
Lung Adenocarcinoma Tumor Origin: A Guide for Personalized Medicine


Abstract
:Simple Summary
Abstract
1. Introduction
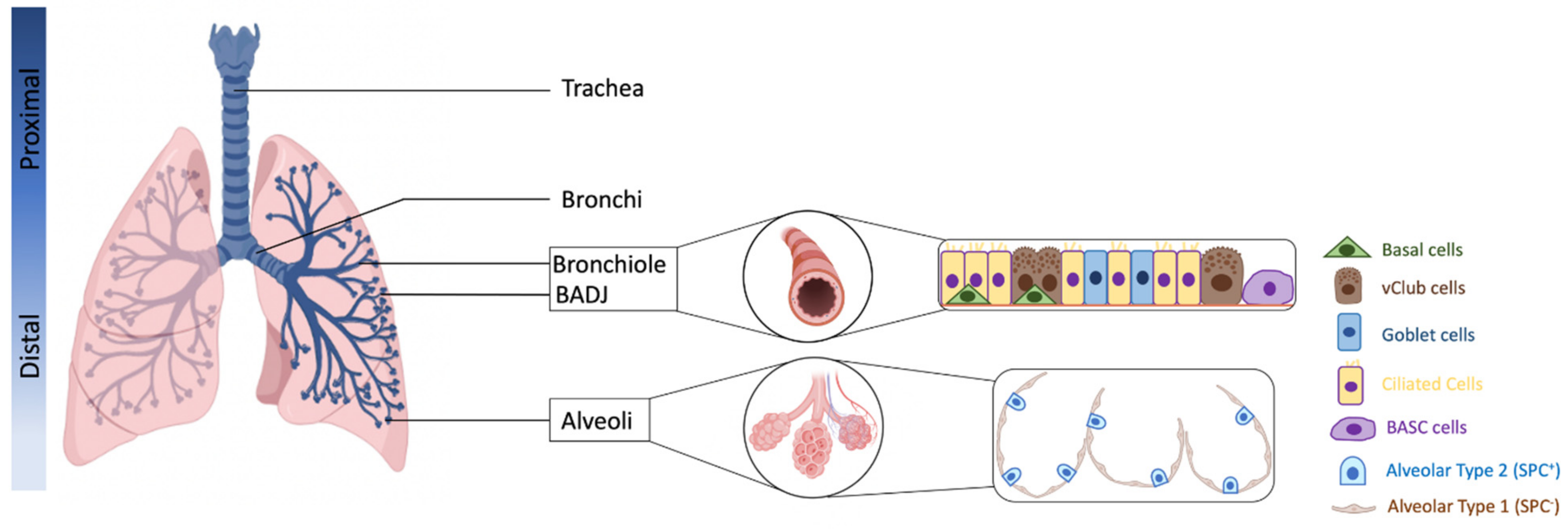
2. Defining Lung Progenitor Cells
3. Cell of Origin of LUAD: Multiple Possibilities
4. Cell of Origin of LUAD: The Influence of Co-Occurring Mutations
5. Cell of Origin of LUAD and CSCs Relationship
6. Cell of Origin of LUAD: The Interplay with the Immune Microenvironment
7. Future Directions/Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.-K. Non-Small-Cell Lung Cancers: A Heterogeneous Set of Diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.R.; Gazdar, A.F.; Clarke, B.E. The Pivotal Role of Pathology in the Management of Lung Cancer. J. Thorac. Dis. 2013, 5, S463–S478. [Google Scholar] [CrossRef] [PubMed]
- Langer, C.J.; Besse, B.; Gualberto, A.; Brambilla, E.; Soria, J.-C. The Evolving Role of Histology in the Management of Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2010, 28, 5311–5320. [Google Scholar] [CrossRef] [PubMed]
- Zito Marino, F.; Bianco, R.; Accardo, M.; Ronchi, A.; Cozzolino, I.; Morgillo, F.; Rossi, G.; Franco, R. Molecular Heterogeneity in Lung Cancer: From Mechanisms of Origin to Clinical Implications. Int. J. Med. Sci. 2019, 16, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef] [Green Version]
- de Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and Temporal Diversity in Genomic Instability Processes Defines Lung Cancer Evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Mayers, J.R.; Torrence, M.E.; Danai, L.V.; Papagiannakopoulos, T.; Davidson, S.M.; Bauer, M.R.; Lau, A.N.; Ji, B.W.; Dixit, P.D.; Hosios, A.M.; et al. Tissue-of-Origin Dictates Branched-Chain Amino Acid Metabolism in Mutant Kras-Driven Cancers. Science 2016, 353, 1161–1165. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Loo, P.V.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational Signatures Associated with Tobacco Smoking in Human Cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct Patterns of Somatic Genome Alterations in Lung Adenocarcinomas and Squamous Cell Carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devarakonda, S.; Morgensztern, D.; Govindan, R. Genomic Alterations in Lung Adenocarcinoma. Lancet Oncol. 2015, 16, e342–e351. [Google Scholar] [CrossRef]
- Román, M.; Baraibar, I.; López, I.; Nadal, E.; Rolfo, C.; Vicent, S.; Gil-Bazo, I. KRAS Oncogene in Non-Small Cell Lung Cancer: Clinical Perspectives on the Treatment of an Old Target. Mol. Cancer 2018, 17, 33. [Google Scholar] [CrossRef] [Green Version]
- Salgia, R.; Pharaon, R.; Mambetsariev, I.; Nam, A.; Sattler, M. The Improbable Targeted Therapy: KRAS as an Emerging Target in Non-Small Cell Lung Cancer (NSCLC). Cell Rep. Med. 2021, 2, 100186. [Google Scholar] [CrossRef] [PubMed]
- Merz, V.; Gaule, M.; Zecchetto, C.; Cavaliere, A.; Casalino, S.; Pesoni, C.; Contarelli, S.; Sabbadini, F.; Bertolini, M.; Mangiameli, D.; et al. Targeting KRAS: The Elephant in the Room of Epithelial Cancers. Front. Oncol. 2021, 11, 638360. [Google Scholar] [CrossRef]
- Desai, T.J. Developmental Insights into Lung Cancer. Annu. Rev. Cancer Biol. 2021, 5, 351–369. [Google Scholar] [CrossRef]
- Bowen, M.E.; McClendon, J.; Long, H.K.; Sorayya, A.; Van Nostrand, J.L.; Wysocka, J.; Attardi, L.D. The Spatiotemporal Pattern and Intensity of P53 Activation Dictates Phenotypic Diversity in P53-Driven Developmental Syndromes. Dev. Cell 2019, 50, 212–228.e6. [Google Scholar] [CrossRef]
- Visvader, J.E. Cells of Origin in Cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer Stem Cells: Current Status and Evolving Complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Hogan, B.L.; Barkauskas, C.E.; Chapman, H.A.; Epstein, J.A.; Jain, R.; Hsia, C.C.; Niklason, L.; Calle, E.; Le, A.; Randell, S.H.; et al. Repair and Regeneration of the Respiratory System: Complexity, Plasticity, and Mechanisms of Lung Stem Cell Function. Cell Stem Cell 2014, 15, 123–138. [Google Scholar] [CrossRef] [Green Version]
- Zepp, J.A.; Morrisey, E.E. Cellular Crosstalk in the Development and Regeneration of the Respiratory System. Nat. Rev. Mol. Cell Biol. 2019, 20, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Jones-Freeman, B.; Starkey, M.R. Bronchioalveolar Stem Cells in Lung Repair, Regeneration and Disease. J. Pathol. 2020, 252, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L.M. Type 2 Alveolar Cells Are Stem Cells in Adult Lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the Lung Alveolus by an Evolutionarily Conserved Epithelial Progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-Cell Wnt Signaling Niches Maintain Stemness of Alveolar Type 2 Cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Park, J.-E.; Tsagkogeorga, G.; Yanagita, M.; Koo, B.-K.; Han, N.; Lee, J.-H. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors That Mediate Alveolar Regeneration. Cell Stem Cell 2020, 27, 366–382.e7. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Lee, R.; Ou, J.; Banovich, N.; Kropski, J.; Tata, P.R. Persistence of a Regeneration-Associated, Transitional Alveolar Epithelial Cell State in Pulmonary Fibrosis. Nat. Cell Biol. 2020, 22, 934–946. [Google Scholar] [CrossRef]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar Regeneration through a Krt8+ Transitional Stem Cell State That Persists in Human Lung Fibrosis. Nat. Commun. 2020, 11, 3559. [Google Scholar] [CrossRef]
- Kathiriya, J.J.; Wang, C.; Zhou, M.; Brumwell, A.; Cassandras, M.; Le Saux, C.J.; Cohen, M.; Alysandratos, K.-D.; Wang, B.; Wolters, P.; et al. Human Alveolar Type 2 Epithelium Transdifferentiates into Metaplastic KRT5+ Basal Cells. Nat. Cell Biol. 2022, 24, 10–23. [Google Scholar] [CrossRef]
- Kim, C.F.B.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of Bronchioalveolar Stem Cells in Normal Lung and Lung Cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Liu, K.; Cui, G.; Huang, X.; Yao, S.; Guo, W.; Qin, Z.; Li, Y.; Yang, R.; Pu, W.; et al. Lung Regeneration by Multipotent Stem Cells Residing at the Bronchioalveolar-Duct Junction. Nat. Genet. 2019, 51, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Salwig, I.; Spitznagel, B.; Vazquez-Armendariz, A.I.; Khalooghi, K.; Guenther, S.; Herold, S.; Szibor, M.; Braun, T. Bronchioalveolar Stem Cells Are a Main Source for Regeneration of Distal Lung Epithelia in Vivo. EMBO J. 2019, 38, e102099. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Herndon, M.E.; Lawler, J. The Cell Biology of Thrombospondin-1. Matrix Biol. 2000, 19, 597–614. [Google Scholar] [CrossRef]
- Lee, J.H.; Bhang, D.H.; Beede, A.; Huang, T.L.; Stripp, B.R.; Bloch, K.D.; Wagers, A.J.; Tseng, Y.H.; Ryeom, S.; Kim, C.F. Lung Stem Cell Differentiation in Mice Directed by Endothelial Cells via a BMP4-NFATc1-Thrombospondin-1 Axis. Cell 2014, 156, 440–455. [Google Scholar] [CrossRef] [Green Version]
- McQualter, J.L.; Yuen, K.; Williams, B.; Bertoncello, I. Evidence of an Epithelial Stem/Progenitor Cell Hierarchy in the Adult Mouse Lung. Proc. Natl. Acad. Sci. USA 2010, 107, 1414–1419. [Google Scholar] [CrossRef] [Green Version]
- Asselin-Labat, M.-L.; Filby, C.E. Adult Lung Stem Cells and Their Contribution to Lung Tumourigenesis. Open Biol. 2012, 2, 120094. [Google Scholar] [CrossRef] [Green Version]
- Chapman, H.A.; Li, X.; Alexander, J.P.; Brumwell, A.; Lorizio, W.; Tan, K.; Sonnenberg, A.; Wei, Y.; Vu, T.H. Integrin A6β4 Identifies an Adult Distal Lung Epithelial Population with Regenerative Potential in Mice. J. Clin. Investig. 2011, 121, 2855–2862. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, A.E.; Brumwell, A.N.; Xi, Y.; Gotts, J.; Brownfield, D.G.; Treutlein, B.; Tan, K.; Tan, V.; Liu, F.; Looney, M.R.; et al. Lineage-Negative Progenitors Mobilize to Regenerate Lung Epithelium after Major Injury. Nature 2015, 517, 621–625. [Google Scholar] [CrossRef]
- Kathiriya, J.J.; Brumwell, A.N.; Jackson, J.R.; Tang, X.; Chapman, H.A. Distinct Airway Epithelial Stem Cells Hide among Club Cells but Mobilize to Promote Alveolar Regeneration. Cell Stem Cell 2020, 26, 346–358.e4. [Google Scholar] [CrossRef]
- Xi, Y.; Kim, T.; Brumwell, A.N.; Driver, I.H.; Wei, Y.; Tan, V.; Jackson, J.R.; Xu, J.; Lee, D.-K.; Gotts, J.E.; et al. Local Lung Hypoxia Determines Epithelial Fate Decisions during Alveolar Regeneration. Nat. Cell Biol. 2017, 19, 904–914. [Google Scholar] [CrossRef]
- Kumar, P.A.; Hu, Y.; Yamamoto, Y.; Hoe, N.B.; Wei, T.S.; Mu, D.; Sun, Y.; Joo, L.S.; Dagher, R.; Zielonka, E.M.; et al. Distal Airway Stem Cells Yield Alveoli in Vitro and during Lung Regeneration Following H1N1 Influenza Infection. Cell 2011, 147, 525–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, W.; Zhang, T.; Wu, D.Z.; Guan, S.P.; Liew, A.-A.; Yamamoto, Y.; Wang, X.; Lim, S.J.; Vincent, M.; Lessard, M.; et al. P63+Krt5+ Distal Airway Stem Cells Are Essential for Lung Regeneration. Nature 2015, 517, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Chiba, N.; Yao, C.; Guan, X.; McConnell, A.M.; Brockway, B.; Que, L.; McQualter, J.L.; Stripp, B.R. Rare SOX2+ Airway Progenitor Cells Generate KRT5+ Cells That Repopulate Damaged Alveolar Parenchyma Following Influenza Virus Infection. Stem Cell Rep. 2016, 7, 817–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, X.; Chen, H. Organoid Models in Lung Regeneration and Cancer. Cancer Lett. 2020, 475, 129–135. [Google Scholar] [CrossRef]
- Zhang, D.-G.; Jiang, A.-G.; Lu, H.-Y.; Zhang, L.-X.; Gao, X.-Y. Isolation, Cultivation and Identification of Human Lung Adenocarcinoma Stem Cells. Oncol. Lett. 2015, 9, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Ferone, G.; Lee, M.C.; Sage, J.; Berns, A. Cells of Origin of Lung Cancers: Lessons from Mouse Studies. Genes Dev. 2020, 34, 1017–1032. [Google Scholar] [CrossRef]
- Xu, X.; Rock, J.R.; Lu, Y.; Futtner, C.; Schwab, B.; Guinney, J.; Hogan, B.L.M.; Onaitis, M.W. Evidence for Type II Cells as Cells of Origin of K-Ras–Induced Distal Lung Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 4910–4915. [Google Scholar] [CrossRef] [Green Version]
- Mainardi, S.; Mijimolle, N.; Francoz, S.; Vicente-Dueñas, C.; Sánchez-García, I.; Barbacid, M. Identification of Cancer Initiating Cells in K-Ras Driven Lung Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2014, 111, 255–260. [Google Scholar] [CrossRef] [Green Version]
- Spella, M.; Lilis, I.; Pepe, M.A.; Chen, Y.; Armaka, M.; Lamort, A.-S.; Zazara, D.E.; Roumelioti, F.; Vreka, M.; Kanellakis, N.I.; et al. Club Cells Form Lung Adenocarcinomas and Maintain the Alveoli of Adult Mice. eLife 2019, 8, e45571. [Google Scholar] [CrossRef]
- Sutherland, K.D.; Song, J.-Y.; Kwon, M.C.; Proost, N.; Zevenhoven, J.; Berns, A. Multiple Cells-of-Origin of Mutant K-Ras–Induced Mouse Lung Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2014, 111, 4952–4957. [Google Scholar] [CrossRef] [Green Version]
- Yin, N.; Liu, Y.; Khoor, A.; Wang, X.; Thompson, E.A.; Leitges, M.; Justilien, V.; Weems, C.; Murray, N.R.; Fields, A.P. Protein Kinase Cι and Wnt/β-Catenin Signaling: Alternative Pathways to Kras/Trp53-Driven Lung Adenocarcinoma. Cancer Cell 2019, 36, 156–167.e7. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, A.S.; Lahtela, J.; Hemmes, A.; Pellinen, T.; Blom, S.; Devlin, J.R.; Salmenkivi, K.; Kallioniemi, O.; Mäyränpää, M.I.; Närhi, K.; et al. Cell of Origin Links Histotype Spectrum to Immune Microenvironment Diversity in Non-Small-Cell Lung Cancer Driven by Mutant Kras and Loss of Lkb1. Cell Rep. 2017, 18, 673–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, S.A.; Ding, S.; Kersbergen, A.; Dong, X.; Song, J.-Y.; Xie, Y.; Reljic, B.; Li, K.; Vince, J.E.; Rathi, V.; et al. Distinct Initiating Events Underpin the Immune and Metabolic Heterogeneity of KRAS-Mutant Lung Adenocarcinoma. Nat. Commun. 2019, 10, 4190. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of Lung Tumor Initiation and Progression Using Conditional Expression of Oncogenic K-Ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.-C.; Lai, C.-Y.; Shao, L.-E.; Yu, J. Identification of Tumorigenic Cells in Kras(G12D)-Induced Lung Adenocarcinoma. Cancer Res. 2011, 71, 7250–7258. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Liu, J.; Flight, R.M.; Naughton, K.J.; Lukyanchuk, A.; Edgin, A.R.; Song, X.; Zhang, H.; Wong, K.-K.; Moseley, H.N.B.; et al. Cellular Origins of EGFR-Driven Lung Cancer Cells Determine Sensitivity to Therapy. Adv. Sci. 2021, 8, 2101999. [Google Scholar] [CrossRef]
- Yoshida, K.; Gowers, K.H.C.; Lee-Six, H.; Chandrasekharan, D.P.; Coorens, T.; Maughan, E.F.; Beal, K. Tobacco Smoking and Somatic Mutations in Human Bronchial Epithelium. Nature 2020, 578, 266–272. [Google Scholar] [CrossRef]
- Zilfou, J.T.; Lowe, S.W. Tumor Suppressive Functions of P53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef]
- Jackson, E.L.; Olive, K.P.; Tuveson, D.A.; Bronson, R.; Crowley, D.; Brown, M.; Jacks, T. The Differential Effects of Mutant P53 Alleles on Advanced Murine Lung Cancer. Cancer Res. 2005, 65, 10280–10288. [Google Scholar] [CrossRef] [Green Version]
- Regala, R.P.; Davis, R.K.; Kunz, A.; Khoor, A.; Leitges, M.; Fields, A.P. Atypical Protein Kinase Cι Is Required for Bronchioalveolar Stem Cell Expansion and Lung Tumorigenesis. Cancer Res. 2009, 69, 7603–7611. [Google Scholar] [CrossRef] [Green Version]
- Calles, A.; Sholl, L.M.; Rodig, S.J.; Pelton, A.K.; Hornick, J.L.; Butaney, M.; Lydon, C.; Dahlberg, S.E.; Oxnard, G.R.; Jackman, D.M.; et al. Immunohistochemical Loss of LKB1 Is a Biomarker for More Aggressive Biology in KRAS-Mutant Lung Adenocarcinoma. Clin. Cancer Res. 2015, 21, 2851–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, M.; Xu, T.; Chang, P. KRAS/LKB1 and KRAS/TP53 Co-Mutations Create Divergent Immune Signatures in Lung Adenocarcinomas. Ther. Adv. Med. Oncol. 2021, 13, 17588359211006950. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 Modulates Lung Cancer Differentiation and Metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Li, F.; Fang, Z.; Gao, Y.; Li, F.; Fang, R.; Yao, S.; Sun, Y.; Li, L.; Zhang, W.; et al. Transdifferentiation of Lung Adenocarcinoma in Mice with Lkb1 Deficiency to Squamous Cell Carcinoma. Nat. Commun. 2014, 5, 3261. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Han, X.; Li, F.; Wang, R.; Wang, H.; Gao, Y.; Wang, X.; Fang, Z.; Zhang, W.; Yao, S.; et al. LKB1 Inactivation Elicits a Redox Imbalance to Modulate Non-Small Cell Lung Cancer Plasticity and Therapeutic Response. Cancer Cell 2015, 27, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Wohlhieter, C.A.; Richards, A.L.; Uddin, F.; Hulton, C.H.; Quintanal-Villalonga, À.; Martin, A.; de Stanchina, E.; Bhanot, U.; Asher, M.; Shah, N.S.; et al. Concurrent Mutations in STK11 and KEAP1 Promote Ferroptosis Protection and SCD1 Dependence in Lung Cancer. Cell Rep. 2020, 33, 108444. [Google Scholar] [CrossRef]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 Loss Promotes Kras-Driven Lung Cancer and Results in Dependence on Glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Eun, K.; Ham, S.W.; Kim, H. Cancer Stem Cell Heterogeneity: Origin and New Perspectives on CSC Targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar Progenitor and Stem Cells in Lung Development, Renewal and Cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef] [Green Version]
- Visvader, J.E.; Lindeman, G.J. Cancer Stem Cells in Solid Tumours: Accumulating Evidence and Unresolved Questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An Integrin Beta(3)-KRAS-RalB Complex Drives Tumour Stemness and Resistance to EGFR Inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; de la Cruz, C.C.; Sayles, L.C.; Alleyne-Chin, C.; Vaka, D.; Knaak, T.D.; Bigos, M.; Xu, Y.; Hoang, C.D.; Shrager, J.B.; et al. A Rare Population of CD24(+)ITGB4(+)Notch(Hi) Cells Drives Tumor Propagation in NSCLC and Requires Notch3 for Self-Renewal. Cancer Cell 2013, 24, 59–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, S.J.; Sinkevicius, K.W.; Li, D.; Lau, A.N.; Roach, R.R.; Zamponi, R.; Woolfenden, A.E.; Kirsch, D.G.; Wong, K.-K.; Kim, C.F. Primary Tumor Genotype Is an Important Determinant in Identification of Lung Cancer Propagating Cells. Cell Stem Cell 2010, 7, 127–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardavella, G.; George, R.; Sethi, T. Lung Cancer Stem Cells—Characteristics, Phenotype. Transl. Lung Cancer Res. 2016, 5, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Marjanovic, N.D.; Hofree, M.; Chan, J.E.; Canner, D.; Wu, K.; Trakala, M.; Hartmann, G.G.; Smith, O.C.; Kim, J.Y.; Evans, K.V.; et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 2020, 38, 229–246.e13. [Google Scholar] [CrossRef]
- Dovey, J.S.; Zacharek, S.J.; Kim, C.F.; Lees, J.A. Bmi1 Is Critical for Lung Tumorigenesis and Bronchioalveolar Stem Cell Expansion. Proc. Natl. Acad. Sci. USA 2008, 105, 11857–11862. [Google Scholar] [CrossRef] [Green Version]
- Park, I.-K.; Morrison, S.J.; Clarke, M.F. Bmi1, Stem Cells, and Senescence Regulation. J. Clin. Investig. 2004, 113, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Sauvageau, M.; Sauvageau, G. Polycomb Group Proteins: Multi-Faceted Regulators of Somatic Stem Cells and Cancer. Cell Stem Cell 2010, 7, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Siddique, H.R.; Saleem, M. Role of BMI1, a Stem Cell Factor, in Cancer Recurrence and Chemoresistance: Preclinical and Clinical Evidences. Stem Cells 2012, 30, 372–378. [Google Scholar] [CrossRef]
- Jia, L.; Zhang, W.; Wang, C.-Y. BMI1 Inhibition Eliminates Residual Cancer Stem Cells after PD1 Blockade and Activates Antitumor Immunity to Prevent Metastasis and Relapse. Cell Stem Cell 2020, 27, 238–253.e6. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.L.; West, D.; Wang, C.; Weiss, H.L.; Gal, T.; Durbin, E.; O’Connor, W.; Chen, M.; O’Connor, K.L. Elevated Integrin A6β4 Expression Is Associated with Venous Invasion and Decreased Overall Survival in Non-Small Cell Lung Cancer. Hum. Pathol. 2016, 54, 174–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and Cancer: Regulators of Cancer Stemness, Metastasis, and Drug Resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-Directed Immune Escape in Lung Cancer Evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef]
- Dawe, D.E.; Harlos, C.H.; Juergens, R.A. Immuno-Oncology—The New Paradigm of Lung Cancer Treatment. Curr. Oncol. 2020, 27, S78–S86. [Google Scholar] [CrossRef]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765.e17. [Google Scholar] [CrossRef] [Green Version]
- Kargl, J.; Busch, S.E.; Yang, G.H.Y.; Kim, K.-H.; Hanke, M.L.; Metz, H.E.; Hubbard, J.J.; Lee, S.M.; Madtes, D.K.; McIntosh, M.W.; et al. Neutrophils Dominate the Immune Cell Composition in Non-Small Cell Lung Cancer. Nat. Commun. 2017, 8, 14381. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Rodriguez-Canales, J.; et al. Co-Occurring Genomic Alterations Define Major Subsets of KRAS–Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [Green Version]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e14. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-Cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK Pathway: Metabolism and Growth Control in Tumour Suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Qi, R.; Ren, J.; Lv, D.; Yang, H. Characterization with KRAS Mutant Is a Critical Determinant in Immunotherapy and Other Multiple Therapies for Non-Small Cell Lung Cancer. Front. Oncol. 2022, 11, 780655. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Aredo, J.V.; Padda, S.K.; Kunder, C.A.; Han, S.S.; Neal, J.W.; Shrager, J.B.; Wakelee, H.A. Impact of KRAS Mutation Subtype and Concurrent Pathogenic Mutations on Non-Small Cell Lung Cancer Outcomes. Lung Cancer 2019, 133, 144–150. [Google Scholar] [CrossRef]
- Shergold, A.L.; Millar, R.; Nibbs, R.J.B. Understanding and Overcoming the Resistance of Cancer to PD-1/PD-L1 Blockade. Pharmacol. Res. 2019, 145, 104258. [Google Scholar] [CrossRef]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive Resistance to Therapeutic PD-1 Blockade Is Associated with Upregulation of Alternative Immune Checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Genetics | Inducer | Target Cells | Inflammation | Tumor Type/Location | Reference |
|---|---|---|---|---|---|
| LSL-KrasG12D | Ad5–Cre | lung epithelial cells | Yes | LUAD/papillary | Kim et al. [30] |
| LSL–KrasG12D; Trp53Flox/+; SPC–CreER; Rosa26R–fGFP | Tam | SPC+ cells (BASC and AT2) | No | LUAD in alveolar space | Xu et al. [47] |
| LSL–KrasG12D; Trp53Flox/+; CC10–CreER; Rosa26R–fGFP | Tam | CC10+ cells (BASC and Club) | No | Hyperplasia in the BADJ | Xu et al. [47] |
| LSL–KrasG12Vg | Tam | lung epithelial cells | No | LUAD in alveolar space | Mainardi et al. [48] |
| LSL–KrasG12Vg; Sca1–Cre | Sca1 expression | Sca1+ cells | no | CC10+ tumors in the bronchiole/BADJ/aleoli | Mainardi et al. [48] |
| GFP; Sftpc–CRE | urethane | lung epithelial cells | yes | LUAD in alveolar space and in the airway | Spella et al. [49] |
| GFP; Ccsp–CRE, | urethane | lung epithelial cells | yes | LUAD in alveolar space and in the airway | Spella et al. [49] |
| LSL–KrasG12D; Trp53F/F | Ad5–SPC–Cre | SPC+ cells (BASC and AT2) | yes | LUAD in alveolar space | Sutherland et al. [50] |
| LSL–KrasG12D; Trp53F/F | Ad5–CC10–Cre | CC10+ cells (BASC and Club) | yes | LUAD in alveolar space and BADJ | Sutherland et al. [50] |
| LSL–KrasG12D; Trp53F/F | Ad5–Cre | lung epithelial cells | yes | LUAD in alveolar space and BADJ | Yin et al. [51] |
| LSL–KrasG12D; Trp53F/F; PRKCiF/F | Ad5–Cre | lung epithelial cells | yes | LUAD in alveolar space | Yin et al. [51] |
| LSL–KrasG12D; STK11F/F | Ad5–SPC–Cre | SPC+ cells (BASC and AT2) | yes | LUAD in alveolar space | Nagaraj et al. [52] |
| LSL–KrasG12D; STK11F/F | Ad5–CC10–Cre | CC10+ cells (BASC and Club) | yes | lung adenosquamous and LUSC | Nagaraj et al. [52] |
| KrasG12D; KEAP1F/F | Ad5–SPC–Cre | SPC+ cells (BASC and AT2) | yes | LUAD in alveolar space | Best et al. [53] |
| KrasG12D; KEAP1F/F | Ad5–CC10–Cre | CC10+ cells (BASC and Club) | yes | LUSC in the airway | Best et al. [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seguin, L.; Durandy, M.; Feral, C.C. Lung Adenocarcinoma Tumor Origin: A Guide for Personalized Medicine. Cancers 2022, 14, 1759. https://doi.org/10.3390/cancers14071759
Seguin L, Durandy M, Feral CC. Lung Adenocarcinoma Tumor Origin: A Guide for Personalized Medicine. Cancers. 2022; 14(7):1759. https://doi.org/10.3390/cancers14071759
Chicago/Turabian StyleSeguin, Laetitia, Manon Durandy, and Chloe C. Feral. 2022. "Lung Adenocarcinoma Tumor Origin: A Guide for Personalized Medicine" Cancers 14, no. 7: 1759. https://doi.org/10.3390/cancers14071759
APA StyleSeguin, L., Durandy, M., & Feral, C. C. (2022). Lung Adenocarcinoma Tumor Origin: A Guide for Personalized Medicine. Cancers, 14(7), 1759. https://doi.org/10.3390/cancers14071759