GTC TTT TCC GTG CTC CTC CA | 61 |
| CASPASE-10 | CCA GGT GAA CTG GAA TGC CT | CCA CTA GCT TTT GGC CCT GA | 60 |
| ERK-1 | ACTCCAAAGCCCTTGACCTG | CTTCAGCCGCTCCTTAGGTA | 60 |
| HIF-1a | GTCTGAGGGGACAGGAGGAT | GCACCAAGCAGGTCATAGGT | 61 |
| MEK-1 | CTTCGCAGAGCGGCTAGG | AGCTCTAGCTCCTCCAGCTT | 61 |
| P21 | AGT ACC CTC TCA GCT CCA GG | TGT CTG ACT CCT TGT TCC GC | 61 |
| P27 | TGG CTT GTC AGG AAC TCG AC | CTA GTC TCC AGG GAG GTG CT | 63 |
| P53 | AGG CCT TGG AAC TCA AGG AT | CCC TTT TTG GAC TTC AGG TG | 58 |
| RAF | GGT TTT GGC GTA GAT TCC CC | ACC TGA AGC AAA GAT GGC GT | 59 |
| RAS | AGCAGGTGGTCATTGATGGG | CCGTTTGATCTGCTCCCTGT | 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guarnaccia, L.; Navone, S.E.; Masseroli, M.M.; Balsamo, M.; Caroli, M.; Valtorta, S.; Moresco, R.M.; Campanella, R.; Schisano, L.; Fiore, G.; et al. Effects of Metformin as Add-On Therapy against Glioblastoma: An Old Medicine for Novel Oncology Therapeutics. Cancers 2022, 14, 1412. https://doi.org/10.3390/cancers14061412
Guarnaccia L, Navone SE, Masseroli MM, Balsamo M, Caroli M, Valtorta S, Moresco RM, Campanella R, Schisano L, Fiore G, et al. Effects of Metformin as Add-On Therapy against Glioblastoma: An Old Medicine for Novel Oncology Therapeutics. Cancers. 2022; 14(6):1412. https://doi.org/10.3390/cancers14061412
Chicago/Turabian StyleGuarnaccia, Laura, Stefania E. Navone, Matteo M. Masseroli, Melissa Balsamo, Manuela Caroli, Silvia Valtorta, Rosa M. Moresco, Rolando Campanella, Luigi Schisano, Giorgio Fiore, and et al. 2022. "Effects of Metformin as Add-On Therapy against Glioblastoma: An Old Medicine for Novel Oncology Therapeutics" Cancers 14, no. 6: 1412. https://doi.org/10.3390/cancers14061412
APA StyleGuarnaccia, L., Navone, S. E., Masseroli, M. M., Balsamo, M., Caroli, M., Valtorta, S., Moresco, R. M., Campanella, R., Schisano, L., Fiore, G., Galiano, V., Garzia, E., Appiani, G. C., Locatelli, M., Riboni, L., & Marfia, G. (2022). Effects of Metformin as Add-On Therapy against Glioblastoma: An Old Medicine for Novel Oncology Therapeutics. Cancers, 14(6), 1412. https://doi.org/10.3390/cancers14061412