
Distinct Genomic Profiles Are Associated with Treatment Response and Survival in Ovarian Cancer

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
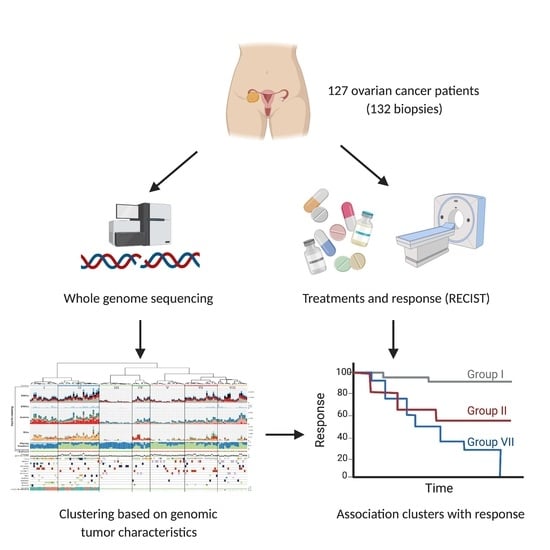
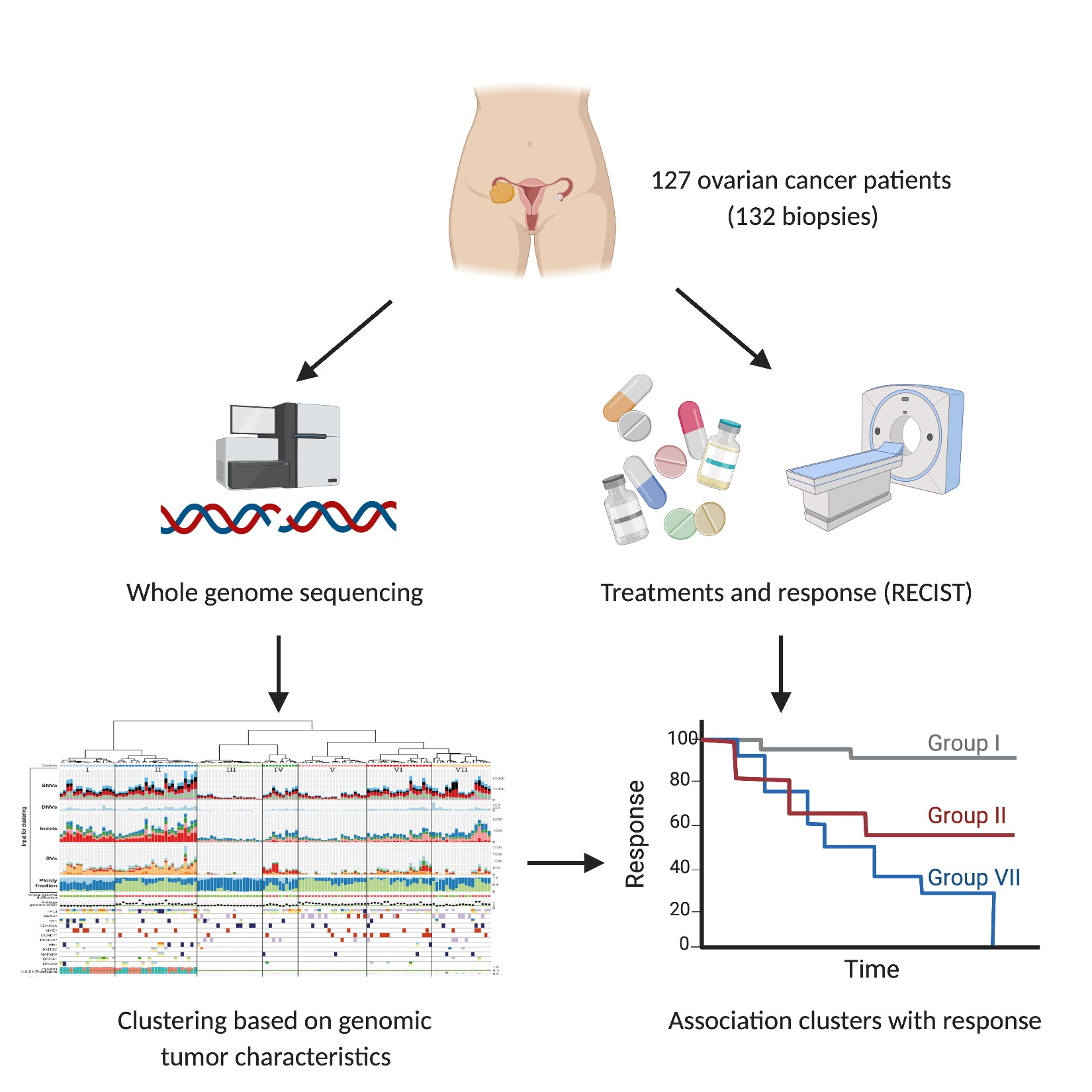
1. Introduction
2. Materials and Methods
2.1. Patient Inclusion, Sample Selection and Clinical Data Collection
2.2. WGS and Data Analysis
2.3. Gene Mutation Burden Analysis
2.4. Mutational Signatures
2.5. Determining HR-Status
2.6. Cluster Analysis
2.7. Actionability Analysis
2.8. Statistics
3. Results
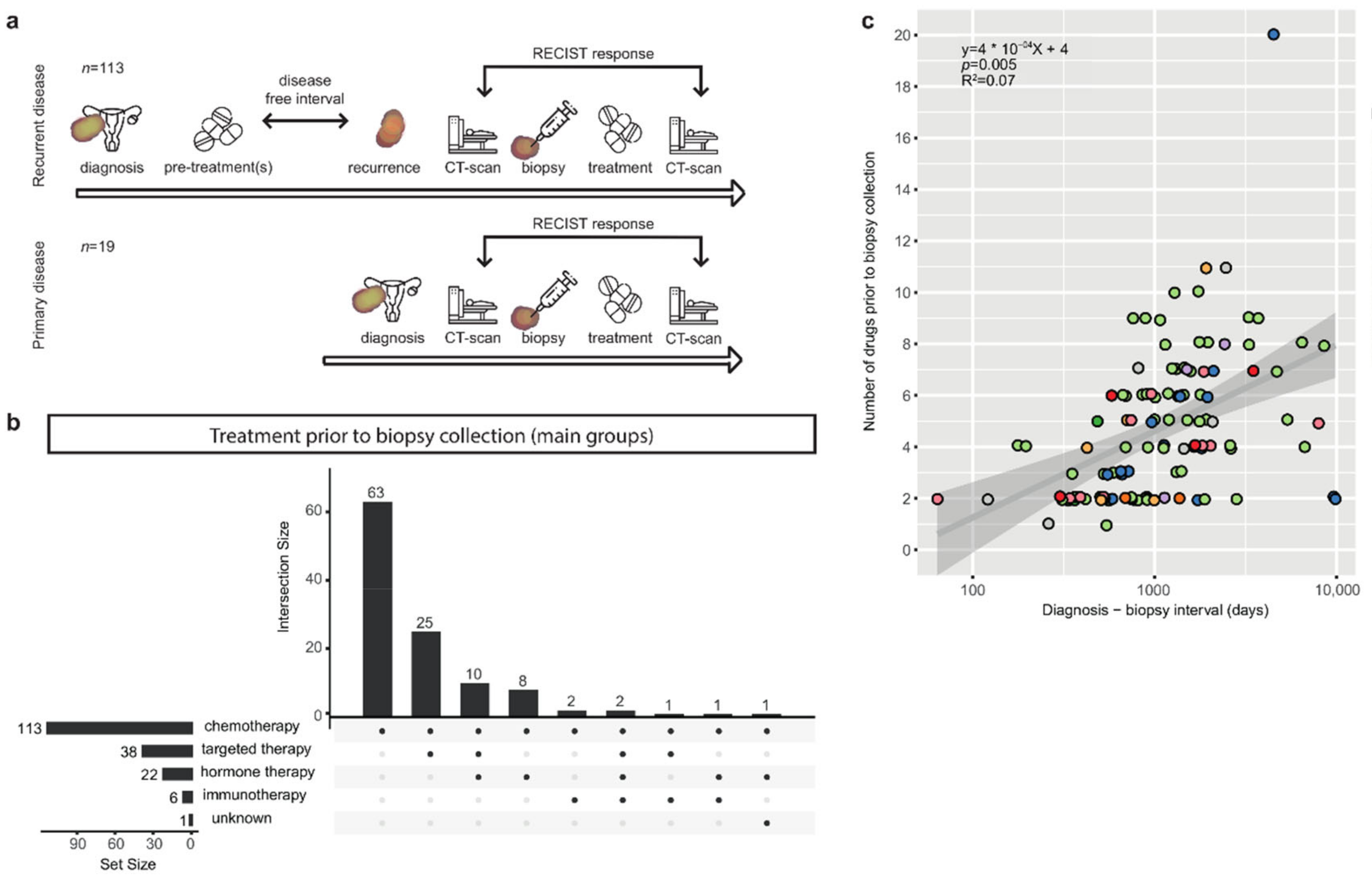
3.1. Patients with Metastatic Ovarian Cancer Have Diverse Treatment Histories
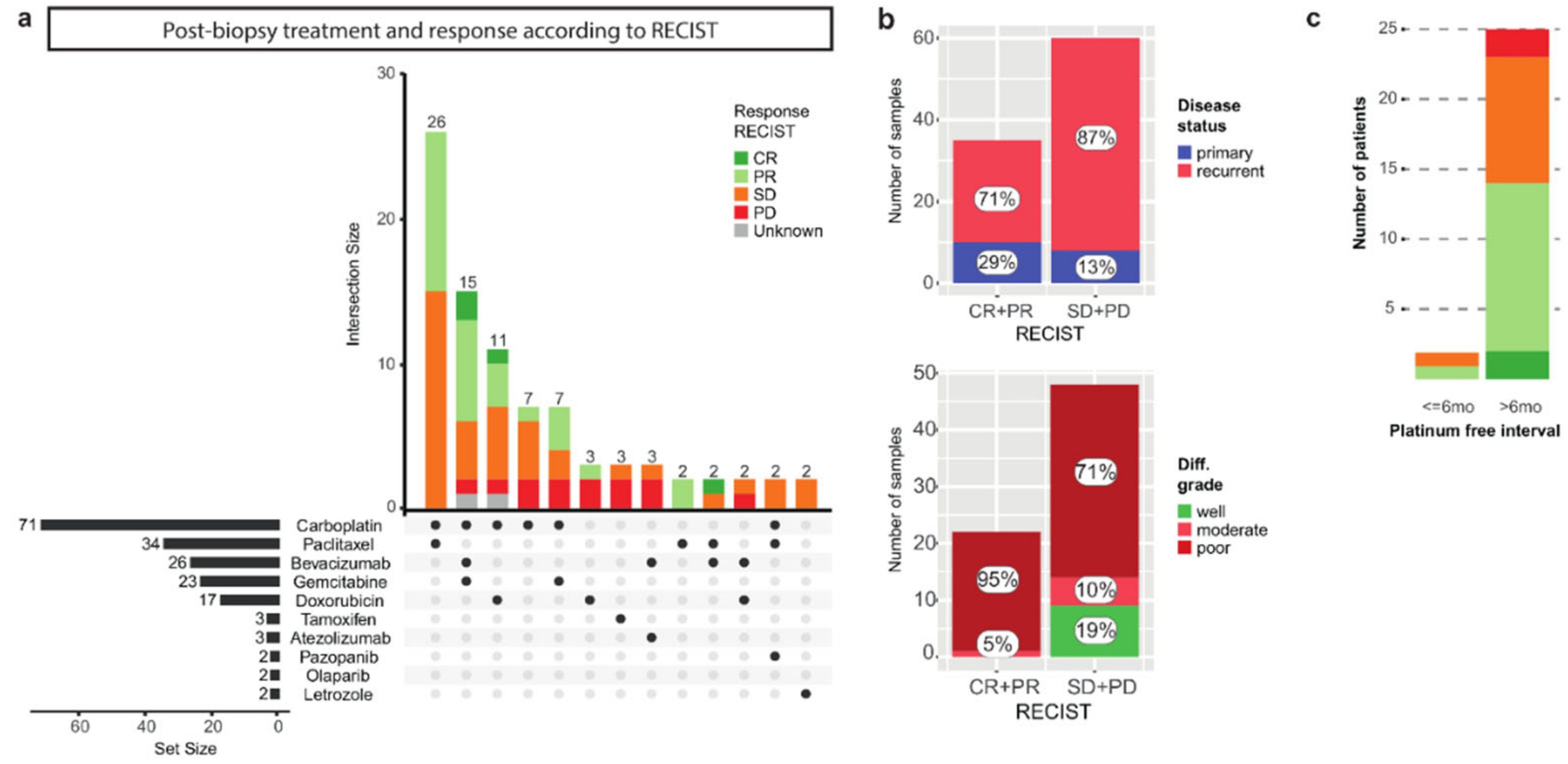
3.2. The Correlation of Known Clinical Determinants and Drug Response
3.3. Primary and Recurrent Samples Have Comparable Mutational Loads
3.4. Copy Number Aberrations Characterize Ovarian Cancer beyond HGSC
3.5. HR-Deficient Samples Harbor High TMB and Are Likely to Be HGSC
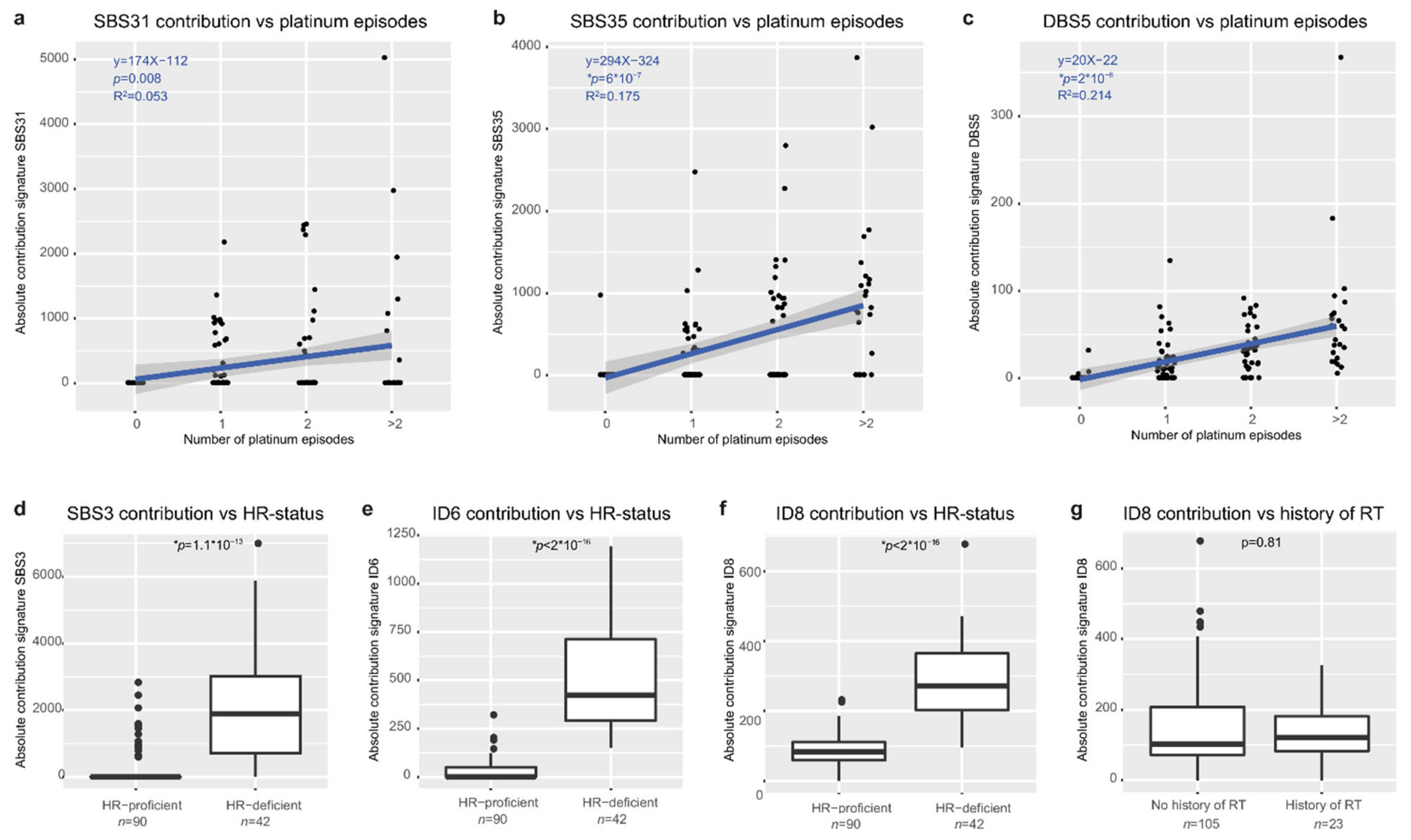
3.6. Mutational Signatures Reflect Treatment History and Tumor Biology
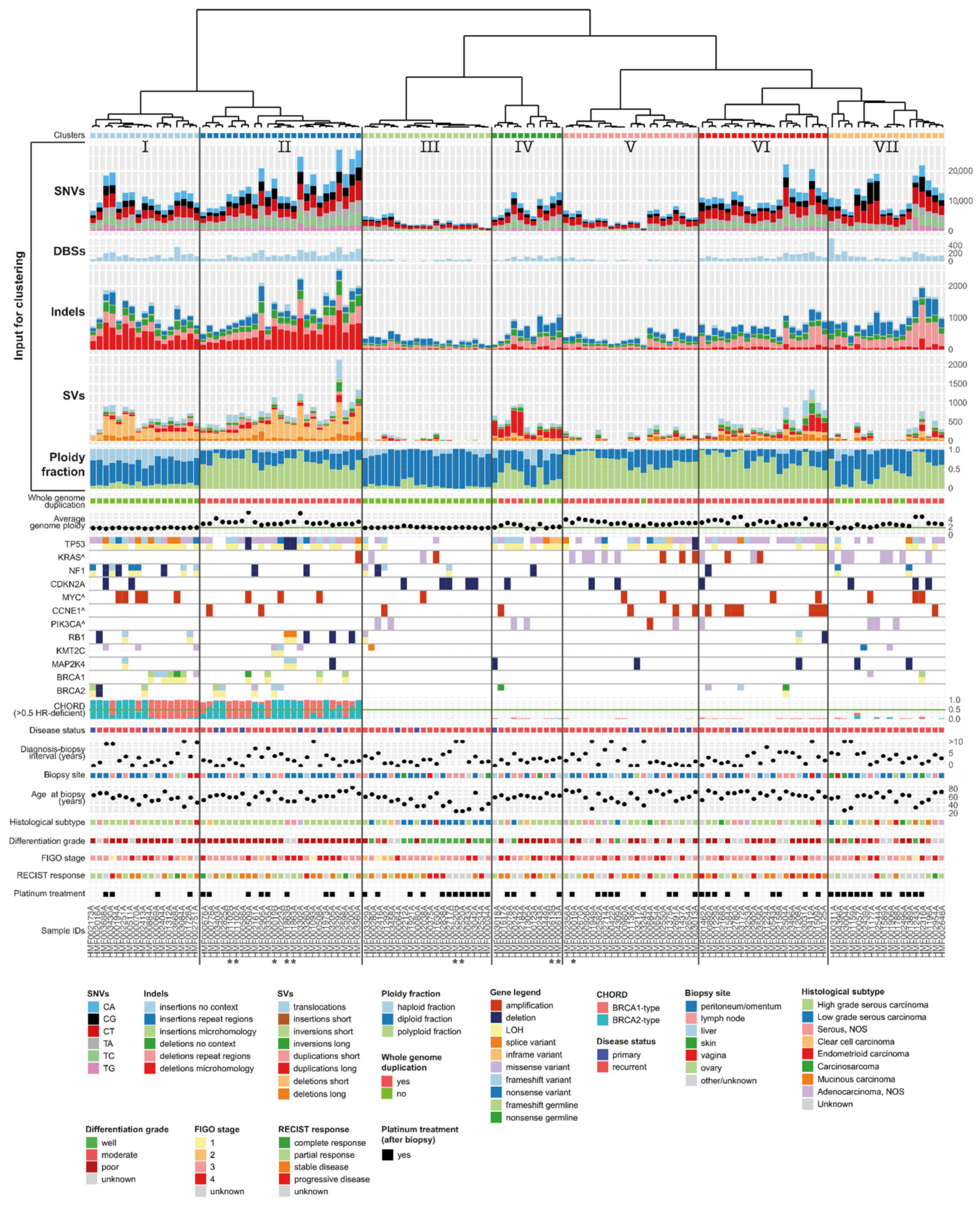
3.7. Unsupervised Clustering Based on Genomic Input Reveals Seven Distinct Clusters
3.8. Clusters with Specific Genomic and Clinical Features Have Potential Clinical Impact
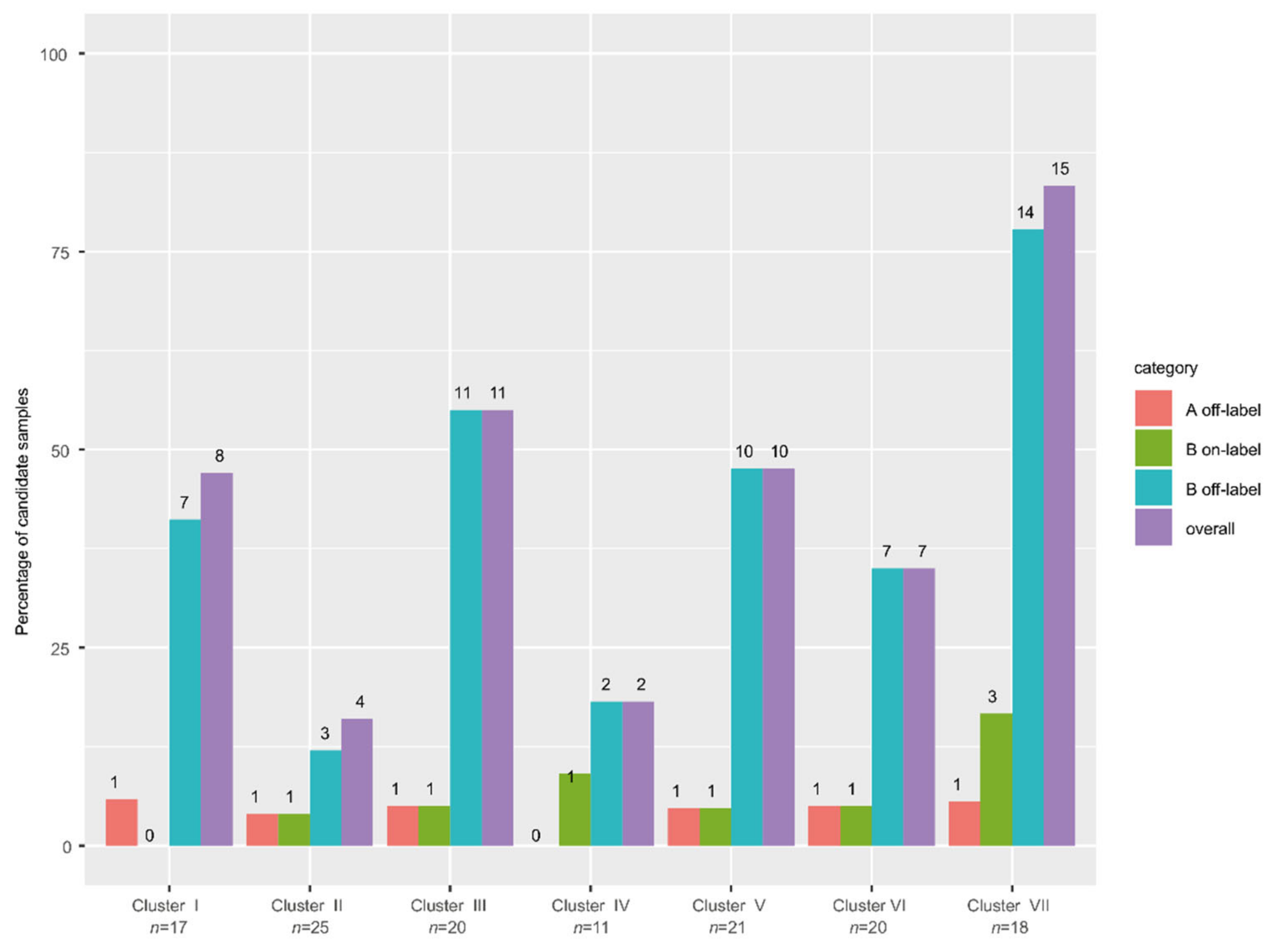
3.9. High Frequency of Actionable Targets in the Poor Response Cluster
3.10. Intrapatient Genomic Stability and Actionability Over Time
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Timmermans, M.; Sonke, G.S.; Van de Vijver, K.K.; van der Aa, M.A.; Kruitwagen, R.F.P.M. No Improvement in Long-Term Survival for Epithelial Ovarian Cancer Patients: A Population-Based Study between 1989 and 2014 in the Netherlands. Eur. J. Cancer 2018, 88, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giornelli, G.H. Management of Relapsed Ovarian Cancer: A Review. SpringerPlus 2016, 5, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giornelli, G.H.; Mandó, P. A Theoretical View of Ovarian Cancer Relapse. EMJ 2017, 2, 128–135. [Google Scholar]
- Colombo, N.; Sessa, C.; du Bois, A.; Ledermann, J.; McCluggage, W.G.; McNeish, I.; Morice, P.; Pignata, S.; Ray-Coquard, I.; Vergote, I.; et al. ESMO-ESGO Consensus Conference Recommendations on Ovarian Cancer: Pathology and Molecular Biology, Early and Advanced Stages, Borderline Tumours and Recurrent Disease. Int. J. Gynecol. Cancer 2019, 30, 672–705. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615, Erratum in 2012, 490, 292. [Google Scholar]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-Genome Characterization of Chemoresistant Ovarian Cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-Cancer Whole-Genome Analyses of Metastatic Solid Tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Angus, L.; Smid, M.; Wilting, S.M.; van Riet, J.; Van Hoeck, A.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.G.; Tjan-Heijnen, V.C.G.; Labots, M.; et al. The Genomic Landscape of Metastatic Breast Cancer Highlights Changes in Mutation and Signature Frequencies. Nat. Genet. 2019, 51, 1450–1458. [Google Scholar] [CrossRef]
- Van Dessel, L.F.; van Riet, J.; Smits, M.; Zhu, Y.; Hamberg, P.; van der Heijden, M.S.; Bergman, A.M.; van Oort, I.M.; de Wit, R.; Voest, E.E.; et al. The Genomic Landscape of Metastatic Castration-Resistant Prostate Cancers Reveals Multiple Distinct Genotypes with Potential Clinical Impact. Nat. Commun. 2019, 10, 5251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macintyre, G.; Goranova, T.E.; De Silva, D.; Ennis, D.; Piskorz, A.M.; Eldridge, M.; Sie, D.; Lewsley, L.-A.; Hanif, A.; Wilson, C.; et al. Copy Number Signatures and Mutational Processes in Ovarian Carcinoma. Nat. Genet. 2018, 50, 1262–1270. [Google Scholar] [CrossRef]
- Nones, K.; Johnson, J.; Newell, F.; Patch, A.M.; Thorne, H.; Kazakoff, S.H.; de Luca, X.M.; Parsons, M.T.; Ferguson, K.; Reid, L.E.; et al. Whole-Genome Sequencing Reveals Clinically Relevant Insights into the Aetiology of Familial Breast Cancers. Ann. Oncol. 2019, 30, 1071–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shale, C.; Baber, J.; Cameron, D.; Wong, M.; Cowley, M.J. Unscrambling Cancer Genomes via Integrated Analysis of Structural Variation and Copy Number. bioRxiv 2020. [Google Scholar] [CrossRef]
- Roepman, P.; de Bruijn, E.; van Lieshout, S.; Schoenmaker, L.; Boelens, M.C.; Dubbink, H.J.; Geurts-Giele, W.R.R.; Groenendijk, F.H.; Huibers, M.M.H.; Kranendonk, M.E.G.; et al. Clinical Validation of Whole Genome Sequencing for Cancer Diagnostics. J. Mol. Diagn. 2021, 23, 816–833. [Google Scholar] [CrossRef]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041.e21. [Google Scholar] [CrossRef]
- Blokzijl, F.; Janssen, R.; van Boxtel, R.; Cuppen, E. MutationalPatterns: Comprehensive Genome-Wide Analysis of Mutational Processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef]
- Nguyen, L.; Martens, J.W.M.; Van Hoeck, A.; Cuppen, E. Pan-Cancer Landscape of Homologous Recombination Deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef]
- Suzuki, R.; Shimodaira, H. Pvclust: An R Package for Assessing the Uncertainty in Hierarchical Clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef]
- Griffith, M.; Spies, N.C.; Krysiak, K.; McMichael, J.F.; Coffman, A.C.; Danos, A.M.; Ainscough, B.J.; Ramirez, C.A.; Rieke, D.T.; Kujan, L.; et al. CIViC Is a Community Knowledgebase for Expert Crowdsourcing the Clinical Interpretation of Variants in Cancer. Nat. Genet. 2017, 49, 170–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer Genome Interpreter Annotates the Biological and Clinical Relevance of Tumor Alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Verweij, J. 11 New Response Evaluation Criteria in Solid Tumors: Recist Guideline Version 1.1. Eur. J. Cancer Suppl. 2009, 7, 5. [Google Scholar] [CrossRef]
- Hanker, L.C.; Loibl, S.; Burchardi, N.; Pfisterer, J.; Meier, W.; Pujade-Lauraine, E.; Ray-Coquard, I.; Sehouli, J.; Harter, P.; du Bois, A.; et al. The Impact of Second to Sixth Line Therapy on Survival of Relapsed Ovarian Cancer after Primary Taxane/Platinum-Based Therapy. Ann. Oncol. 2012, 23, 2605–2612. [Google Scholar] [CrossRef] [PubMed]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation-Positive Women with Ovarian Cancer: A Report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [Green Version]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The Evolutionary History of 2,658 Cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 Facilitates Sensitive and Confident Localization of the Targets of Focal Somatic Copy-Number Alteration in Human Cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledermann, J.A.; Drew, Y.; Kristeleit, R.S. Homologous Recombination Deficiency and Ovarian Cancer. Eur. J. Cancer 2016, 60, 49–58. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Borrego-Soto, G.; Ortiz-López, R.; Rojas-Martínez, A. Ionizing Radiation-Induced DNA Injury and Damage Detection in Patients with Breast Cancer. Genet. Mol. Biol. 2015, 38, 420–432. [Google Scholar] [CrossRef] [PubMed]
- McCluggage, W.G. Morphological Subtypes of Ovarian Carcinoma: A Review with Emphasis on New Developments and Pathogenesis. Pathology 2011, 43, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Menghi, F.; Barthel, F.P.; Yadav, V.; Tang, M.; Ji, B.; Tang, Z.; Carter, G.W.; Ruan, Y.; Scully, R.; Verhaak, R.G.W.; et al. The Tandem Duplicator Phenotype Is a Prevalent Genome-Wide Cancer Configuration Driven by Distinct Gene Mutations. Cancer Cell 2018, 34, 197–210.e5. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Frankum, J.R.; Punta, M.; Lise, S.; Alexander, J.; Chen, Y.; Yap, T.A.; Haider, S.; Tutt, A.N.J.; Lord, C.J. Clinical Reversion Analysis Identifies Hotspot Mutations and Predicted Neoantigens Associated with Therapy Resistance. Cancer Discov. 2020, 10, 1475–1488. [Google Scholar] [CrossRef]
- Etemadmoghadam, D.; deFazio, A.; Beroukhim, R.; Mermel, C.; George, J.; Getz, G.; Tothill, R.; Okamoto, A.; Raeder, M.B.; Harnett, P.; et al. Integrated Genome-Wide DNA Copy Number and Expression Analysis Identifies Distinct Mechanisms of Primary Chemoresistance in Ovarian Carcinomas. Clin. Cancer Res. 2009, 15, 1417–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorski, J.W.; Ueland, F.R.; Kolesar, J.M. CCNE1 Amplification as a Predictive Biomarker of Chemotherapy Resistance in Epithelial Ovarian Cancer. Diagnostics 2020, 10, 279. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel Molecular Subtypes of Serous and Endometrioid Ovarian Cancer Linked to Clinical Outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.P.; Eljanne, M.; Harris, L.; Malik, S.; Seibel, N.L. National Cancer Institute Basket/Umbrella Clinical Trials: MATCH, LungMAP, and Beyond. Cancer J. 2019, 25, 272–281. [Google Scholar] [CrossRef]
- Nangalia, J.; Campbell, P.J. Genome Sequencing during a Patient’s Journey through Cancer. N. Engl. J. Med. 2019, 381, 2145–2156. [Google Scholar] [CrossRef]
- Mandelker, D.; Zhang, L.; Kemel, Y.; Stadler, Z.K.; Joseph, V.; Zehir, A.; Pradhan, N.; Arnold, A.; Walsh, M.F.; Li, Y.; et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. JAMA 2017, 318, 825–835. [Google Scholar] [CrossRef]
- Dion, L.; Carton, I.; Jaillard, S.; Nyangoh Timoh, K.; Henno, S.; Sardain, H.; Foucher, F.; Levêque, J.; de la Motte Rouge, T.; Brousse, S.; et al. The Landscape and Therapeutic Implications of Molecular Profiles in Epithelial Ovarian Cancer. J. Clin. Med. Res. 2020, 9, 2239. [Google Scholar] [CrossRef] [PubMed]
- deFazio, A.; Gao, B.; Mapagu, C.; Moujaber, T.; Harnett, P.R. Chapter 1—Epithelial Ovarian Cancer: Genomic Landscape and Evolving Precision Treatment. In Overcoming Ovarian Cancer Chemoresistance; Samimi, G., Annunziata, C., Eds.; Academic Press: Cambridge, MA, USA, 2021; Volume 11, pp. 1–23. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Median/n | Range/% |
|---|---|---|
| Age at biopsy | 63 | 31–85 |
| Disease status | ||
| primary disease | 19 | 14% |
| recurrent disease | 113 | 86% |
| Biopsy Site | ||
| Peritoneum/omentum | 63 | 48% |
| Lymph node | 33 | 25% |
| Liver | 14 | 11% |
| Skin | 6 | 5% |
| Vagina | 5 | 4% |
| Ovary | 4 | 3% |
| Other/unknown | 7 | 5% |
| Histopathological subtype (at diagnosis) | ||
| High grade serous carcinoma | 74 | 56% |
| Low grade serous carcinoma | 16 | 12% |
| Serous carcinoma, NOS 1 | 13 | 10% |
| Adenocarcinoma, NOS 1 | 6 | 5% |
| Clear cell carcinoma | 5 | 4% |
| Endometrioid carcinoma | 5 | 4% |
| Carcinosarcoma | 3 | 2% |
| Mucinous carcinoma | 2 | 2% |
| Unknown | 8 | 6% |
| Differentiation grade (at diagnosis) | ||
| Well | 18 | 14% |
| Moderate | 13 | 10% |
| Poor | 72 | 55% |
| Unknown | 29 | 22% |
| FIGO stage (at diagnosis) | ||
| I | 4 | 3% |
| II | 9 | 7% |
| III | 69 | 52% |
| IV | 40 | 30% |
| unknown | 10 | 8% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Witte, C.J.; Kutzera, J.; van Hoeck, A.; Nguyen, L.; Boere, I.A.; Jalving, M.; Ottevanger, P.B.; van Schaik-van de Mheen, C.; Stevense, M.; Kloosterman, W.P.; et al. Distinct Genomic Profiles Are Associated with Treatment Response and Survival in Ovarian Cancer. Cancers 2022, 14, 1511. https://doi.org/10.3390/cancers14061511
de Witte CJ, Kutzera J, van Hoeck A, Nguyen L, Boere IA, Jalving M, Ottevanger PB, van Schaik-van de Mheen C, Stevense M, Kloosterman WP, et al. Distinct Genomic Profiles Are Associated with Treatment Response and Survival in Ovarian Cancer. Cancers. 2022; 14(6):1511. https://doi.org/10.3390/cancers14061511
Chicago/Turabian Stylede Witte, Chris J., Joachim Kutzera, Arne van Hoeck, Luan Nguyen, Ingrid A. Boere, Mathilde Jalving, Petronella B. Ottevanger, Christa van Schaik-van de Mheen, Marion Stevense, Wigard P. Kloosterman, and et al. 2022. "Distinct Genomic Profiles Are Associated with Treatment Response and Survival in Ovarian Cancer" Cancers 14, no. 6: 1511. https://doi.org/10.3390/cancers14061511
APA Stylede Witte, C. J., Kutzera, J., van Hoeck, A., Nguyen, L., Boere, I. A., Jalving, M., Ottevanger, P. B., van Schaik-van de Mheen, C., Stevense, M., Kloosterman, W. P., Zweemer, R. P., Cuppen, E., & Witteveen, P. O. (2022). Distinct Genomic Profiles Are Associated with Treatment Response and Survival in Ovarian Cancer. Cancers, 14(6), 1511. https://doi.org/10.3390/cancers14061511