
Targeting an MDM2/MYC Axis to Overcome Drug Resistance in Multiple Myeloma

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
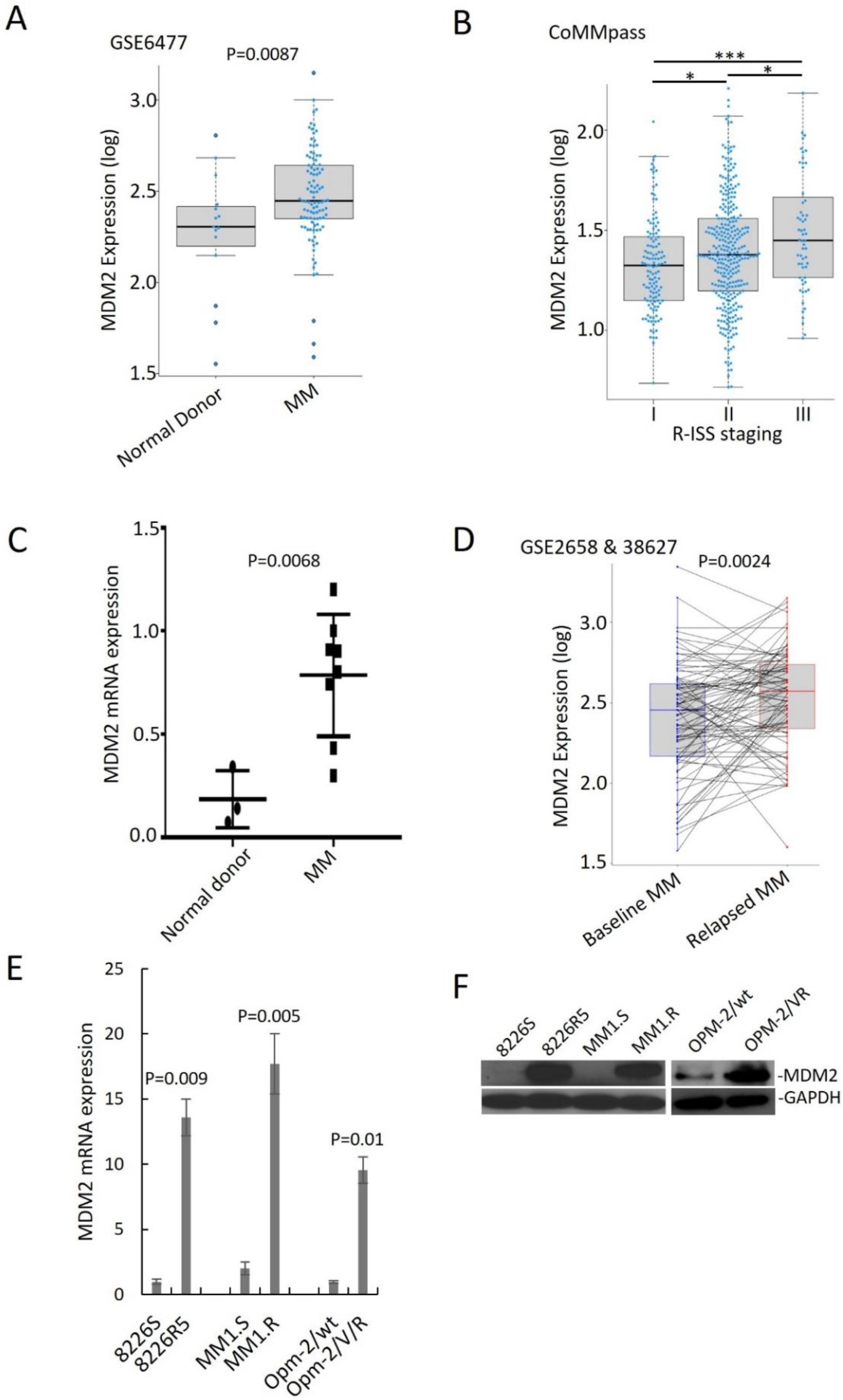
3.1. MDM2 Contributes Chemoresistance in MM
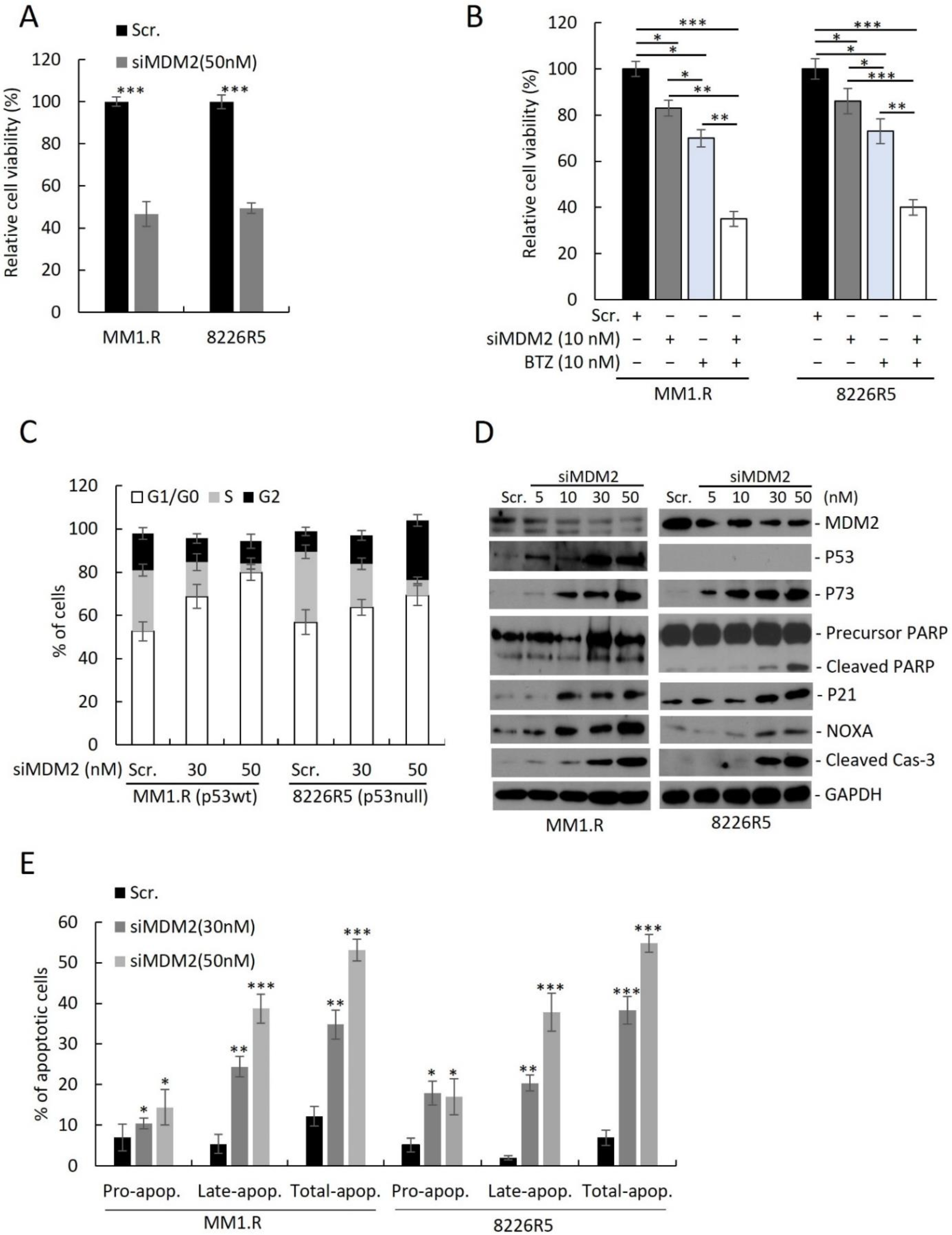
3.2. Silencing MDM2 Induces Apoptosis via p53-Dependent/Independent Pathways in Drug-resistant MM Cells and Re-Sensitizes MM Cells to Conventional Chemotherapy
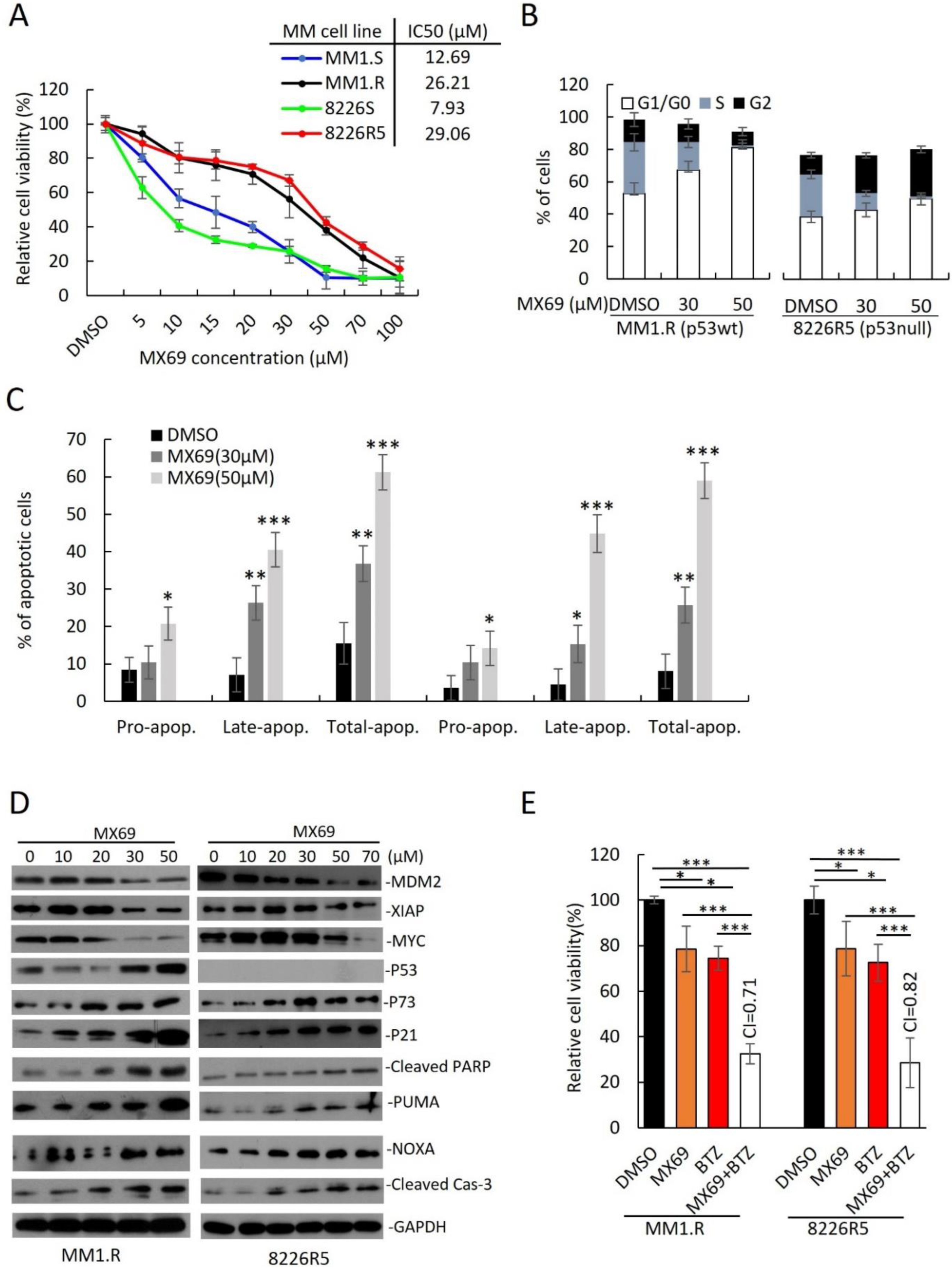
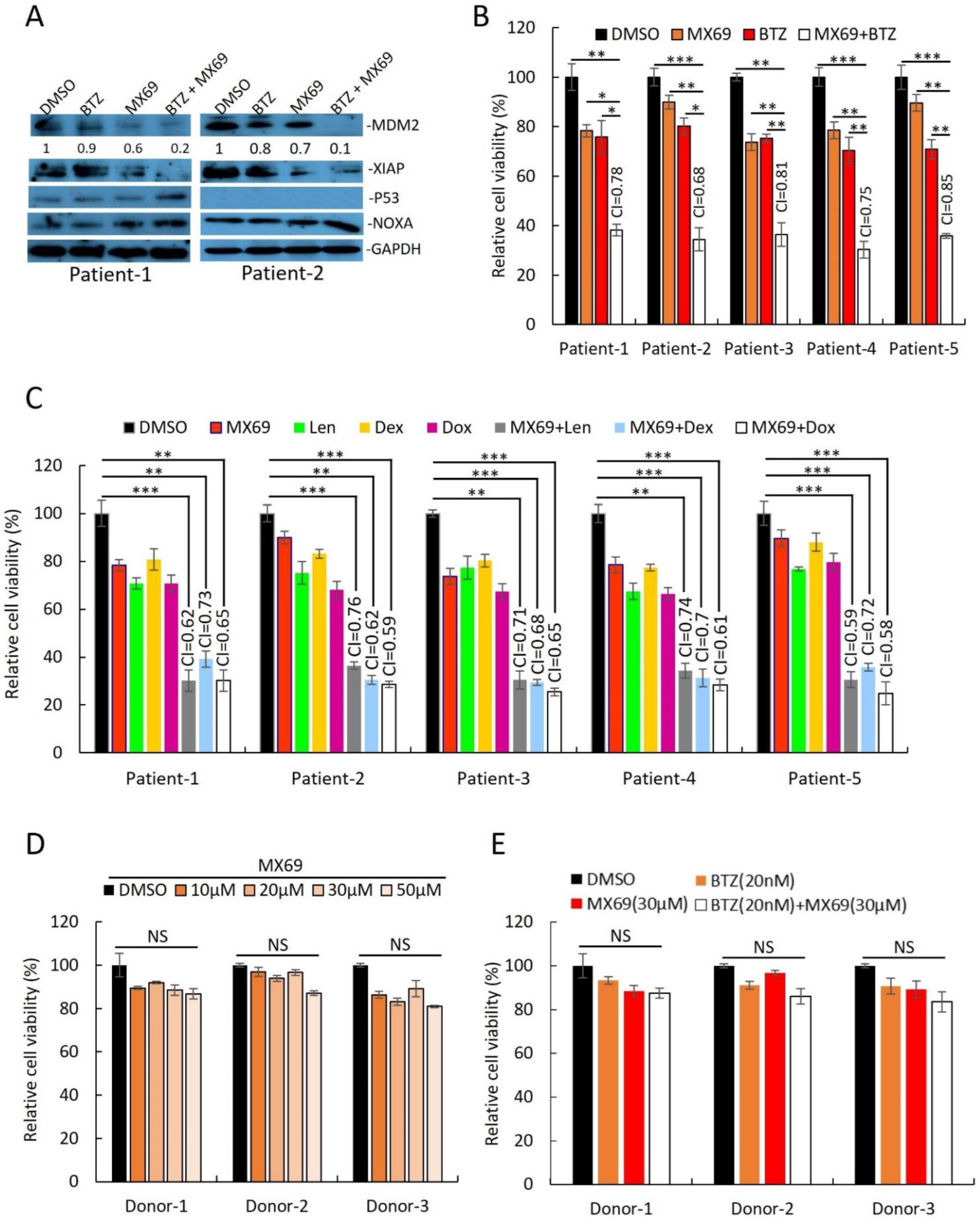
3.3. MX69 Inhibits the Growth of Drug-Resistant MM Cells through Induction of p53-Dependent and Independent Pathways
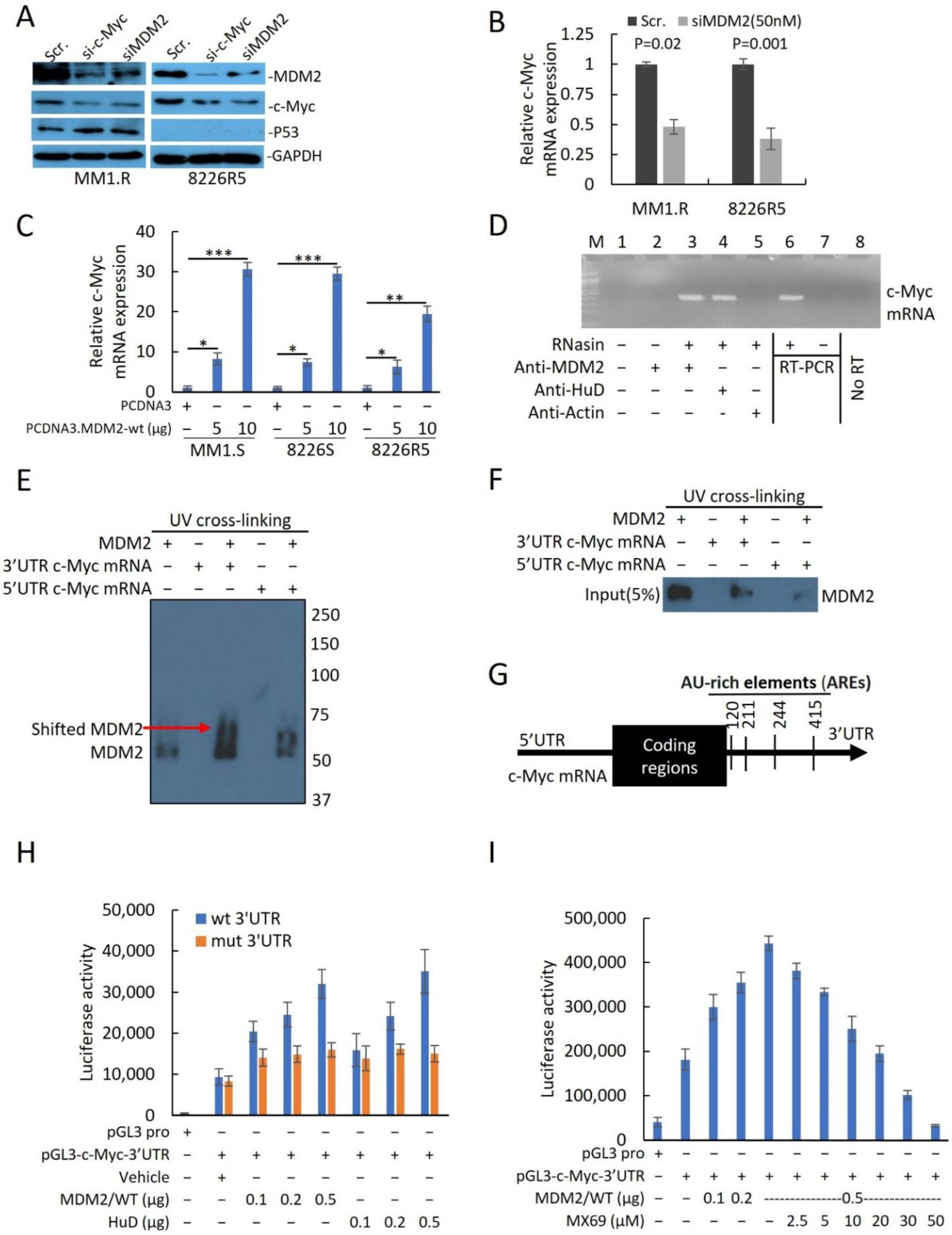
3.4. MX69 Inhibits c-Myc mRNA Potentially by Attenuating MDM2-cMYC mRNA Interaction
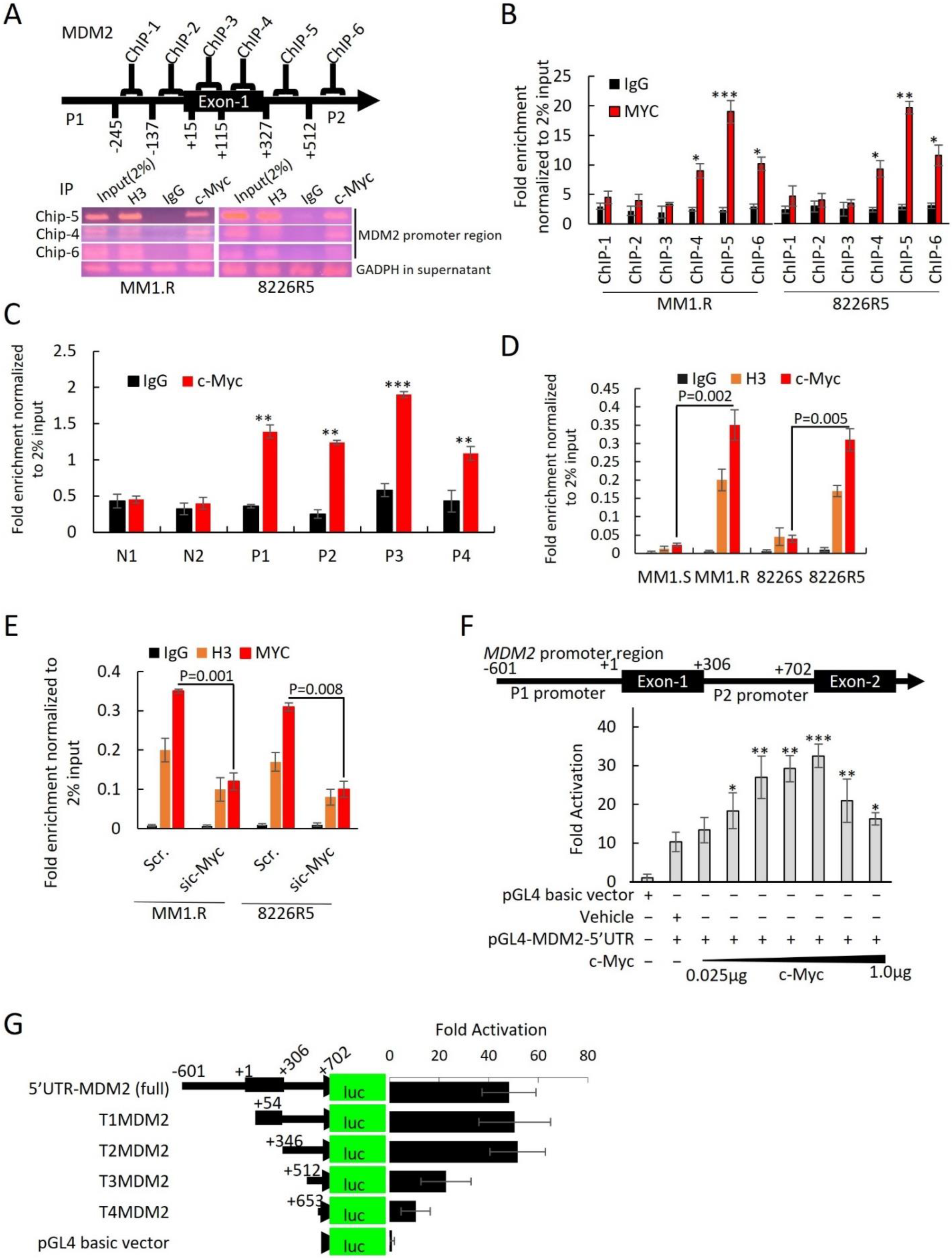
3.5. MDM2 Is a Direct Transcriptional Target of c-Myc in MM
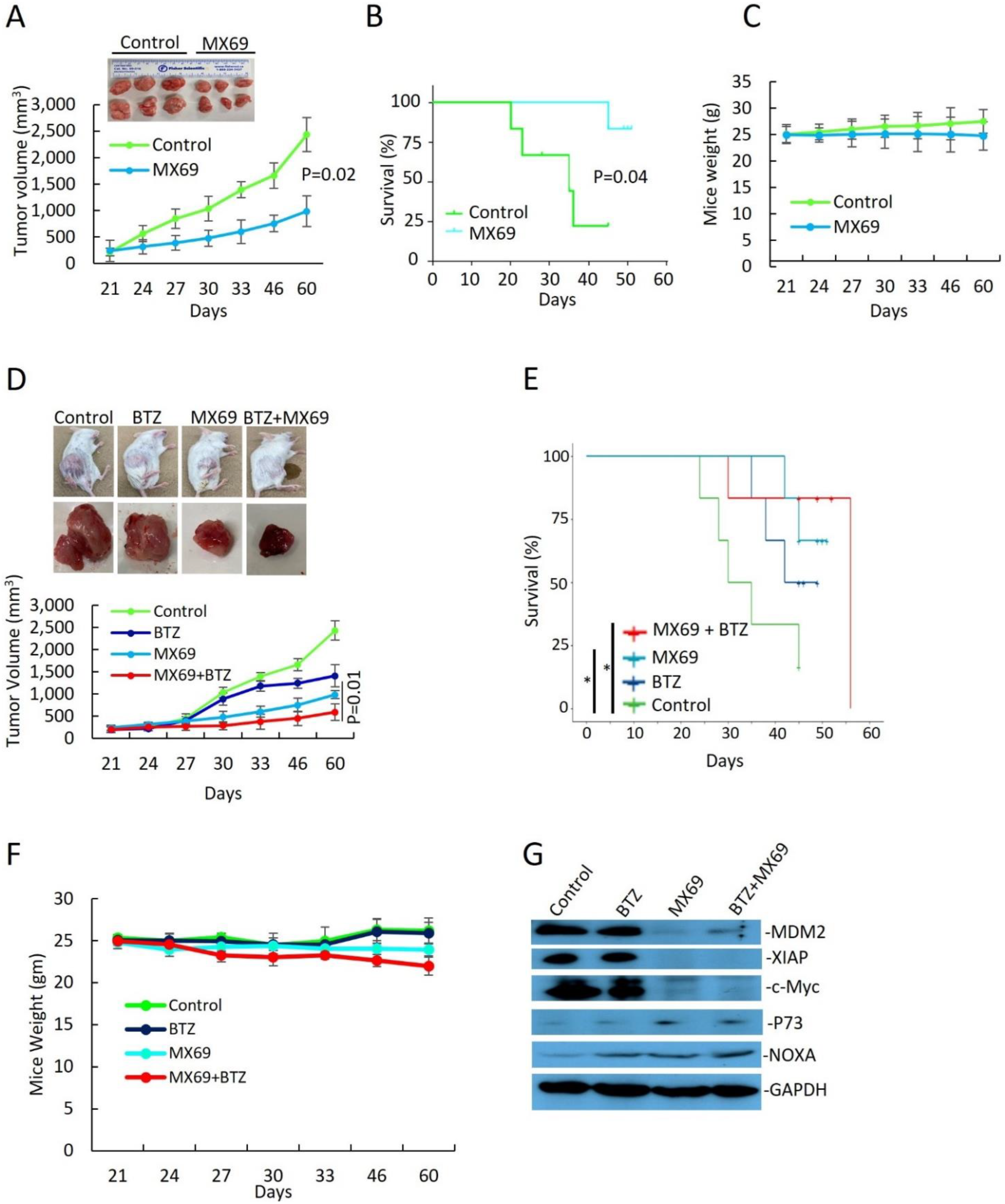
3.6. MX69 Inhibits Tumorigenesis in a MM Xenograft Model
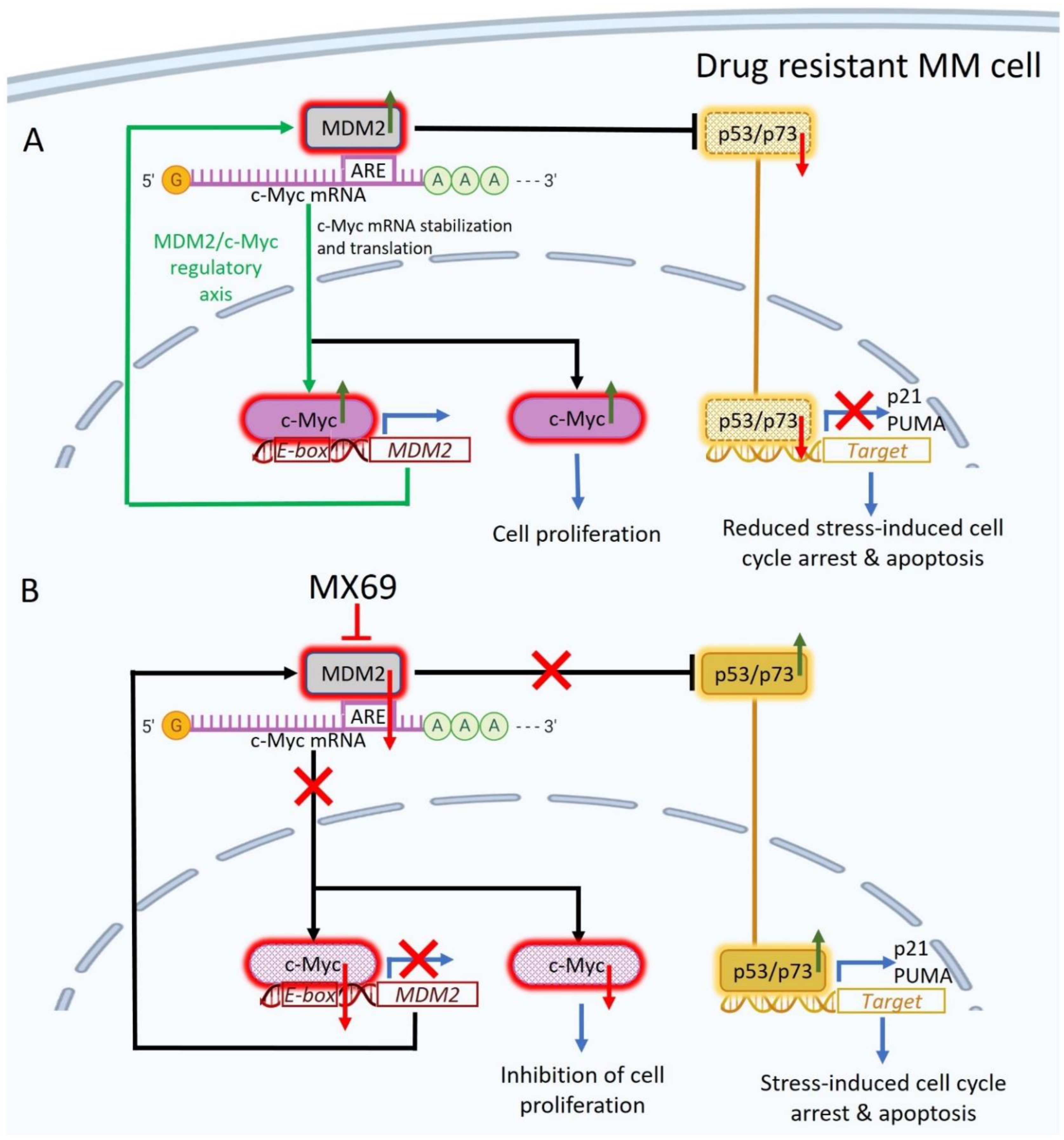
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rajkumar, S.V. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping Dou, Q.; Zonder, J.A. Overview of proteasome inhibitor-based anti-cancer therapies: Perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr. Cancer Drug Targets 2014, 14, 517–536. [Google Scholar]
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, J.J.; Orlowski, R.Z. Proteasome inhibitors in the treatment of multiple myeloma. Leukemia 2009, 23, 1964–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nag, S.; Zhang, X.; Srivenugopal, K.S.; Wang, M.H.; Wang, W.; Zhang, R. Targeting MDM2-p53 interaction for cancer therapy: Are we there yet? Curr. Med. Chem. 2014, 21, 553–574. [Google Scholar] [CrossRef] [PubMed]
- Teoh, G.; Urashima, M.; Ogata, A.; Chauhan, D.; DeCaprio, J.A.; Treon, S.P.; Schlossman, R.L.; Anderson, K.C. MDM2 protein overexpression promotes proliferation and survival of multiple myeloma cells. Blood J. Am. Soc. Hematol. 1997, 90, 1982–1992. [Google Scholar] [CrossRef] [Green Version]
- Stühmer, T.; Chatterjee, M.; Hildebrandt, M.; Herrmann, P.; Gollasch, H.; Gerecke, C.; Theurich, S.; Cigliano, L.; Manz, R.A.; Daniel, P.T.; et al. Nongenotoxic activation of the p53 pathway as a therapeutic strategy for multiple myeloma. Blood 2005, 106, 3609–3617. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.N.; Jiang, H.; Jayakar, J.; Reece, D.; Branch, D.R.; Chang, H. MDM2 antagonist nutlin plus proteasome inhibitor velcade combination displays a synergistic anti-myeloma activity. Cancer Biol. Ther. 2010, 9, 936–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, M.N.; Jiang, H.; Chang, H. Molecular mechanisms of nutlin-induced apoptosis in multiple myeloma: Evidence for p53-transcription-dependent and-independent pathways. Cancer Biol. Ther. 2010, 10, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Saha, M.N.; Jiang, H.; Mukai, A.; Chang, H. RITA inhibits multiple myeloma cell growth through induction of p53-mediated caspase-dependent apoptosis and synergistically enhances nutlin-induced cytotoxic responses. Mol. Cancer Ther. 2010, 9, 3041–3051. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2–p53 protein–protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment: Miniperspective. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zawacka-Pankau, J. The p53/MDM2/MDMX-targeted therapies—A clinical synopsis. Cell Death Dis. 2020, 11, 237. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Yeung, J.; Qi, C.; Xu, W. Aberrant nuclear p53 protein expression detected by immunohistochemistry is associated with hemizygous P53 deletion and poor survival for multiple myeloma. Br. J. Haematol. 2007, 138, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Qi, C.X.; Saha, M.N.; Chang, H. p53 nuclear expression correlates with hemizygous TP53 deletion and predicts an adverse outcome for patients with relapsed/refractory multiple myeloma treated with lenalidomide. Am. J. Clin. Pathol. 2012, 137, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, M.; Barbieri, M.; Manzoni, M.; Fabris, S.; Bandini, C.; Todoerti, K.; Nozza, F.; Rossi, D.; Musto, P.; Baldini, L.; et al. Molecular spectrum of TP53 mutations in plasma cell dyscrasias by next generation sequencing: An Italian cohort study and overview of the literature. Oncotarget 2016, 7, 21353. [Google Scholar] [CrossRef] [Green Version]
- Flynt, E.; Bisht, K.; Sridharan, V.; Ortiz, M.; Towfic, F.; Thakurta, A. Prognosis, biology, and targeting of TP53 dysregulation in multiple myeloma. Cells 2020, 9, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Leng, R.P. MDM2 mediates p73 ubiquitination: A new molecular mechanism for suppression of p73 function. Oncotarget 2015, 6, 21479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Zhang, H.; He, J.; Li, J.; Huang, M.; Zhou, M. MDM2 regulates MYCN mRNA stabilization and translation in human neuroblastoma cells. Oncogene 2012, 31, 1342–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Zhu, N.; Zhang, H.; Durden, D.L.; Feng, Y.; Zhou, M. Regulation of XIAP translation and induction by MDM2 following irradiation. Cancer Cell 2009, 15, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Gu, L.; He, J.; Zhang, H.; Zhou, M. MDM2 regulates vascular endothelial growth factor mRNA stabilization in hypoxia. Mol. Cell. Biol. 2011, 31, 4928–4937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Kim, J.; Park, J.K.; Hwang, S.G.; Moon, S.K.; Kim, W.J.; Um, H.D. Mdm2 increases cellular invasiveness by binding to and stabilizing the Slug mRNA. Cancer Lett. 2013, 335, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhang, H.; Liu, T.; Zhou, S.; Du, Y.; Xiong, J.; Yi, S.; Qu, C.K.; Fu, H.; Zhou, M. Discovery of dual inhibitors of MDM2 and XIAP for cancer treatment. Cancer Cell 2016, 30, 623–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzzeo, R.; Enkemann, S.; Nimmanapalli, R.; Alsina, M.; Lichtenheld, M.G.; Dalton, W.S.; Beaupre, D.M. Characterization of a R115777-resistant human multiple myeloma cell line with cross-resistance to PS-341. Clin. Cancer Res. 2005, 11, 6057–6064. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Chen, Y.; Saha, M.N.; Chen, J.; Evans, K.; Qiu, L.; Reece, D.; Chen, G.A.; Chang, H. Targeting phospho-MARCKS overcomes drug-resistance and induces antitumor activity in preclinical models of multiple myeloma. Leukemia 2015, 29, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, A.; Mortensen, J.M.; Winnenburg, R.; Liu, C.; Alessi, D.T.; Swamy, V.; Vallania, F.; Lofgren, S.; Haynes, W.; Shah, N.H.; et al. Interpretation of biological experiments changes with evolution of the Gene Ontology and its annotations. Sci. Rep. 2018, 8, 5115. [Google Scholar] [CrossRef]
- Holien, T.; Våtsveen, T.K.; Hella, H.; Waage, A.; Sundan, A. Addiction to c-MYC in multiple myeloma. Blood J. Am. Soc. Hematol. 2012, 120, 2450–2453. [Google Scholar] [CrossRef] [Green Version]
- Lai, W.S.; Arvola, R.M.; Goldstrohm, A.C.; Blackshear, P.J. Inhibiting transcription in cultured metazoan cells with actinomycin D to monitor mRNA turnover. Methods 2019, 155, 77–87. [Google Scholar] [CrossRef]
- Deschênes-Furry, J.; Perrone-Bizzozero, N.; Jasmin, B.J. The RNA-binding protein HuD: A regulator of neuronal differentiation, maintenance and plasticity. Bioessays 2006, 28, 822–833. [Google Scholar] [CrossRef]
- Srivastava, R.; Micanovic, R.; El-Achkar, T.M.; Janga, S.C. An intricate network of conserved DNA upstream motifs and associated transcription factors regulate the expression of uromodulin gene. J. Urol. 2014, 192, 981–989. [Google Scholar] [CrossRef]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef]
- Pelengaris, S.; Littlewood, T.; Khan, M.; Elia, G.; Evan, G. Reversible activation of c-Myc in skin: Induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol. Cell 1999, 3, 565–577. [Google Scholar] [CrossRef]
- Chin, M.; Sive, J.I.; Allen, C.; Roddie, C.; Chavda, S.J.; Smith, D.; Blombery, P.; Jones, K.; Ryland, G.L.; Popat, R.; et al. Prevalence and timing of TP53 mutations in del (17p) myeloma and effect on survival. Blood Cancer J. 2017, 7, e610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, H.; Sun, D.; Zhang, X. The role of MDM2 amplification and overexpression in therapeutic resistance of malignant tumors. Cancer Cell Int. 2019, 19, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayburn, E.; Zhang, R.; He, J.; Wang, H. MDM2 and human malignancies: Expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr. Cancer Drug Targets 2005, 5, 27–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovanović, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting MYC in multiple myeloma. Leukemia 2018, 32, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.G.; Gang, A.O.; Pedersen, M.Ø.; Poulsen, T.S.; Klausen, T.W.; Nørgaard, P. Overexpression of c-myc is associated with adverse clinical features and worse overall survival in multiple myeloma. Leuk. Lymphoma 2016, 57, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Huang, G.F.; Chung, T.H.; Ng, S.B.; Gonzalez-Paz, N.; Troska-Price, T.; Mulligan, G.; Chesi, M.; Bergsagel, P.L.; Fonseca, R. Clinical and biological implications of MYC activation: A common difference between MGUS and newly diagnosed multiple myeloma. Leukemia 2011, 25, 1026–1035. [Google Scholar] [CrossRef] [Green Version]
- Tran, H.N.; Singh, H.P.; Guo, W.; Cambier, L.; Riggan, L.; Shackleford, G.M.; Thornton, M.E.; Grubbs, B.H.; Erdreich-Epstein, A.; Qi, D.L.; et al. Reciprocal induction of MDM2 and MYCN in neural and neuroendocrine cancers. Front. Oncol. 2020, 10, 2933. [Google Scholar] [CrossRef]
- Chen, C.Y.; Shyu, A.B. AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995, 20, 465–470. [Google Scholar] [CrossRef]
- Lazarova, D.L.; Spengler, B.A.; Biedler, J.L.; Ross, R.A. HuD, a neuronal-specific RNA-binding protein, is a putative regulator of N-myc pre-mRNA processing/stability in malignant human neuroblasts. Oncogene 1999, 18, 2703–2710. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hall, T.M. Structural basis for recognition of AU-rich element RNA by the HuD protein. Nat. Struct. Biol. 2001, 8, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Eisenman, R.N. Myc target genes. Trends Biochem. Sci. 1997, 22, 177–181. [Google Scholar] [CrossRef]
- Amati, B.; Brooks, M.W.; Levy, N.; Littlewood, T.D.; Evan, G.I.; Land, H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell 1993, 72, 233–245. [Google Scholar] [CrossRef]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faruq, O.; Zhao, D.; Shrestha, M.; Vecchione, A.; Zacksenhaus, E.; Chang, H. Targeting an MDM2/MYC Axis to Overcome Drug Resistance in Multiple Myeloma. Cancers 2022, 14, 1592. https://doi.org/10.3390/cancers14061592
Faruq O, Zhao D, Shrestha M, Vecchione A, Zacksenhaus E, Chang H. Targeting an MDM2/MYC Axis to Overcome Drug Resistance in Multiple Myeloma. Cancers. 2022; 14(6):1592. https://doi.org/10.3390/cancers14061592
Chicago/Turabian StyleFaruq, Omar, Davidson Zhao, Mariusz Shrestha, Andrea Vecchione, Eldad Zacksenhaus, and Hong Chang. 2022. "Targeting an MDM2/MYC Axis to Overcome Drug Resistance in Multiple Myeloma" Cancers 14, no. 6: 1592. https://doi.org/10.3390/cancers14061592
APA StyleFaruq, O., Zhao, D., Shrestha, M., Vecchione, A., Zacksenhaus, E., & Chang, H. (2022). Targeting an MDM2/MYC Axis to Overcome Drug Resistance in Multiple Myeloma. Cancers, 14(6), 1592. https://doi.org/10.3390/cancers14061592