
From Biology to Treatment of Monoclonal Gammopathies of Neurological Significance

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
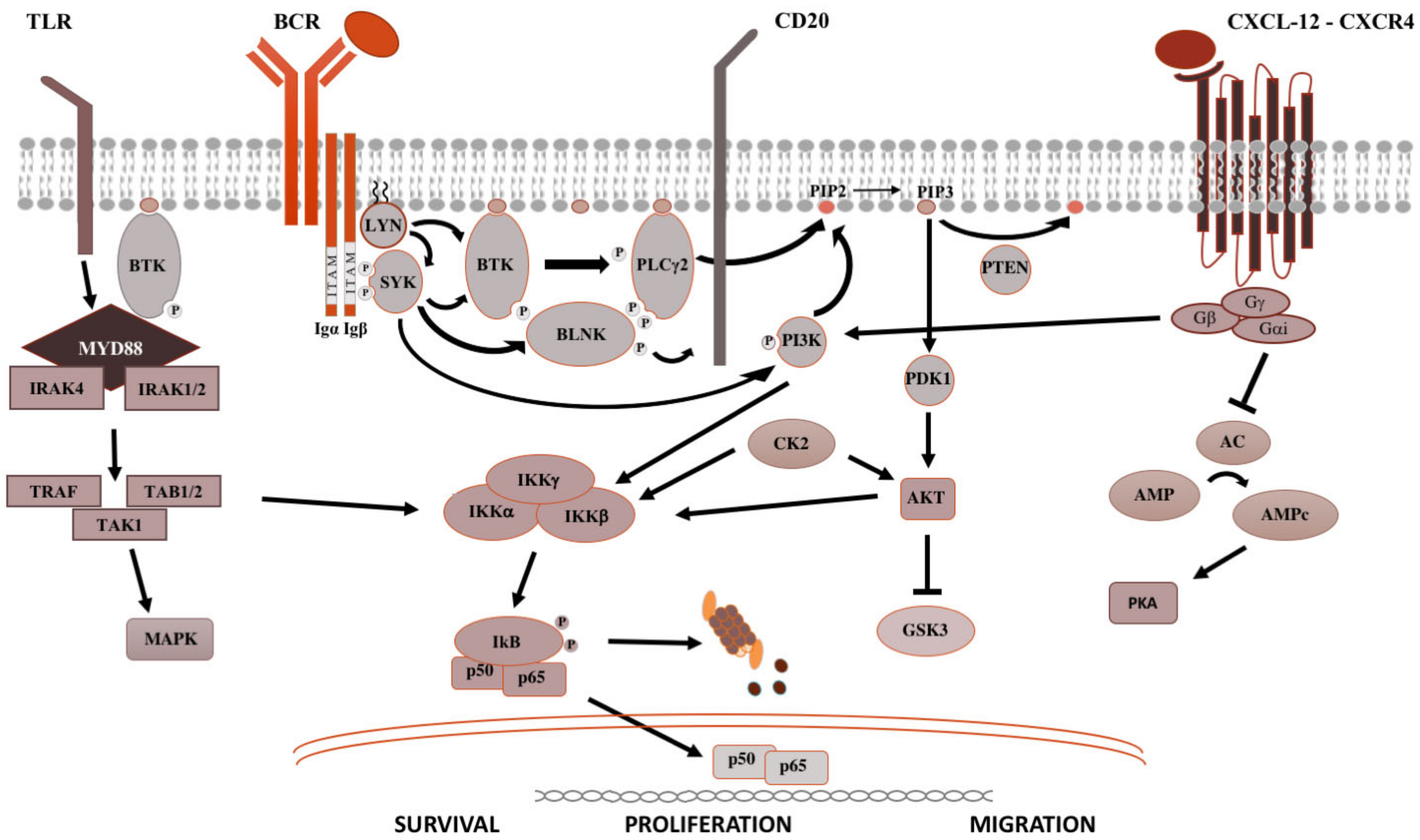
2. Signaling Pathways in B-Cell Malignancies

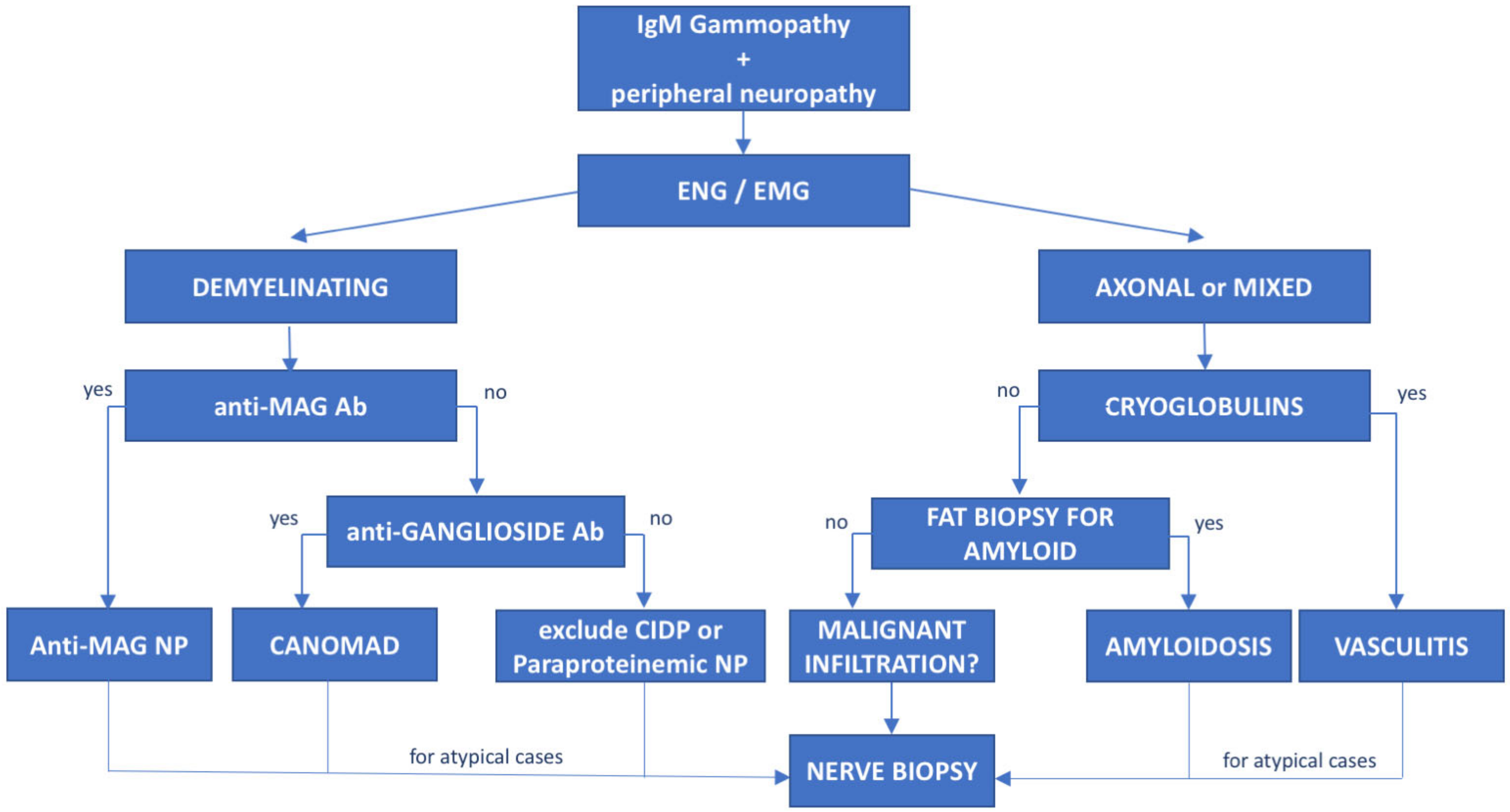
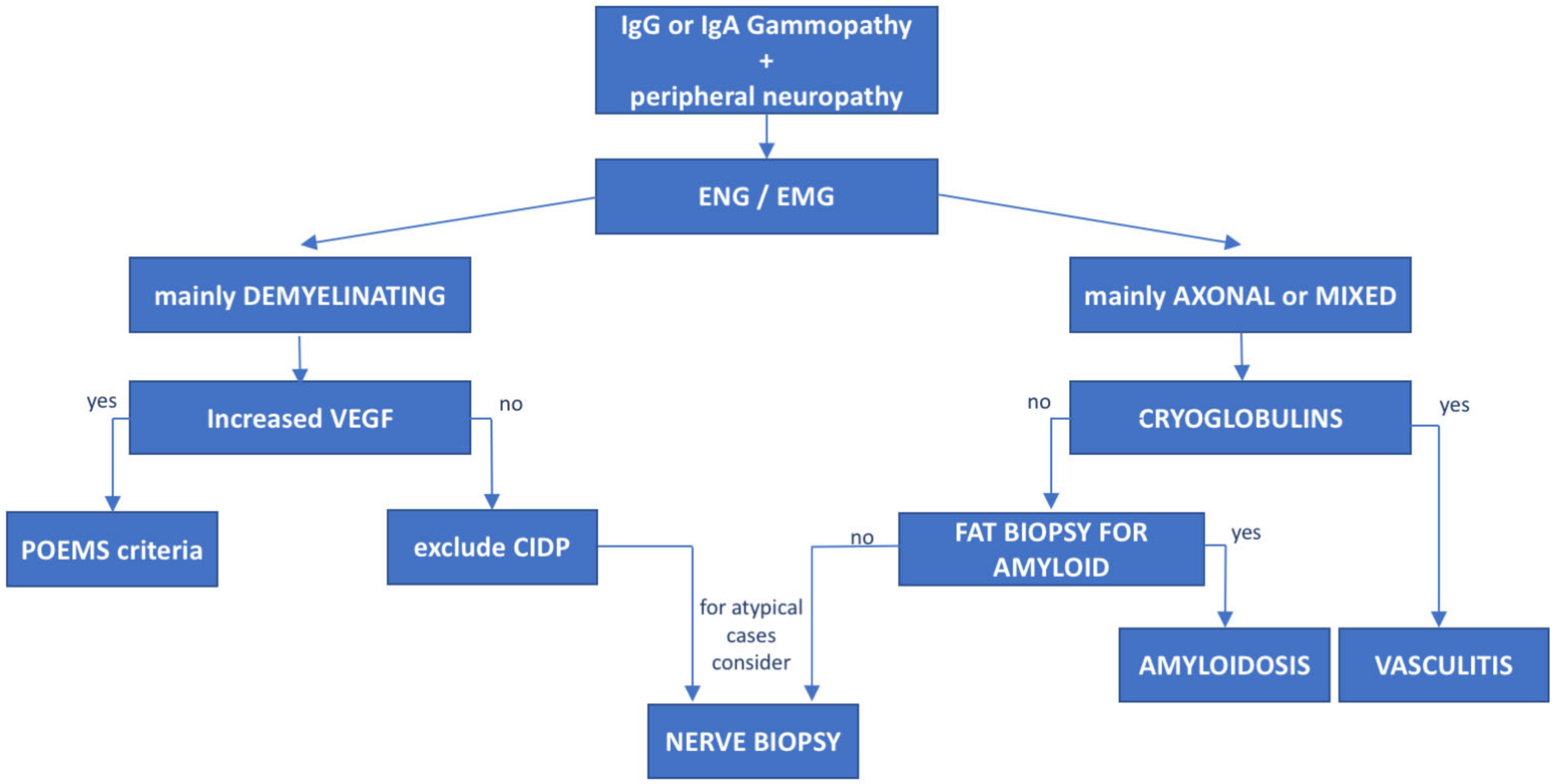
3. Monoclonal Gammopathies of Neurological Significance
4. Monoclonal Gammopathies of Undetermined Significance
5. Waldenström’s Macroglobulinemia/Lymphoplasmacytic Lymphoma
6. Anti-Myelin-Associated Glycoprotein (MAG) Antibody Neuropathy
7. Immunoglobulin Light-Chain (AL) Amyloidosis
8. POEMS Syndrome
9. Castleman’s Disease
10. Non-Hodgkin Lymphoma-Related Peripheral Neuropathies
10.1. Chronic Lymphocytic Leukemia
10.2. Intravascular Lymphoma
10.3. Neurolymphomatosis
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Briani, C.; Visentin, A.; Campagnolo, M.; Salvalaggio, A.; Ferrari, S.; Cavallaro, T.; Manara, R.; Gasparotti, R.; Piazza, F. Peripheral nervous system involvement in lymphomas. J. Peripher. Nerv. Syst. 2019, 24, 5–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piazza, F.; Manni, S.; Arjomand, A.; Visentin, A.; Trentin, L.; Semenzato, G. New responsibilities for aged kinases in B-lymphomas. Hematol. Oncol. 2020, 38, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Diepstraten, S.T.; Anderson, M.A.; Czabotar, P.E.; Lessene, G.; Strasser, A.; Kelly, G.L. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat. Rev. Cancer 2022, 22, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [Green Version]
- De Groen, R.A.L.; Schrader, A.M.R.; Kersten, M.J.; Pals, S.T.; Vermaat, J.S.P. MYD88 in the driver’s seat of B-cell lymphomagenesis: From molecular mechanisms to clinical implications. Haematologica 2019, 104, 2337–2348. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Li, W.; Deng, Q.; Li, L.; Hsi, E.D.; Young, K.H.; Zhang, M.; Li, Y. MYD88 L265P Mutation in Lymphoid Malignancies. Cancer Res. 2018, 78, 2457–2462. [Google Scholar] [CrossRef] [Green Version]
- Dubois, S.; Viailly, P.-J.; Bohers, E.; Bertrand, P.; Ruminy, P.; Marchand, V.; Maingonnat, C.; Mareschal, S.; Picquenot, J.-M.; Penther, D.; et al. Biological and Clinical Relevance of Associated Genomic Alterations in MYD88 L265P and non-L265P-Mutated Diffuse Large B-Cell Lymphoma: Analysis of 361 Cases. Clin. Cancer Res. 2017, 23, 2232–2244. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-C.; Lo, Y.-C.; Wu, H. Helical assembly in the MyD88–IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Severin, F.; Frezzato, F.; Visentin, A.; Martini, V.; Trimarco, V.; Carraro, S.; Tibaldi, E.; Brunati, A.M.; Piazza, F.; Semenzato, G.; et al. In Chronic Lymphocytic Leukemia the JAK2/STAT3 Pathway Is Constitutively Activated and Its Inhibition Leads to CLL Cell Death Unaffected by the Protective Bone Marrow Microenvironment. Cancers 2019, 11, 1939. [Google Scholar] [CrossRef] [Green Version]
- Frezzato, F.; Raggi, F.; Martini, V.; Severin, F.; Trimarco, V.; Visentin, A.; Scomazzon, E.; Accordi, B.; Bresolin, S.; Piazza, F.; et al. HSP70/HSF1 axis, regulated via a PI3K/AKT pathway, is a druggable target in chronic lymphocytic leukemia. Int. J. Cancer 2019, 145, 3089–3100. [Google Scholar] [CrossRef]
- Martini, V.; Gattazzo, C.; Frezzato, F.; Trimarco, V.; Pizzi, M.; Chiodin, G.; Severin, F.; Scomazzon, E.; Guzzardo, V.; Saraggi, D.; et al. Cortactin, a Lyn substrate, is a checkpoint molecule at the intersection of BCR and CXCR4 signalling pathway in chronic lymphocytic leukaemia cells. Br. J. Haematol. 2017, 178, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treon, S.P.; Xu, L.; Liu, X.; Hunter, Z.R.; Yang, G.; Castillo, J.J. Genomic Landscape of Waldenström Macroglobulinemia. Hematol. Oncol. Clin. N. Am. 2018, 32, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Ohwada, C.; Takeuchi, M.; Takeda, Y.; Tsukamoto, S.; Mimura, N.; Nagisa, O.-H.; Sugita, Y.; Tanaka, H.; Wakita, H.; et al. Detection of MYD88 L265P mutation by next-generation deep sequencing in peripheral blood mononuclear cells of Waldenström’s macroglobulinemia and IgM monoclonal gammopathy of undetermined significance. PLoS ONE 2019, 14, e0221941. [Google Scholar] [CrossRef] [PubMed]
- Bagratuni, T.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Mavrianou-Koutsoukou, N.; Liacos, C.; Patseas, D.; Kanellias, N.; Migkou, M.; Ziogas, D.C.; Eleutherakis-Papaiakovou, E.; et al. Detection of MYD88 and CXCR4 mutations in cell-free DNA of patients with IgM monoclonal gammopathies. Leukemia 2018, 32, 2617–2625. [Google Scholar] [CrossRef]
- Carroll, A.S.; Lunn, M.P.T. Paraproteinaemic neuropathy: MGUS and beyond. Pract. Neurol. 2021, 21, 492–503. [Google Scholar] [CrossRef]
- Kelly, J.J., Jr.; Kyle, R.A.; O’Brien, P.C.; Dyck, P.J. Prevalence of monoclonal protein in peripheral neuropathy. Neurology 1981, 31, 1480–1483. [Google Scholar] [CrossRef]
- Hanewinckel, R.; Drenthen, J.; van Oijen, M.; Hofman, A.; van Doorn, P.A.; Ikram, M.A. Prevalence of polyneuropathy in the general middle-aged and elderly population. Neurology 2016, 87, 1892–1898. [Google Scholar] [CrossRef]
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2018, 378, 241–249. [Google Scholar] [CrossRef]
- Varettoni, M.; Zibellini, S.; Boveri, E.; Klersy, C.; Candido, C.; Rattotti, S.; Ferretti, V.V.; DeFrancesco, I.; Mangiacavalli, S.; Nizzoli, M.E.; et al. A risk-stratification model based on the initial concentration of the serum monoclonal protein and MYD 88 mutation status identifies a subset of patients with IgM monoclonal gammopathy of undetermined significance at high risk of progression to Waldenström macroglobulinaemia or other lymphoproliferative disorders. Br. J. Haematol. 2019, 187, 441–446. [Google Scholar] [CrossRef]
- Steck, A.J. Anti-MAG neuropathy: From biology to clinical management. J. Neuroimmunol. 2021, 361, 577725. [Google Scholar] [CrossRef]
- Latov, N. Antibody testing in neuropathy associated with anti-Myelin-Associated Glycoprotein antibodies: Where we are after 40 years. Curr. Opin. Neurol. 2021, 34, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Boso, F.; Ruggero, S.; Giannotta, C.; Benedetti, L.; Marfia, G.A.; Ermani, M.; Campagnolo, M.; Salvalaggio, A.; Gallia, F.; De Michelis, C.; et al. Anti-sulfatide/galactocerebroside antibodies in immunoglobulin M paraproteinemic neuropathies. Eur. J. Neurol. 2017, 24, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Niermeijer, J.; Fischer, K.; Eurelings, M.; Franssen, H.; Wokke, J.H.; Notermans, N.C. Prognosis of polyneuropathy due to IgM monoclonal gammopathy: A prospective cohort study. Neurology 2010, 74, 406–412. [Google Scholar] [CrossRef] [PubMed]
- LaRue, S.; Bombelli, F.; Viala, K.; Neil, J.; Maisonobe, T.; Bouche, P.; Musset, L.; Fournier, E.; Léger, J.M. Non-anti-MAG DADS neuropathy as a variant of CIDP: Clinical, electrophysiological, laboratory features and response to treatment in 10 cases. Eur. J. Neurol. 2011, 18, 899–905. [Google Scholar] [CrossRef]
- Frustaci, A.M.; Rusconi, C.; Picardi, P.; Veronese, S.M.; Montillo, M.; Cairoli, R.; Tedeschi, A. Bing Neel Syndrome in a Previously Untreated Patient with Waldenström’s Macroglobulinemia: Contribution of MYD88 L265P Mutation on Cerebrospinal Fluid. Clin. Lymphoma Myeloma Leuk. 2016, 16, e7–e9. [Google Scholar] [CrossRef]
- Castillo, J.J.; Itchaki, G.; Paludo, J.; Varettoni, M.; Buske, C.; Eyre, T.A.; Chavez, J.C.; Shain, K.H.; Issa, S.; Palomba, M.L.; et al. Ibrutinib for the treatment of Bing-Neel syndrome: A multicenter study. Blood 2019, 133, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Campagnolo, M.; Ferrari, S.; Dalla Torre, C.; Cabrini, I.; Cacciavillani, M.; Lucchetta, M.; Ruggero, S.; Toffanin, E.; Cavallaro, T.; Briani, C. Polyneuropathy with anti-sulfatide and anti-MAG antibodies: Clinical, neurophysiological, pathological features and response to treatment. J. Neuroimmunol. 2015, 281, 1–4. [Google Scholar] [CrossRef]
- Le Cann, M.; Bouhour, F.; Viala, K.; Simon, L.; Tard, C.; Rossi, C.; Morel, G.; Lagrange, E.; Magy, L.; Créange, A.; et al. CANOMAD: A neurological monoclonal gammopathy of clinical significance that benefits from B-cell-targeted therapies. Blood 2020, 136, 2428–2436. [Google Scholar] [CrossRef]
- Muchtar, E.; Magen, H.; Gertz, M.A. How I treat cryoglobulinemia. Blood 2017, 129, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Latov, N. Diagnosis and treatment of chronic acquired demyelinating polyneuropathies. Nat. Rev. Neurol. 2014, 10, 435–446. [Google Scholar] [CrossRef]
- Campagnolo, M.; Ruiz, M.; Falzone, Y.M.; Ermani, M.; Bianco, M.; Martinelli, D.; Cerri, F.; Quattrini, A.; Salvalaggio, A.; Castellani, F.; et al. Limitations in daily activities and general perception of quality of life: Long term follow-up in patients with anti-myelin-glycoprotein antibody polyneuropathy. J. Peripher. Nerv. Syst. 2019, 24, 276–282. [Google Scholar] [CrossRef]
- Lunn, M.P.; Nobile-Orazio, E. Immunotherapy for IgM anti-myelin-associated glycoprotein paraprotein-associated peripheral neuropathies. Cochrane Database Syst. Rev. 2010, 2016, CD002827. [Google Scholar] [CrossRef] [Green Version]
- Dalakas, M.C.; Rakocevic, G.; Salajegheh, M.; Dambrosia, J.M.; Hahn, A.F.; Raju, R.; McElroy, B. Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein antibody demyelinating neuropathy. Ann. Neurol. 2009, 65, 286–293. [Google Scholar] [CrossRef]
- Léger, J.-M.; Viala, K.; Nicolas, G.; Créange, A.; Vallat, J.-M.; Pouget, J.; Clavelou, P.; Vial, C.; Steck, A.; Musset, L.; et al. Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein neuropathy. Neurology 2013, 80, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Campagnolo, M.; Zambello, R.; Nobile-Orazio, E.; Benedetti, L.; Marfia, G.A.; Riva, N.; Castellani, F.; Bianco, M.; Salvalaggio, A.; Garnero, M.; et al. IgM MGUS and Waldenstrom-associated anti-MAG neuropathies display similar response to rituximab therapy. J. Neurol. Neurosurg. Psychiatry 2017, 88, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Hospital, M.-A.; Viala, K.; Dragomir, S.; Levy, V.; Cohen-Aubart, F.; Neil, J.; Musset, L.; Choquet, S.; Leger, J.-M.; Leblond, V. Immunotherapy-based regimen in anti-MAG neuropathy: Results in 45 patients. Haematologica 2013, 98, e155–e157. [Google Scholar] [CrossRef] [PubMed]
- Gruson, B.; Ghomari, K.; Beaumont, M.; Garidi, R.; Just, A.; Merle, P.; Merlusca, L.; Marolleau, J.P.; Royer, B. Long-term response to rituximab and fludarabine combination in IgM anti-myelin-associated glycoprotein neuropathy. J. Peripher. Nerv. Syst. 2011, 16, 180–185. [Google Scholar] [CrossRef]
- Massa, F.; Zuppa, A.; Pesce, G.; Demichelis, C.; Bergamaschi, M.; Garnero, M.; Briani, C.; Ferrari, S.; Schenone, A.; Benedetti, L. Bendamustine-rituximab (BR) combined therapy for treatment of immuno-mediated neuropathies associated with hematologic malignancy. J. Neurol. Sci. 2020, 413, 116777. [Google Scholar] [CrossRef]
- Rakocevic, G.; Martinez-Outschoorn, U.; Dalakas, M.C. Obinutuzumab, a potent anti-B-cell agent, for rituximab-unresponsive IgM anti-MAG neuropathy. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e460. [Google Scholar] [CrossRef] [Green Version]
- Briani, C.; Visentin, A.; Salvalaggio, A.; Cacciavillani, M.; Trentin, L. Obinutuzumab, a new anti-CD20 antibody, and chlorambucil are active and effective in anti-myelin-associated glycoprotein antibody polyneuropathy. Eur. J. Neurol. 2019, 26, 371–375. [Google Scholar] [CrossRef]
- Castellani, F.; Visentin, A.; Campagnolo, M.; Salvalaggio, A.; Cacciavillani, M.; Candiotto, C.; Bertorelle, R.; Trentin, L.; Briani, C. The Bruton tyrosine kinase inhibitor ibrutinib improves anti-MAG antibody polyneuropathy. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, R.G.; McCarthy, H.; Rule, S.; D’Sa, S.; Thomas, S.K.; Tournilhac, O.; Forconi, F.; Kersten, M.J.; Zinzani, P.L.; Iyengar, S.; et al. Acalabrutinib monotherapy in patients with Waldenström macroglobulinemia: A single-arm, multicentre, phase 2 study. Lancet Haematol. 2020, 7, e112–e121. [Google Scholar] [CrossRef]
- Tam, C.S.; Opat, S.; D’Sa, S.; Jurczak, W.; Lee, H.-P.; Cull, G.; Owen, R.G.; Marlton, P.; Wahlin, B.E.; Sanz, R.G.; et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: The ASPEN study. Blood 2020, 136, 2038–2050. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Dimopoulos, M.A.; Garcia-Sanz, R.; Trotman, J.; Opat, S.; Roberts, A.W.; Owen, R.G.; Song, Y.; Xu, W.; Zhu, J.; et al. Pooled safety analysis of zanubrutinib monotherapy in patients with B-cell malignancies. Blood Adv. 2022, 6, 1296–1308. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Sanz, R.G.; Lee, H.-P.; Trneny, M.; Varettoni, M.; Opat, S.; D’Sa, S.; Owen, R.G.; Cull, G.; Mulligan, S.; et al. Zanubrutinib for the treatment of MYD88 wild-type Waldenström macroglobulinemia: A substudy of the phase 3 ASPEN trial. Blood Adv. 2020, 4, 6009–6018. [Google Scholar] [CrossRef]
- Kater, A.P.; Wu, J.Q.; Kipps, T.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Robak, T.; de la Serna, J.; et al. Venetoclax Plus Rituximab in Relapsed Chronic Lymphocytic Leukemia: 4-Year Results and Evaluation of Impact of Genomic Complexity and Gene Mutations from the MURANO Phase III Study. J. Clin. Oncol. 2020, 38, 4042–4054. [Google Scholar] [CrossRef]
- Castillo, J.J.; Allan, J.N.; Siddiqi, T.; Advani, R.H.; Meid, K.; Leventoff, C.; White, T.P.; Flynn, C.A.; Sarosiek, S.; Branagan, A.R.; et al. Venetoclax in Previously Treated Waldenström Macroglobulinemia. J. Clin. Oncol. 2022, 40, 63–71. [Google Scholar] [CrossRef]
- Gertz, M.A.; Dispenzieri, A. Systemic Amyloidosis Recognition, Prognosis, and Therapy: A Systematic Review. JAMA 2020, 324, 79–89. [Google Scholar] [CrossRef]
- Milani, P.; Merlini, G. Monoclonal IgM-related AL amyloidosis. Best Pract. Res. Clin. Haematol. 2016, 29, 241–248. [Google Scholar] [CrossRef]
- Briani, C.; Ferrari, S.; Campagnolo, M.; Tagliapietra, M.; Castellani, F.; Salvalaggio, A.; Mariotto, S.; Visentin, A.; Cavallaro, T. Mechanisms of Nerve Damage in Neuropathies Associated with Hematological Diseases: Lesson from Nerve Biopsies. Brain Sci. 2021, 11, 132. [Google Scholar] [CrossRef]
- Merlini, G.; Dispenzieri, A.; Sanchorawala, V.; Schönland, S.O.; Palladini, G.; Hawkins, P.N.; Gertz, M.A. Systemic immunoglobulin light chain amyloidosis. Nat. Rev. Dis. Primers 2018, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Obici, L.; Adams, D. Acquired and inherited amyloidosis: Knowledge driving patients’ care. J. Peripher. Nerv. Syst. 2020, 25, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Manni, S.; Brancalion, A.; Mandato, E.; Tubi, L.Q.; Colpo, A.; Pizzi, M.; Cappellesso, R.; Zaffino, F.; Di Maggio, S.A.; Cabrelle, A.; et al. Protein Kinase CK2 Inhibition Down Modulates the NF-κB and STAT3 Survival Pathways, Enhances the Cellular Proteotoxic Stress and Synergistically Boosts the Cytotoxic Effect of Bortezomib on Multiple Myeloma and Mantle Cell Lymphoma Cells. PLoS ONE 2013, 8, e75280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastritis, E.; Palladini, G.; Minnema, M.C.; Wechalekar, A.D.; Jaccard, A.; Lee, H.C.; Sanchorawala, V.; Gibbs, S.; Mollee, P.; Venner, C.P.; et al. Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N. Engl. J. Med. 2021, 385, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A. POEMS syndrome: 2021 Update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2021, 96, 872–888. [Google Scholar] [CrossRef]
- Briani, C.; Campagnolo, M.; Luigetti, M.; Lessi, F.; Adami, F. POEMS syndrome. In Dysimmune Neuropathies; Academic Press: Cambridge, MA, USA, 2020; pp. 129–143. [Google Scholar] [CrossRef]
- Briani, C.; Fabrizi, G.M.; Ruggero, S.; Torre, C.D.; Ferrarini, M.; Campagnolo, M.; Cavallaro, T.; Ferrari, S.; Scarlato, M.; Taioli, F.; et al. Vascular endothelial growth factor helps differentiate neuropathies in rare plasma cell dyscrasias. Muscle Nerve 2011, 43, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Briani, C.; Torre, C.D.; Lessi, F.; Cavallaro, T.; Scarlato, M.; Ferrari, S.; Campagnolo, M.; Lucchetta, M.; Cabrini, I.; Morbin, M.; et al. Pentraxin-3 and VEGF in POEMS syndrome: A 2-year longitudinal study. J. Neuroimmunol. 2014, 277, 189–192. [Google Scholar] [CrossRef]
- Scarlato, M.; Previtali, S.C.; Carpo, M.; Pareyson, D.; Briani, C.; Del Bo, R.; Nobile-Orazio, E.; Quattrini, A.; Comi, G.P. Polyneuropathy in POEMS syndrome: Role of angiogenic factors in the pathogenesis. Brain 2005, 128, 1911–1920. [Google Scholar] [CrossRef] [Green Version]
- Nagao, Y.; Mimura, N.; Takeda, J.; Yoshida, K.; Shiozawa, Y.; Oshima, M.; Aoyama, K.; Saraya, A.; Koide, S.; Rizq, O.; et al. Genetic and transcriptional landscape of plasma cells in POEMS syndrome. Leukemia 2019, 33, 1723–1735. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kikuchi, J. Molecular basis of clonal evolution in multiple myeloma. Int. J. Hematol. 2020, 111, 496–511. [Google Scholar] [CrossRef] [Green Version]
- Briani, C.; Fedrigo, M.; Manara, R.; Castellani, C.; Zambello, R.; Citton, V.; Campagnolo, M.; Torre, C.D.; Lucchetta, M.; Orvieto, E.; et al. Pachymeningeal involvement in POEMS syndrome: MRI and histopathological study. J. Neurol. Neurosurg. Psychiatry 2012, 83, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Ziff, O.J.; Hoskote, C.; Keddie, S.; D’Sa, S.; Davangnanam, I.; Lunn, M.P.T. Frequent central nervous system, pachymeningeal and plexus MRI changes in POEMS syndrome. J. Neurol. 2019, 266, 1067–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briani, C.; Manara, R.; Lessi, F.; Citton, V.; Zambello, R.; Adami, F. Pachymeningeal involvement in POEMS syndrome: Dramatic cerebral MRI improvement after lenalidomide therapy. Am. J. Hematol. 2012, 87, 539–541. [Google Scholar] [CrossRef]
- Guibert, C.; Richard, L.; Durand, S.; Maquin, F.; Demiot, C.; Vallat, J.-M.; Jaccard, A.; Magy, L.; Duchesne, M. Skin and Nerve Neovascularization in POEMS Syndrome: Insights from a Small Cohort. J. Neuropathol. Exp. Neurol. 2020, 79, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Cerri, F.; Falzone, Y.M.; Riva, N.; Quattrini, A. An update on the diagnosis and management of the polyneuropathy of POEMS syndrome. J. Neurol. 2019, 266, 258–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karam, C.; Klein, C.J.; Dispenzieri, A.; Dyck, P.J.B.; Mandrekar, J.; D’Souza, A.; Mauermann, M.L. Polyneuropathy improvement following autologous stem cell transplantation for POEMS syndrome. Neurology 2015, 84, 1981–1987. [Google Scholar] [CrossRef] [Green Version]
- Autore, F.; Piccirillo, N.; Nozza, A.; Innocenti, I.; Putzulu, R.; Chiusolo, P.; Sora, F.; Zini, G.; Bacigalupo, A.; Castagna, L.; et al. Peripheral Blood Hemopoietic Stem Cell Mobilization Regimens in POEMS Syndrome: A Retrospective Study at 2 Hematologic Italian Centers. Biol. Blood Marrow Transplant. 2019, 25, 2514–2516. [Google Scholar] [CrossRef]
- Zhao, H.; Huang, X.-F.; Gao, X.-M.; Cai, H.; Zhang, L.; Feng, J.; Cao, X.-X.; Zhou, D.-B.; Li, J. What is the best first-line treatment for POEMS syndrome: Autologous transplantation, melphalan and dexamethasone, or lenalidomide and dexamethasone? Leukemia 2019, 33, 1023–1029. [Google Scholar] [CrossRef]
- Kuwabara, S.; Misawa, S.; Kanai, K.; Suzuki, Y.; Kikkawa, Y.; Sawai, S.; Hattori, T.; Nishimura, M.; Nakaseko, C. Neurologic improvement after peripheral blood stem cell transplantation in POEMS syndrome. Neurology 2008, 71, 1691–1695. [Google Scholar] [CrossRef]
- Autore, F.; Innocenti, I.; Luigetti, M.; Piccirillo, N.; Sora, F.; Chiusolo, P.; Sica, S.; Bacigalupo, A.; Laurenti, L. Autologous peripheral blood stem cell transplantation and the role of lenalidomide in patients affected by poems syndrome. Hematol. Oncol. 2018, 36, 392–398. [Google Scholar] [CrossRef]
- Nozza, A.; Terenghi, F.; Gallia, F.; Adami, F.; Briani, C.; Merlini, G.; Giordano, L.; Santoro, A.; Nobile-Orazio, E. Lenalidomide and dexamethasone in patients with POEMS syndrome: Results of a prospective, open-label trial. Br. J. Haematol. 2017, 179, 748–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, K.; Yang, J.-R.; Li, J.; Wei, Q.; Yang, Y.-M.; Liu, T.; Niu, T. Effective Induction Therapy with Subcutaneous Administration of Bortezomib for Newly Diagnosed POEMS Syndrome: A Case Report and a Review of the Literature. Acta Haematol. 2013, 129, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Riva, M.; Lessi, F.; Berno, T.; Visentin, A.; Campagnolo, M.; Semenzato, G.; Adami, F.; Briani, C. Bortezomib-based regimens in patients with POEMS syndrome: A case series in newly diagnosed and relapsed patients. Leuk. Lymphoma 2019, 60, 2067–2070. [Google Scholar] [CrossRef]
- Khan, M.; Stone, K.; van Rhee, F. Daratumumab for POEMS Syndrome. Mayo Clin. Proc. 2018, 93, 542–544. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, Q.; Xu, H.; Gu, C.; Jiang, L.; Wang, J.; Wang, D.; Xu, B.; Mao, X.; Wang, J.; et al. Anti-BCMA CAR-T cells for treatment of plasma cell dyscrasia: Case report on POEMS syndrome and multiple myeloma. J. Hematol. Oncol. 2018, 11, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rhee, F.; Greenway, A.; Stone, K. Treatment of Idiopathic Castleman Disease. Hematol. Oncol. Clin. N. Am. 2018, 32, 89–106. [Google Scholar] [CrossRef]
- Liu, A.; Nabel, C.S.; Finkelman, B.; Ruth, J.R.; Kurzrock, R.; van Rhee, F.; Krymskaya, V.P.; Kelleher, D.; Rubenstein, A.H.; Fajgenbaum, D.C. Idiopathic multicentric Castleman’s disease: A systematic literature review. Lancet Haematol. 2016, 3, e163–e175. [Google Scholar] [CrossRef]
- Naddaf, E.; Dispenzieri, A.; Mandrekar, J.; Mauermann, M.L. Clinical spectrum of Castleman disease-associated neuropathy. Neurology 2016, 87, 2457–2462. [Google Scholar] [CrossRef] [Green Version]
- van Rhee, F.; Wong, R.; Munshi, N.; Rossi, J.-F.; Ke, X.-Y.; Fosså, A.; Simpson, D.; Capra, M.; Liu, T.; Hsieh, R.K.; et al. Siltuximab for multicentric Castleman’s disease: A randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014, 15, 966–974. [Google Scholar] [CrossRef]
- Murakami, M.; Johkoh, T.; Hayashi, S.; Ohshima, S.; Mizuki, M.; Nakatsuka, S.-I.; Tomobe, M.; Kuroyanagi, K.; Nakasone, A.; Nishimoto, N. Clinicopathologic characteristics of 342 patients with multicentric Castleman disease in Japan. Mod. Rheumatol. 2020, 30, 843–851. [Google Scholar] [CrossRef] [Green Version]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visentin, A.; Facco, M.; Frezzato, F.; Castelli, M.; Trimarco, V.; Martini, V.; Gattazzo, C.; Severin, F.; Chiodin, G.; Martines, A.; et al. Integrated CLL Scoring System, a New and Simple Index to Predict Time to Treatment and Overall Survival in Patients with Chronic Lymphocytic Leukemia. Clin. Lymphoma Myeloma Leuk. 2015, 15, 612–620.e5. [Google Scholar] [CrossRef] [PubMed]
- Raponi, S.; Del Giudice, I.; Marinelli, M.; Wang, J.; Cafforio, L.; Ilari, C.; Piciocchi, A.; Messina, M.; Bonina, S.; Tavolaro, S.; et al. Genetic landscape of ultra-stable chronic lymphocytic leukemia patients. Ann. Oncol. 2018, 29, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Visentin, A.; Facco, M.; Gurrieri, C.; Pagnin, E.; Martini, V.; Imbergamo, S.; Frezzato, F.; Trimarco, V.; Severin, F.; Raggi, F.; et al. Prognostic and Predictive Effect of IGHV Mutational Status and Load in Chronic Lymphocytic Leukemia: Focus on FCR and BR Treatments. Clin. Lymphoma Myeloma Leuk. 2019, 19, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Visentin, A.; Bonaldi, L.; Rigolin, G.M.; Mauro, F.R.; Martines, A.; Frezzato, F.; Imbergamo, S.; Scomazzon, E.; Pravato, S.; Bardi, M.A.; et al. The combination of complex karyotype subtypes and IGHV mutational status identifies new prognostic and predictive groups in chronic lymphocytic leukaemia. Br. J. Cancer 2019, 121, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Visentin, A.; Bonaldi, L.; Rigolin, G.M.; Mauro, F.R.; Martines, A.; Frezzato, F.; Pravato, S.; Gargarella, L.R.; Bardi, M.A.; Cavallari, M.; et al. The complex karyotype landscape in chronic lymphocytic leukemia allows to refine the risk of Richter syndrome transformation. Haematologica 2021. [Google Scholar] [CrossRef]
- Mauro, F.R.; Caputo, M.D.; Rosati, S.; Pepe, S.; De Benedittis, D.; De Luca, M.L.; Foà, R. Balancing efficacy and toxicity of targeted agents currently used for the treatment of patients with chronic lymphocytic leukemia. Expert Rev. Hematol. 2018, 11, 601–611. [Google Scholar] [CrossRef]
- Visentin, A.; Mauro, F.R.; Cibien, F.; Vitale, C.; Reda, G.; Fresa, A.; Ciolli, S.; Pietrasanta, D.; Marchetti, M.; Murru, R.; et al. Continuous treatment with Ibrutinib in 100 untreated patients with TP 53 disrupted chronic lymphocytic leukemia: A real-life campus CLL study. Am. J. Hematol. 2021, 1–5. [Google Scholar] [CrossRef]
- Corbingi, A.; Innocenti, I.; Tomasso, A.; Pasquale, R.; Visentin, A.; Varettoni, M.; Flospergher, E.; Autore, F.; Morelli, F.; Trentin, L.; et al. Monoclonal gammopathy and serum immunoglobulin levels as prognostic factors in chronic lymphocytic leukaemia. Br. J. Haematol. 2020, 190, 901–908. [Google Scholar] [CrossRef]
- Briani, C.; Visentin, A.; Salvalaggio, A.; Imbergamo, S.; Piazza, F.; Cacciavillani, M.; Campagnolo, M.; Frezzato, F.; Semenzato, G.; Trentin, L. Peripheral neuropathies in chronic lymphocytic leukemia: A single center experience on 816 patients. Haematologica 2017, 102, e140–e143. [Google Scholar] [CrossRef] [Green Version]
- Briani, C.; Visentin, A.; Cavallaro, T.; Cacciavillani, M.; Cabrini, I.; Ferrari, S.; Zambello, R.; Trentin, L. Primary neurolymphomatosis as clinical onset of chronic lymphocytic leukemia. Ann. Hematol. 2016, 96, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Visentin, A.; Briani, C.; Imbergamo, S.; Frezzato, F.; Angelini, A.; Fedrigo, M.; Cacciavillani, M.; Altinier, S.; Piazza, F.; Semenzato, G.; et al. Idelalisib plus rituximab is effective in systemic AL amyloidosis secondary to chronic lymphocytic leukaemia. Hematol. Oncol. 2018, 36, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, P.Y.K.; Doorn, P.A.; Hadden, R.D.M.; Avau, B.; Vankrunkelsven, P.; Allen, J.A.; Attarian, S.; Blomkwist-Markens, P.H.; Cornblath, D.R.; Eftimov, F.; et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint Task Force—Second revision. J. Peripher. Nerv. Syst. 2021, 26, 242–268. [Google Scholar] [CrossRef] [PubMed]
- Visentin, A.; Deodato, M.; Mauro, F.R.; Autore, F.; Reda, G.; Vitale, C.; Molica, S.; Rigolin, G.M.; Piazza, F.; Cesini, L.; et al. A scoring system to predict the risk of atrial fibrillation in chronic lymphocytic leukemia. Hematol. Oncol. 2019, 37, 508–512. [Google Scholar] [CrossRef]
- Wrotnowski, U.; Mills, S.E.; Cooper, P.H. Malignant Angioendotheliomatosis: An Angiotropic Lymphoma? Am. J. Clin. Pathol. 1985, 83, 244–248. [Google Scholar] [CrossRef] [Green Version]
- Ponzoni, M.; Campo, E.; Nakamura, S. Intravascular large B-cell lymphoma: A chameleon with multiple faces and many masks. Blood 2018, 132, 1561–1567. [Google Scholar] [CrossRef]
- Hundsberger, T.; Cogliatti, S.; Kleger, G.-R.; Fretz, C.; Gähler, A.; Anliker, M.; Fournier, J.-Y.; Von Moos, R.; Tettenborn, B.; Driessen, C. Intravascular Lymphoma Mimicking Cerebral Stroke: Report of Two Cases. Case Rep. Neurol. 2011, 3, 278–283. [Google Scholar] [CrossRef] [Green Version]
- Schrader, A.M.R.; Jansen, P.M.; Willemze, R.; Vermeer, M.; Cleton-Jansen, A.-M.; Somers, S.F.; Veelken, H.; van Eijk, R.; Kraan, W.; Kersten, M.J.; et al. High prevalence of MYD88 and CD79B mutations in intravascular large B-cell lymphoma. Blood 2018, 131, 2086–2089. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.L.; Pytel, P.; Rowin, J. Disseminated intravascular large-cell lymphoma with initial presentation mimicking Guillain-Barre syndrome. Muscle Nerve 2010, 42, 133–136. [Google Scholar] [CrossRef]
- Lynch, K.M.; Katz, J.D.; Weinberg, D.H.; Lin, D.; Folkerth, R.D. Isolated Mononeuropathy Multiplex—A Rare Manifestation of Intravascular Large B-Cell Lymphoma. J. Clin. Neuromuscul. Dis. 2012, 14, 17–20. [Google Scholar] [CrossRef]
- Fukami, Y.; Koike, H.; Iijima, M.; Hagita, J.; Niwa, H.; Nishi, R.; Kawagashira, Y.; Katsuno, M. Demyelinating Neuropathy Due to Intravascular Large B-cell Lymphoma. Intern. Med. 2020, 59, 435–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grommes, C.; Pastore, A.; Palaskas, N.; Tang, S.S.; Campos, C.; Schartz, D.; Codega, P.; Nichol, D.; Clark, O.; Hsieh, W.-Y.; et al. Ibrutinib Unmasks Critical Role of Bruton Tyrosine Kinase in Primary CNS Lymphoma. Cancer Discov. 2017, 7, 1018–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lhermitte, J.; Trelles, J. Neurolymphomatose périphérique humaine. Presse Med. 1934, 42, 289–292. [Google Scholar]
- Baehring, J.M.; Damek, D.; Martin, E.C.; Betensky, R.A.; Hochberg, F.H. Neurolymphomatosis. Neuro Oncol. 2003, 5, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Campagnolo, M.; Cacciavillani, M.; Cavallaro, T.; Ferrari, S.; Gasparotti, R.; Zambello, R.; Briani, C. Neurolymphomatosis, a rare manifestation of peripheral nerve involvement in lymphomas: Suggestive features and diagnostic challenges. J. Peripher. Nerv. Syst. 2020, 25, 312–315. [Google Scholar] [CrossRef]
- Odabasi, Z.; Parrott, J.H.; Reddy, V.; Oh, S.J. Neurolymphomatosis associated with muscle and cerebral involvement caused by natural killer cell lymphoma: A case report and review of literature. J. Peripher. Nerv. Syst. 2001, 6, 197–203. [Google Scholar] [CrossRef]
- Tomita, M.; Koike, H.; Kawagashira, Y.; Iijima, M.; Adachi, H.; Taguchi, J.; Abe, T.; Sako, K.; Tsuji, Y.; Nakagawa, M.; et al. Clinicopathological features of neuropathy associated with lymphoma. Brain 2013, 136, 2563–2578. [Google Scholar] [CrossRef] [Green Version]
- Gordon, P.H.; Younger, D.S. Neurolymphomatosis. Neurology 1996, 46, 1191–1192. [Google Scholar] [CrossRef]
- van den Bent, M.J.; De Bruin, H.G.; Beun, G.; Vecht, C.J. Neurolymphomatosis of the median nerve. Neurology 1995, 45, 1403–1405. [Google Scholar] [CrossRef]
- Moore, K.R.; Blumenthal, D.T.; Smith, A.G.; Ward, J.H. Neurolymphomatosis of the lumbar plexus: High-resolution MR neurography findings. Neurology 2001, 57, 740–742. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| DISEASES | TREATMENTS | |
|---|---|---|
| Anti-MAG antibody neuropathy | IgM MGUS | Rituximab |
| CLL | Obinutuzumab BTKis or venetoclax ± rituximab | |
| WM/MZL | Rituximab ± chemotherapy BTKis or venetoclax | |
| Paraproteinemic neuropathy (anti-MAG antibody negative) Cryoglobulinemia | IgM MGUS | Rituximab |
| CLL/WM/MZL | Rituximab ± chemotherapy ± bortezomib BTKis or venetoclax | |
| POEMS syndrome AL amyloidosis | IgG or IgA MGUS | Bortezomib Lenalidomide |
| IgG or IgA MM | Daratumumab ± bortezomib ± IMIDs Autologous stem cell transplant | |
| Castleman’s diseases | Siltuximab | |
| Intravascular lymphoma neurolymphomatosis | Rituximab-based chemotherapy | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Visentin, A.; Pravato, S.; Castellani, F.; Campagnolo, M.; Angotzi, F.; Cavarretta, C.A.; Cellini, A.; Ruocco, V.; Salvalaggio, A.; Tedeschi, A.; et al. From Biology to Treatment of Monoclonal Gammopathies of Neurological Significance. Cancers 2022, 14, 1562. https://doi.org/10.3390/cancers14061562
Visentin A, Pravato S, Castellani F, Campagnolo M, Angotzi F, Cavarretta CA, Cellini A, Ruocco V, Salvalaggio A, Tedeschi A, et al. From Biology to Treatment of Monoclonal Gammopathies of Neurological Significance. Cancers. 2022; 14(6):1562. https://doi.org/10.3390/cancers14061562
Chicago/Turabian StyleVisentin, Andrea, Stefano Pravato, Francesca Castellani, Marta Campagnolo, Francesco Angotzi, Chiara Adele Cavarretta, Alessandro Cellini, Valeria Ruocco, Alessandro Salvalaggio, Alessandra Tedeschi, and et al. 2022. "From Biology to Treatment of Monoclonal Gammopathies of Neurological Significance" Cancers 14, no. 6: 1562. https://doi.org/10.3390/cancers14061562
APA StyleVisentin, A., Pravato, S., Castellani, F., Campagnolo, M., Angotzi, F., Cavarretta, C. A., Cellini, A., Ruocco, V., Salvalaggio, A., Tedeschi, A., Trentin, L., & Briani, C. (2022). From Biology to Treatment of Monoclonal Gammopathies of Neurological Significance. Cancers, 14(6), 1562. https://doi.org/10.3390/cancers14061562