
Multiplex Droplet Digital PCR Assay for Detection of MET and HER2 Genes Amplification in Non-Small Cell Lung Cancer

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples and DNA Extraction
2.2. Detection of Common EGFR Gene Mutations
2.3. Positive Controls
2.4. Fluorescence In Situ Hybridization
2.5. Quantitative PCR
2.6. Droplet Digital PCR
3. Results
3.1. Selection of References, Primers, and Probes
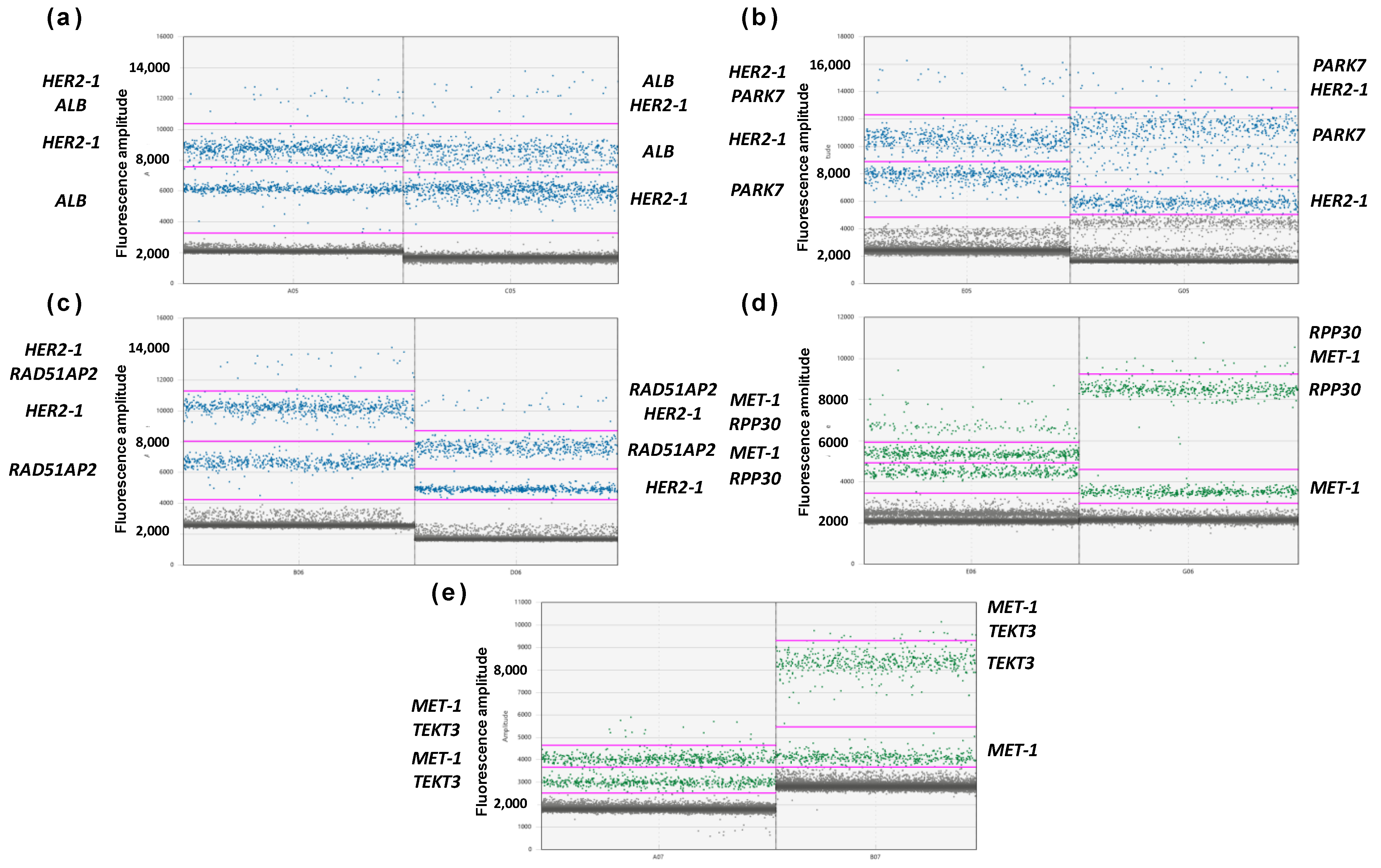
3.2. Optimization of Multiplex ddPCR Assay
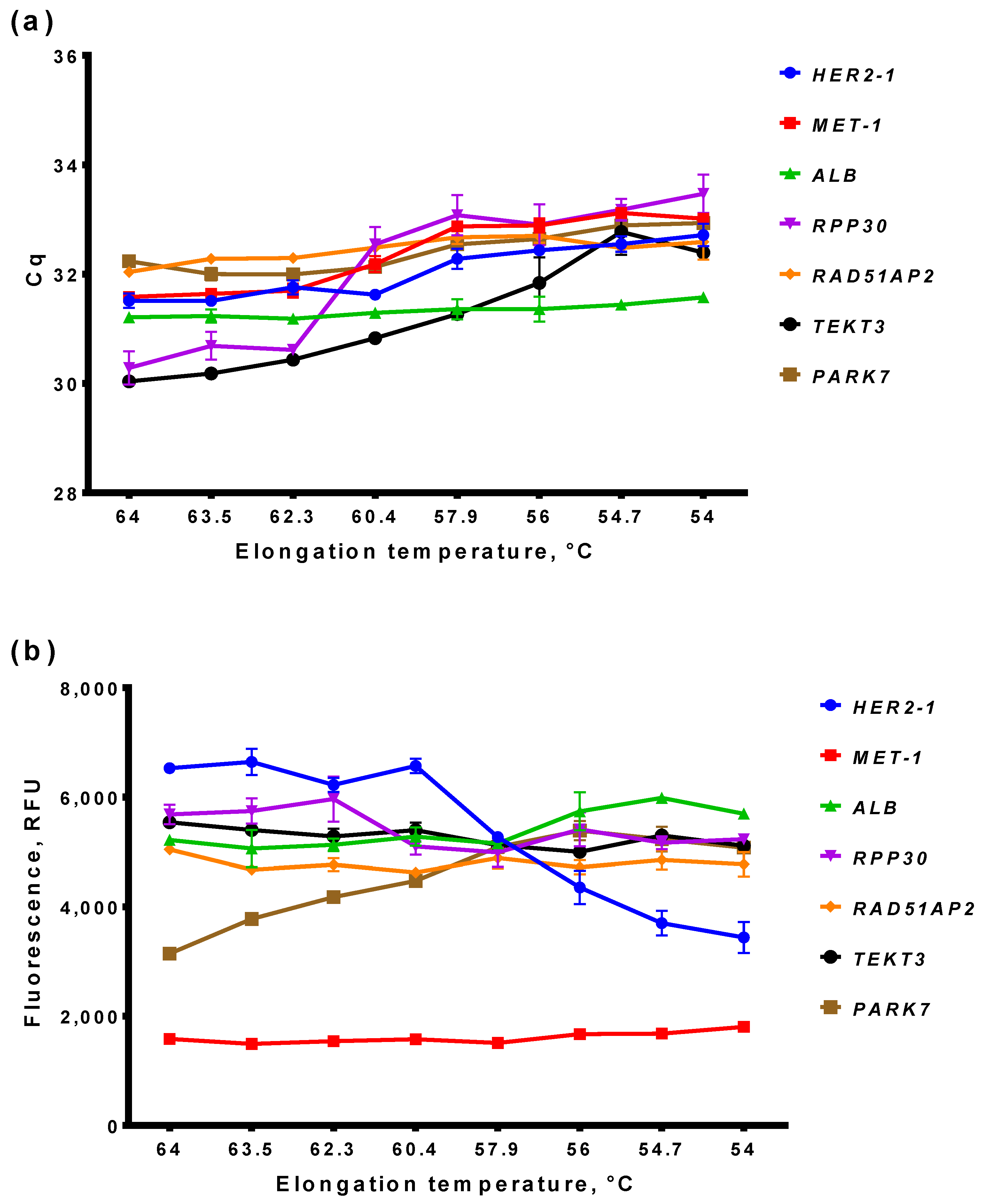
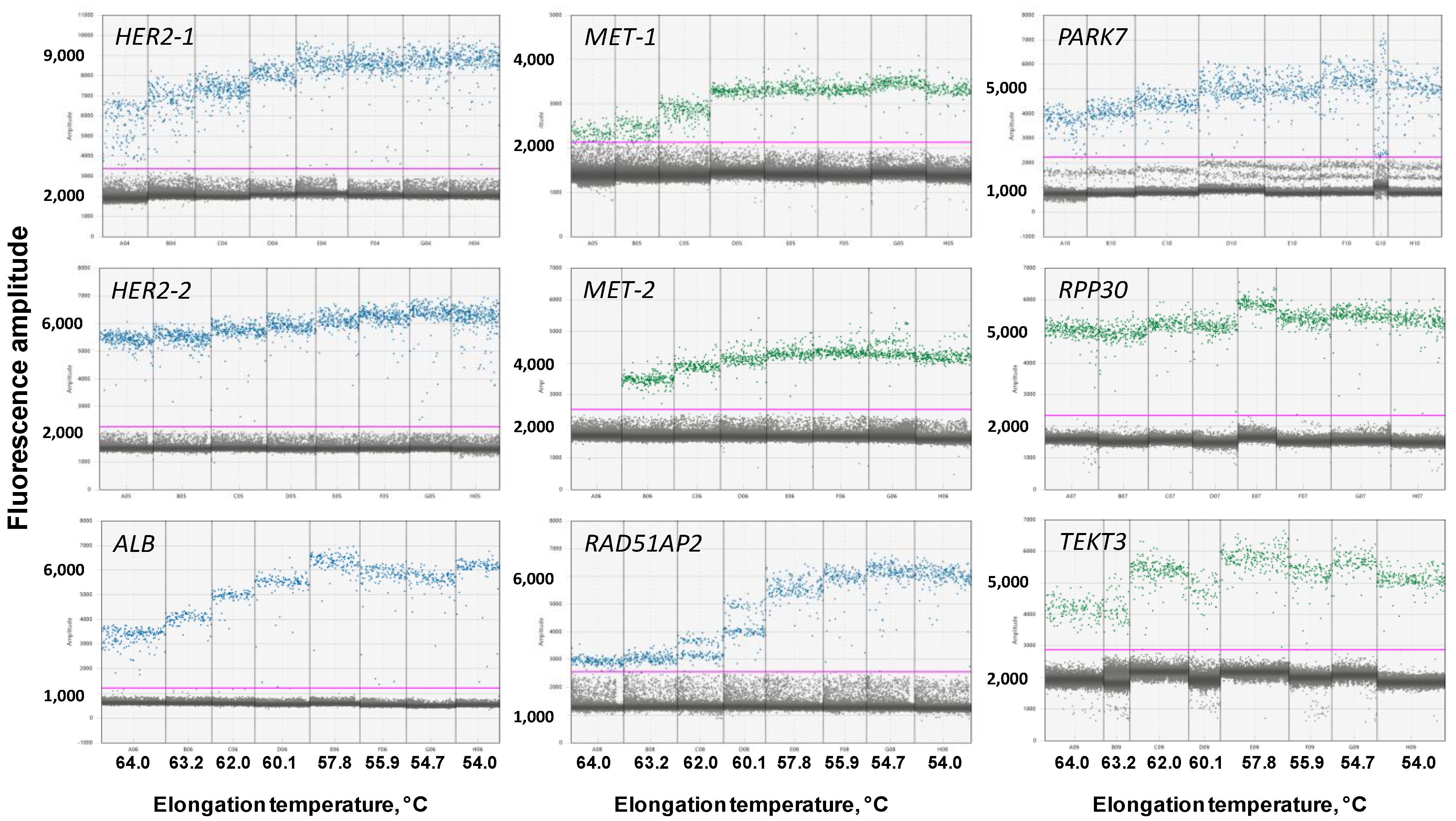
3.2.1. Optimal Elongation Temperature of ddPCR
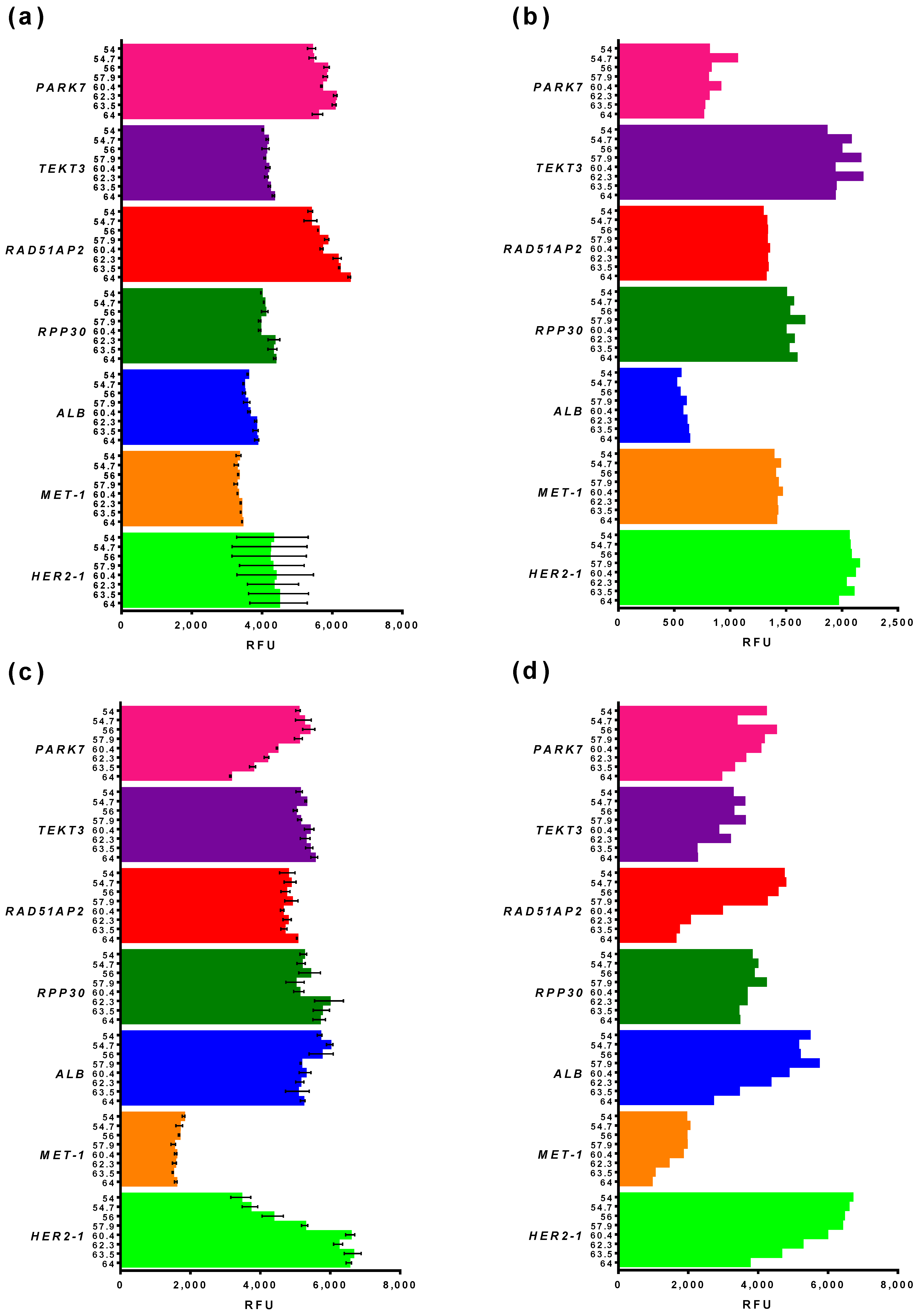
3.2.2. Basic and End-Point Fluorescence in qPCR and ddPCR
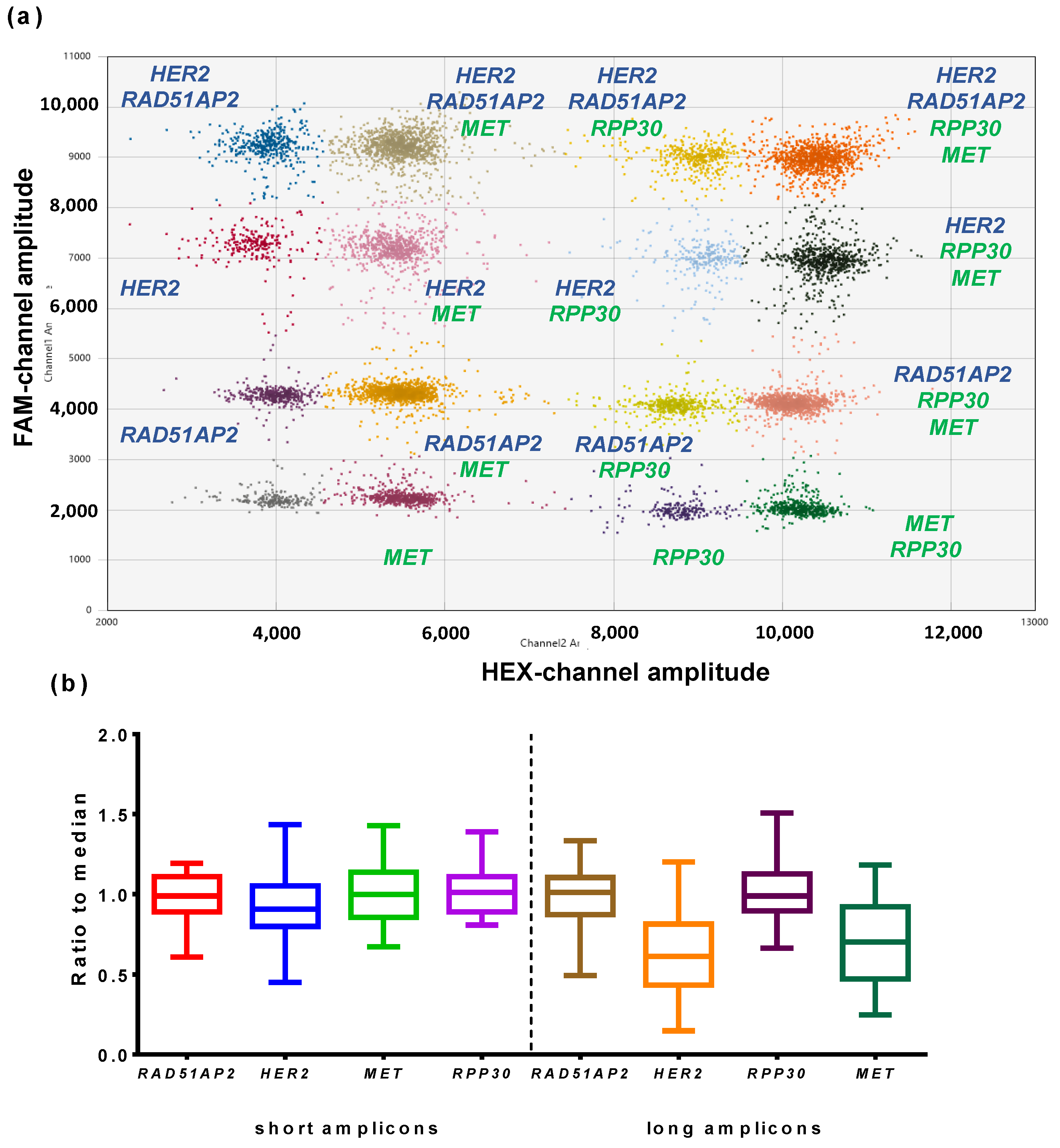
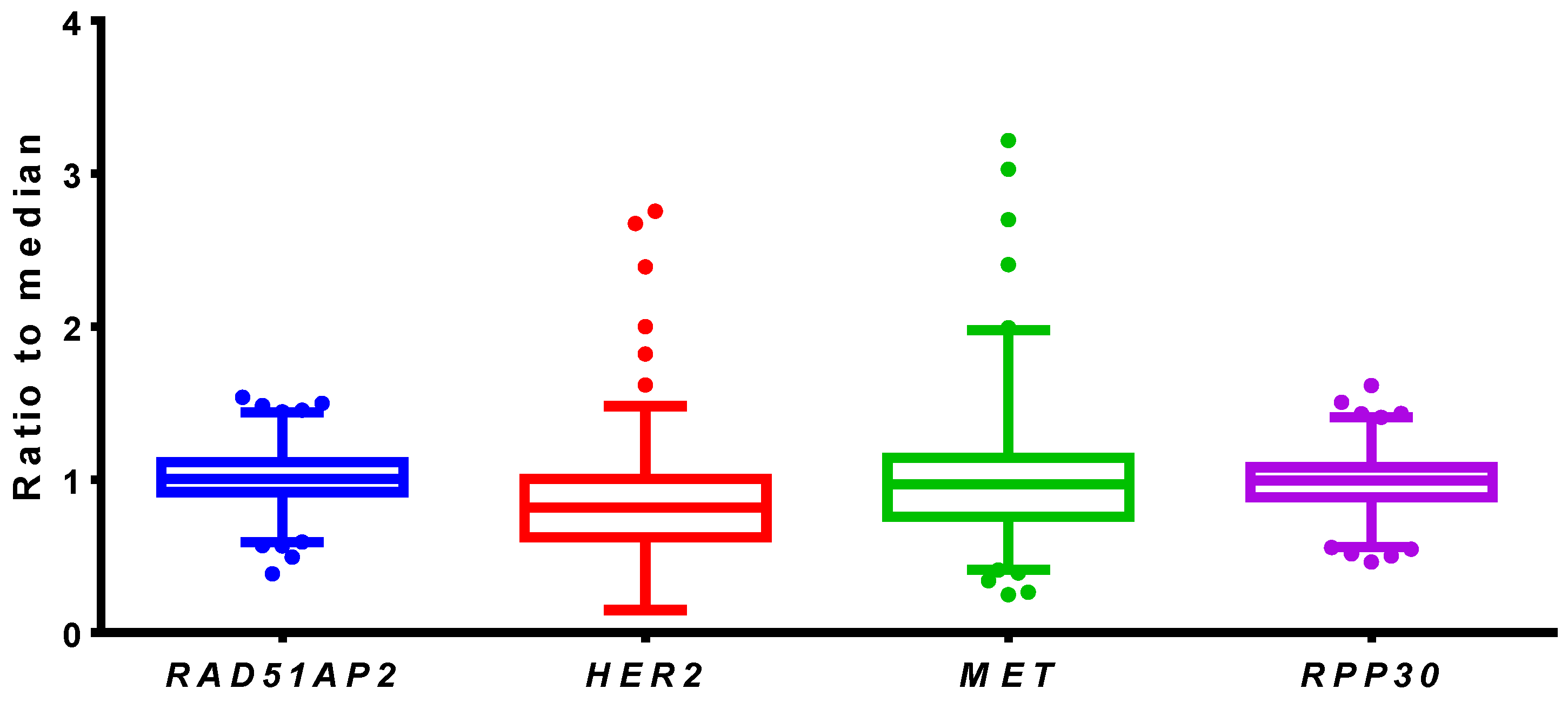
3.2.3. Reference Gene Selection
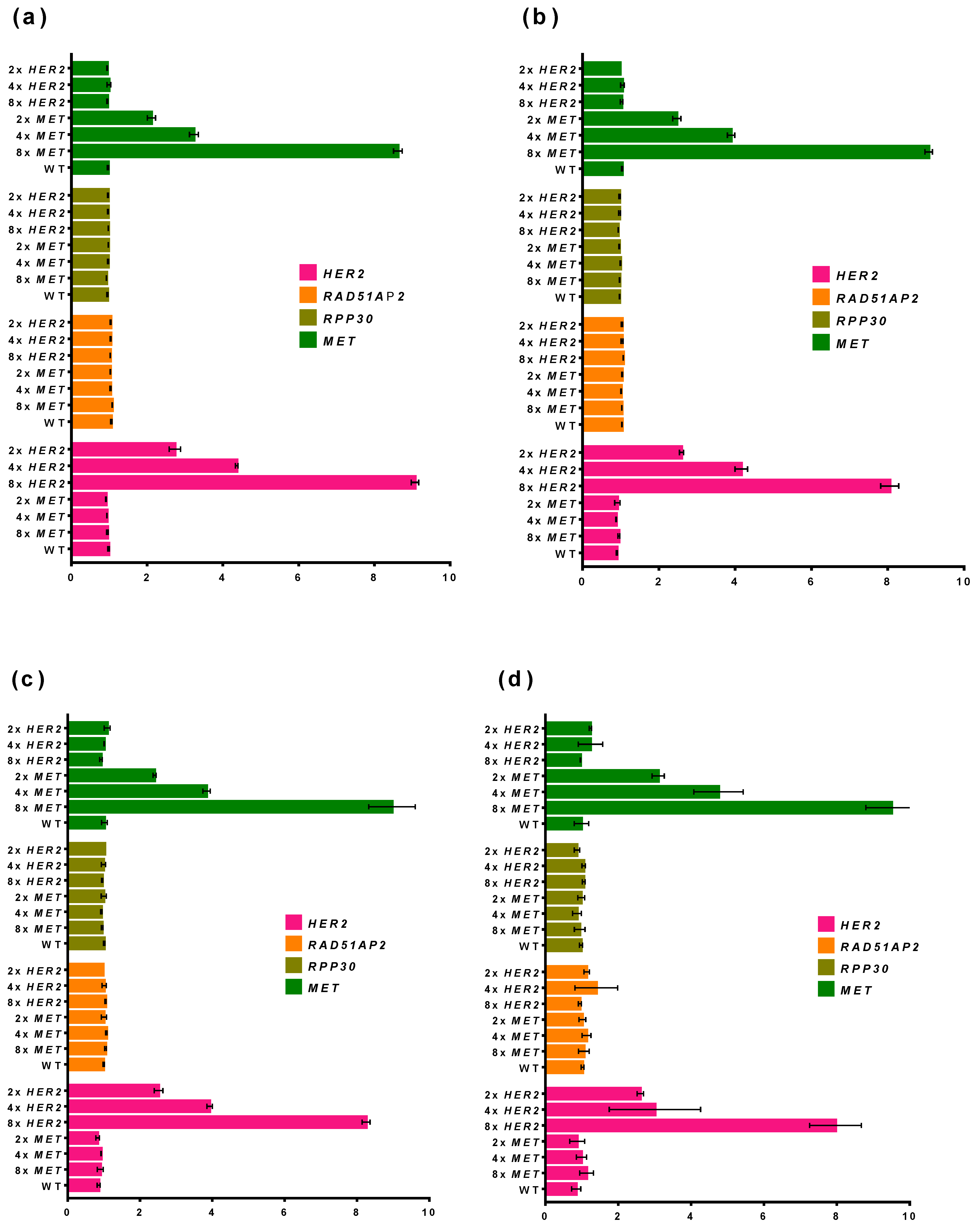
3.2.4. Optimization of MET and HER2 Primers
3.3. Validation of Multiplex ddPCR Assay
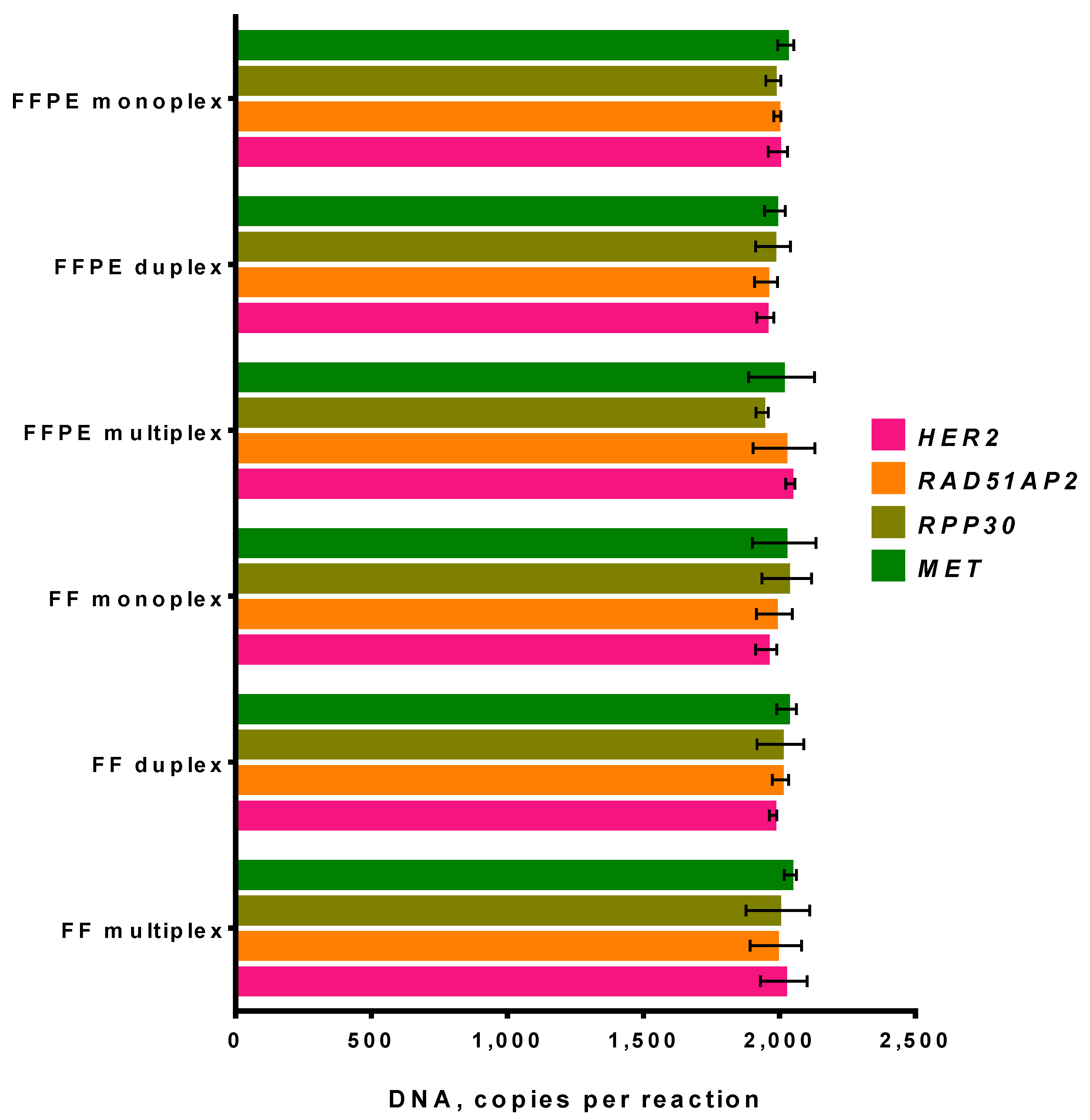
3.3.1. Fresh-Frozen Tissue and FFPE Sample in ddPCR
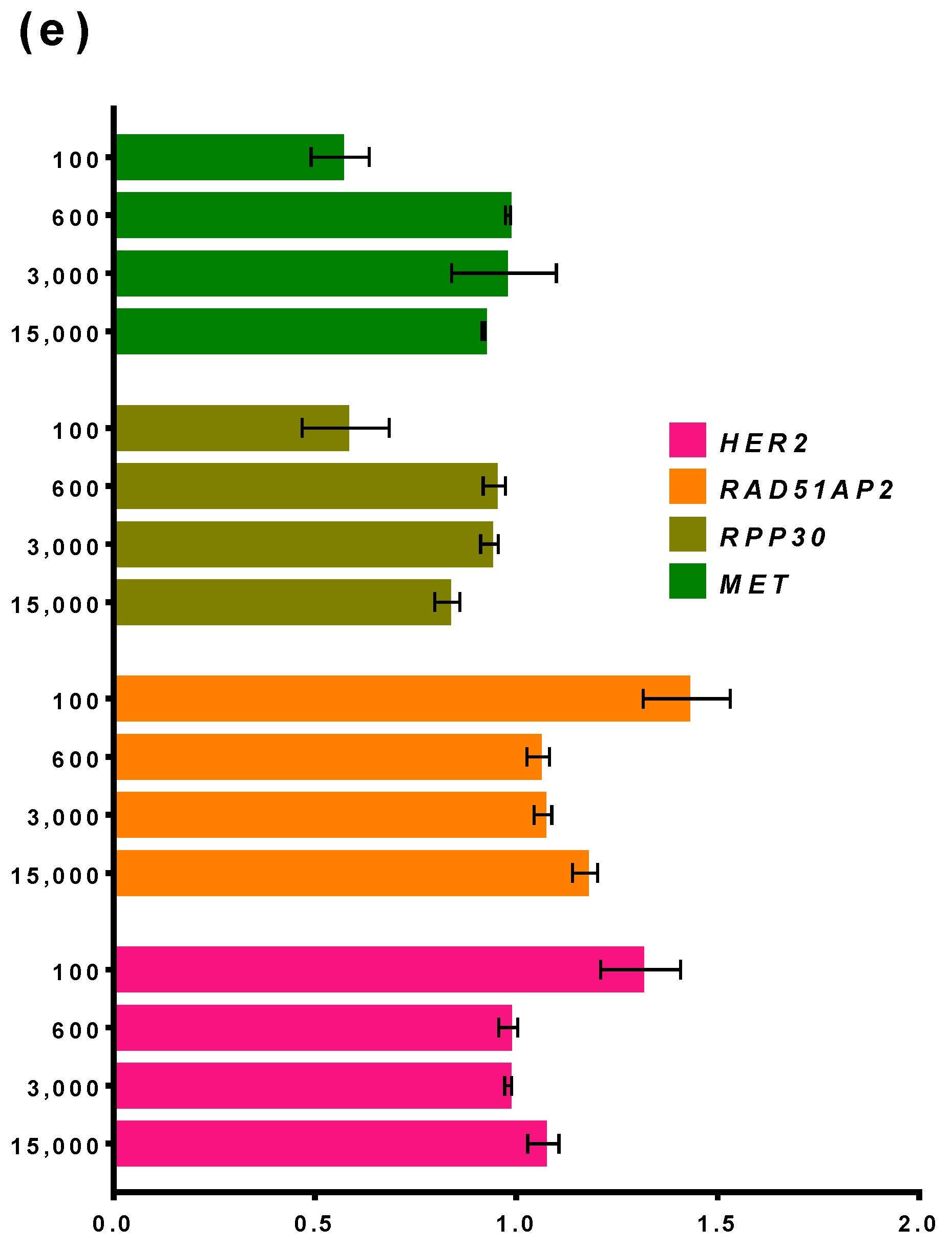
3.3.2. Influence of DNA Concentration on the Performance of ddPCR
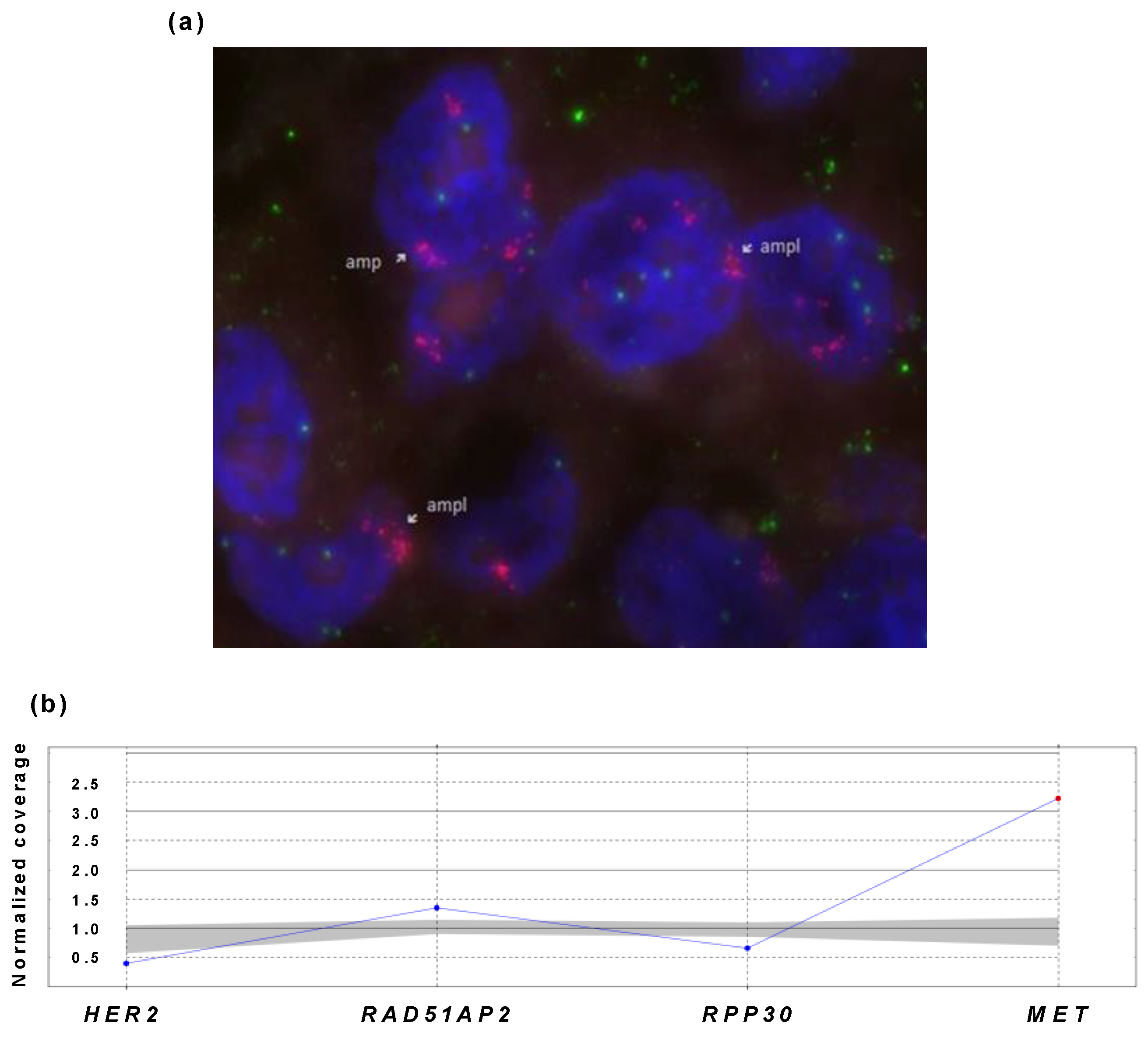
3.3.3. Testing of MET-Positive Sample
3.4. Testing of Clinical Samples
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung cancer. Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Primers 2015, 1, 15009. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.-J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.-H.I.; Dezube, B.J.; Jänne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Melosky, B.; Wheatley-Price, P.; Juergens, R.A.; Sacher, A.; Leighl, N.B.; Tsao, M.-S.; Cheema, P.; Snow, S.; Liu, G.; Card, P.B.; et al. The rapidly evolving landscape of novel targeted therapies in advanced non-small cell lung cancer. Lung Cancer 2021, 160, 136–151. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS G12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Park, K.; Haura, E.B.; Leighl, N.B.; Mitchell, P.; Shu, C.A.; Girard, N.; Viteri, S.; Han, J.-Y.; Kim, S.-W.; Lee, C.K.; et al. Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J. Clin. Oncol. 2021, 39, 3391–3402. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef]
- Fujino, T.; Suda, K.; Mitsudomi, T. Emerging MET tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. Expert Opin. Emerg. Drugs 2020, 25, 229–249. [Google Scholar] [CrossRef]
- Friedlaender, A.; Drilon, A.; Banna, G.L.; Peters, S.; Addeo, A. The METeoric rise of MET in lung cancer. Cancer 2020, 126, 4826–4837. [Google Scholar] [CrossRef]
- Malik, S.M.; Maher, V.E.; Bijwaard, K.E.; Becker, R.L.; Zhang, L.; Tang, S.W.; Song, P.; Liu, Q.; Marathe, A.; Gehrke, B.; et al. U.S. Food and Drug Administration approval: Crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin. Cancer Res. 2014, 20, 2029–2034. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef]
- Albanell, J.; Baselga, J. Trastuzumab, a humanized anti-HER2 monoclonal antibody, for the treatment of breast cancer. Drugs Today (Barc) 1999, 35, 931–946. [Google Scholar]
- Blumenthal, G.M.; Scher, N.S.; Cortazar, P.; Chattopadhyay, S.; Tang, S.; Song, P.; Liu, Q.; Ringgold, K.; Pilaro, A.M.; Tilley, A.; et al. First FDA approval of dual anti-HER2 regimen: Pertuzumab in combination with trastuzumab and docetaxel for HER2-positive metastatic breast cancer. Clin. Cancer Res. 2013, 19, 4911–4916. [Google Scholar] [CrossRef] [Green Version]
- Royce, M.; Osgood, C.L.; Amatya, A.K.; Fiero, M.H.; Chang, C.J.G.; Ricks, T.K.; Shetty, K.A.; Kraft, J.; Qiu, J.; Song, P.; et al. FDA Approval Summary: Margetuximab plus Chemotherapy for Advanced or Metastatic HER2-Positive Breast Cancer. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Jebbink, M.; de Langen, A.J.; Boelens, M.C.; Monkhorst, K.; Smit, E.F. The force of HER2—A druggable target in NSCLC? Cancer Treat. Rev. 2020, 86, 101996. [Google Scholar] [CrossRef]
- Zhao, J.; Xia, Y. Targeting HER2 Alterations in Non–Small-Cell Lung Cancer: A Comprehensive Review. JCO Precis. Oncol. 2020, 4, 411–425. [Google Scholar] [CrossRef]
- Nagasaka, M.; Singh, V.; Baca, Y.; Sukari, A.; Kim, C.; Mamdani, H.; Spira, A.I.; Uprety, D.; Bepler, G.; Kim, E.S.; et al. The Effects of HER2 Alterations in EGFR Mutant Non-small Cell Lung Cancer. Clin. Lung Cancer 2022, 23, 52–59. [Google Scholar] [CrossRef]
- Li, B.T.; Smit, E.F.; Goto, Y.; Nakagawa, K.; Udagawa, H.; Mazières, J.; Nagasaka, M.; Bazhenova, L.; Saltos, A.N.; Felip, E.; et al. Trastuzumab Deruxtecan in HER2-Mutant Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2022, 386, 241–251. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, E.-T.; Du, Z. Detection of MET Gene Copy Number in Cancer Samples Using the Droplet Digital PCR Method. PLoS ONE 2016, 11, e0146784. [Google Scholar] [CrossRef]
- Ishii, H.; Azuma, K.; Sakai, K.; Kawahara, A.; Yamada, K.; Tokito, T.; Okamoto, I.; Nishio, K.; Hoshino, T. Digital PCR analysis of plasma cell-free DNA for non-invasive detection of drug resistance mechanisms in EGFR mutant NSCLC: Correlation with paired tumor samples. Oncotarget 2015, 6, 30850–30858. [Google Scholar] [CrossRef] [Green Version]
- Oddo, D.; Siravegna, G.; Gloghini, A.; Vernieri, C.; Mussolin, B.; Morano, F.; Crisafulli, G.; Berenato, R.; Corti, G.; Volpi, C.C.; et al. Emergence of MET hyper-amplification at progression to MET and BRAF inhibition in colorectal cancer. Br. J. Cancer 2017, 117, 347–352. [Google Scholar] [CrossRef]
- He, H.-J.; Das, B.; Cleveland, M.H.; Chen, L.; Camalier, C.E.; Liu, L.-C.; Norman, K.L.; Fellowes, A.P.; McEvoy, C.R.; Lund, S.P.; et al. Development and interlaboratory evaluation of a NIST Reference Material RM 8366 for EGFR and MET gene copy number measurements. Clin. Chem. Lab. Med. 2019, 57, 1142–1152. [Google Scholar] [CrossRef]
- Chen, W.; Zheng, J.; Wu, C.; Liu, S.; Chen, Y.; Liu, X.; Du, J.; Wang, J. Breast Cancer Subtype Classification Using 4-Plex Droplet Digital PCR. Clin. Chem. 2019, 65, 1051–1059. [Google Scholar] [CrossRef]
- Gevensleben, H.; Garcia-Murillas, I.; Graeser, M.K.; Schiavon, G.; Osin, P.; Parton, M.; Smith, I.E.; Ashworth, A.; Turner, N.C. Noninvasive detection of HER2 amplification with plasma DNA digital PCR. Clin. Cancer Res. 2013, 19, 3276–3284. [Google Scholar] [CrossRef] [Green Version]
- Belgrader, P.; Tanner, S.C.; Regan, J.F.; Koehler, R.; Hindson, B.J.; Brown, A.S. Droplet Digital PCR Measurement of HER2 Copy Number Alteration in Formalin-Fixed Paraffin-Embedded Breast Carcinoma Tissue. Clin. Chem. 2013, 59, 991–994. [Google Scholar] [CrossRef] [Green Version]
- Heredia, N.J.; Belgrader, P.; Wang, S.; Koehler, R.; Regan, J.; Cosman, A.M.; Saxonov, S.; Hindson, B.; Tanner, S.C.; Brown, A.S.; et al. Droplet DigitalTM PCR quantitation of HER2 expression in FFPE breast cancer samples. Methods 2013, 59, S20–S23. [Google Scholar] [CrossRef]
- Turner, N.C.; Kingston, B.; Kilburn, L.S.; Kernaghan, S.; Wardley, A.M.; Macpherson, I.R.; Baird, R.D.; Roylance, R.; Stephens, P.; Oikonomidou, O.; et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): A multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 2020, 21, 1296–1308. [Google Scholar] [CrossRef]
- He, H.-J.; Almeida, J.L.; Lund, S.P.; Steffen, C.R.; Choquette, S.; Cole, K.D. Development of NIST standard reference material 2373: Genomic DNA standards for HER2 measurements. Biomol. Detect. Quantif. 2016, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Camidge, D.R.; Otterson, G.A.; Clark, J.W.; Ignatius Ou, S.-H.; Weiss, J.; Ades, S.; Shapiro, G.I.; Socinski, M.A.; Murphy, D.A.; Conte, U.; et al. Crizotinib in Patients With MET-Amplified NSCLC. J. Thorac. Oncol. 2021, 16, 1017–1029. [Google Scholar] [CrossRef]
- Jones, M.; Williams, J.; Gärtner, K.; Phillips, R.; Hurst, J.; Frater, J. Low copy target detection by Droplet Digital PCR through application of a novel open access bioinformatic pipeline, “definetherain”. J. Virol. Methods 2014, 202, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Della Starza, I.; Nunes, V.; Cavalli, M.; De Novi, L.A.; Ilari, C.; Apicella, V.; Vitale, A.; Testi, A.M.; Del Giudice, I.; Chiaretti, S.; et al. Comparative analysis between RQ-PCR and digital-droplet-PCR of immunoglobulin/T-cell receptor gene rearrangements to monitor minimal residual disease in acute lymphoblastic leukaemia. Br. J. Haematol. 2016, 174, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Oscorbin, I.; Kechin, A.; Boyarskikh, U.; Filipenko, M. Multiplex ddPCR assay for screening copy number variations in BRCA1 gene. Breast Cancer Res. Treat. 2019, 178, 545–555. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.-C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29, viii740. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.A.; Wu, Y.-L.; Han, J.-Y.; Ahn, M.-J.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.C.-H.; Bulusu, K.C.; Laus, G.; et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29, viii741. [Google Scholar] [CrossRef]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.-L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Bean, J.; Brennan, C.; Shih, J.-Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA. 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients with EGFR -Mutant Lung Cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Berry, L.D.; Aisner, D.L.; Sheren, J.; Boyle, T.; Bunn, P.A.; Johnson, B.E.; Kwiatkowski, D.J.; Drilon, A.; Sholl, L.M.; et al. MET IHC Is a Poor Screen for MET Amplification or MET Exon 14 Mutations in Lung Adenocarcinomas: Data from a Tri-Institutional Cohort of the Lung Cancer Mutation Consortium. J. Thorac. Oncol. 2019, 14, 1666–1671. [Google Scholar] [CrossRef]
- Nitta, H.; Kelly, B.D.; Allred, C.; Jewell, S.; Banks, P.; Dennis, E.; Grogan, T.M. The assessment of HER2 status in breast cancer: The past, the present, and the future. Pathol. Int. 2016, 66, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Heydt, C.; Becher, A.-K.; Wagener-Ryczek, S.; Ball, M.; Schultheis, A.M.; Schallenberg, S.; Rüsseler, V.; Büttner, R.; Merkelbach-Bruse, S. Comparison of in situ and extraction-based methods for the detection of MET amplifications in solid tumors. Comput. Struct. Biotechnol. J. 2019, 17, 1339–1347. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene, Amplicon Length | Oligonucleotide | 5′-Sequence-3′ |
|---|---|---|
| HER2 set 1 100 bp | HER2-F1 | CATCTTCCACAAGAACAACCAG |
| HER2-R1 | GTCTGAGAGAAGAGGCAGCA | |
| HER2-P | FAM-AGACACCAACCGCTCTCGGGC-BHQ1 | |
| HER2 set 2 72 bp | HER2-F2 | CAGCTGGCTCTCACACTGA |
| HER2-R2 | AAGAGGCAGCAGGGAGG | |
| HER2-P | FAM-AGACACCAACCGCTCTCGGGC-BHQ1 | |
| MET set 1 100 bp | MET-F1 | AAGTGGGGAACTGATGTGACTTA |
| MET-R1 | CAAGATCGTCAACAAAAACAATG | |
| MET-P | HEX-CGGACCCAATCATGAGCACTG-BHQ1 | |
| MET set 2 77 bp | MET-F2 | CAATGTGAGATGTCTCCAGCAT |
| MET-R2 | GGGAACTGATGTGACTTACCCTA | |
| MET-P | HEX-CGGACCCAATCATGAGCACTG-BHQ1 | |
| ALB 94 bp | ALB-U | GACTTGCCAAGACATATGAAACC |
| ALB-FAM | FAM–TGCTGTGCCGCTGCAGATCC-BHQ1 | |
| ALB-R | TCCAACAATAAACCTACCACTTTG | |
| PARK7 74 bp | PARK7-F | CGTTGCAGGCCTGGCTGGAAA |
| PARK7-R | CAAGGCTGGCATCAGGACAAATG | |
| PARK7-P | FAM-CCCAGTACAGTGTAGCCGTGATGTGG-BHQ1 | |
| RAD51AP2 74 bp | RAD51AP2-U | TGGTGACTTTTGCCCATATTAG |
| RAD51AP2-R | GTGGTCAACAAAATACGTGCA | |
| RAD51AP2-P | FAM-CCTGCTATAGTATCATGGAACGAGG-BHQ1 | |
| RPP30 61 bp | RPP30-F | CAGATTTGGACCTGCGAGCG |
| RPP30-R | GAGCGGCTGTCTCCACAAGT | |
| RP30-HEX | HEX-TCTGACCTGAAGGCTCTGCGCG-BHQ2 | |
| TEKT3 67 bp | TEKT3-F | GCGTGTGCCTGACTTACGTTG |
| TEKT3-R | ACATCAGACGGTGTCGGCTAC | |
| TEKT3-P | HEX-CGACCCTCTCCACTCCGCG-BHQ2 | |
| Beta-lactamase | Lac-U | CGTCGTTTGGTATGGCTTCATTC |
| Lac-R | AGGACCGAAGGAGCTAACCG | |
| Lac-P | HEX-CGGTTCCCAACGATCAAGGCGAG-BHQ2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oscorbin, I.P.; Smertina, M.A.; Pronyaeva, K.A.; Voskoboev, M.E.; Boyarskikh, U.A.; Kechin, A.A.; Demidova, I.A.; Filipenko, M.L. Multiplex Droplet Digital PCR Assay for Detection of MET and HER2 Genes Amplification in Non-Small Cell Lung Cancer. Cancers 2022, 14, 1458. https://doi.org/10.3390/cancers14061458
Oscorbin IP, Smertina MA, Pronyaeva KA, Voskoboev ME, Boyarskikh UA, Kechin AA, Demidova IA, Filipenko ML. Multiplex Droplet Digital PCR Assay for Detection of MET and HER2 Genes Amplification in Non-Small Cell Lung Cancer. Cancers. 2022; 14(6):1458. https://doi.org/10.3390/cancers14061458
Chicago/Turabian StyleOscorbin, Igor P., Maria A. Smertina, Ksenia A. Pronyaeva, Mikhail E. Voskoboev, Ulyana A. Boyarskikh, Andrey A. Kechin, Irina A. Demidova, and Maxim L. Filipenko. 2022. "Multiplex Droplet Digital PCR Assay for Detection of MET and HER2 Genes Amplification in Non-Small Cell Lung Cancer" Cancers 14, no. 6: 1458. https://doi.org/10.3390/cancers14061458
APA StyleOscorbin, I. P., Smertina, M. A., Pronyaeva, K. A., Voskoboev, M. E., Boyarskikh, U. A., Kechin, A. A., Demidova, I. A., & Filipenko, M. L. (2022). Multiplex Droplet Digital PCR Assay for Detection of MET and HER2 Genes Amplification in Non-Small Cell Lung Cancer. Cancers, 14(6), 1458. https://doi.org/10.3390/cancers14061458