
Isolation of Neoantigen-Specific Human T Cell Receptors from Different Human and Murine Repertoires

 , , , , , ,
, , , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Patient Material
2.2. Cell Cultures
2.3. Identification of Neoepitope Candidates
2.4. TIL Cultures and TMG Stimulation
2.5. Generation of Neoepitope-Specific T Cells from PB
2.6. Immunization of ABabDII Mice
2.7. TCRα/β Chain Identification
2.8. Virus Production and Transduction of PBLs
2.9. Transfection of Cell Lines
2.10. CTL Screening and Functional Assays
2.11. Statistical Analysis
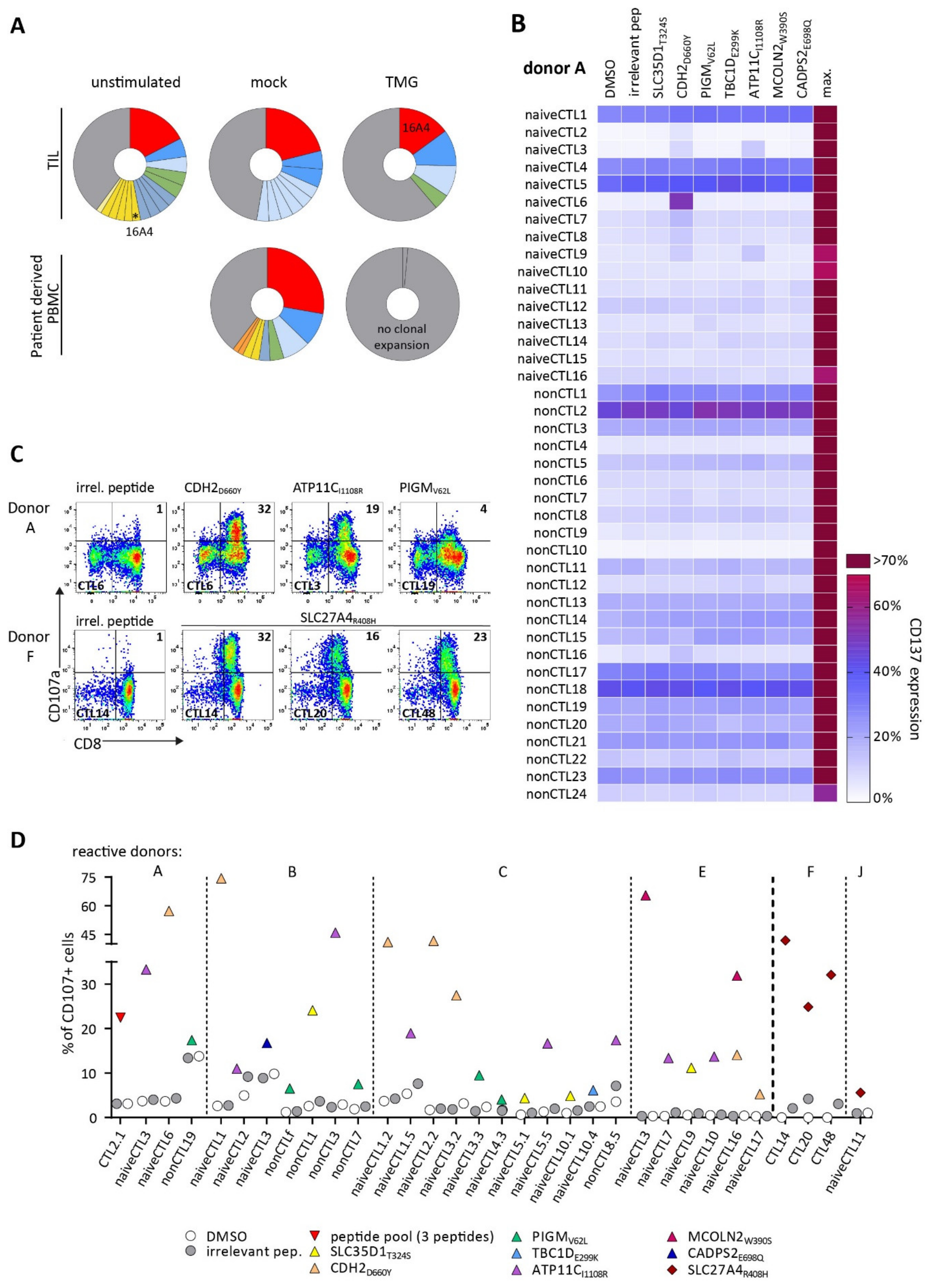
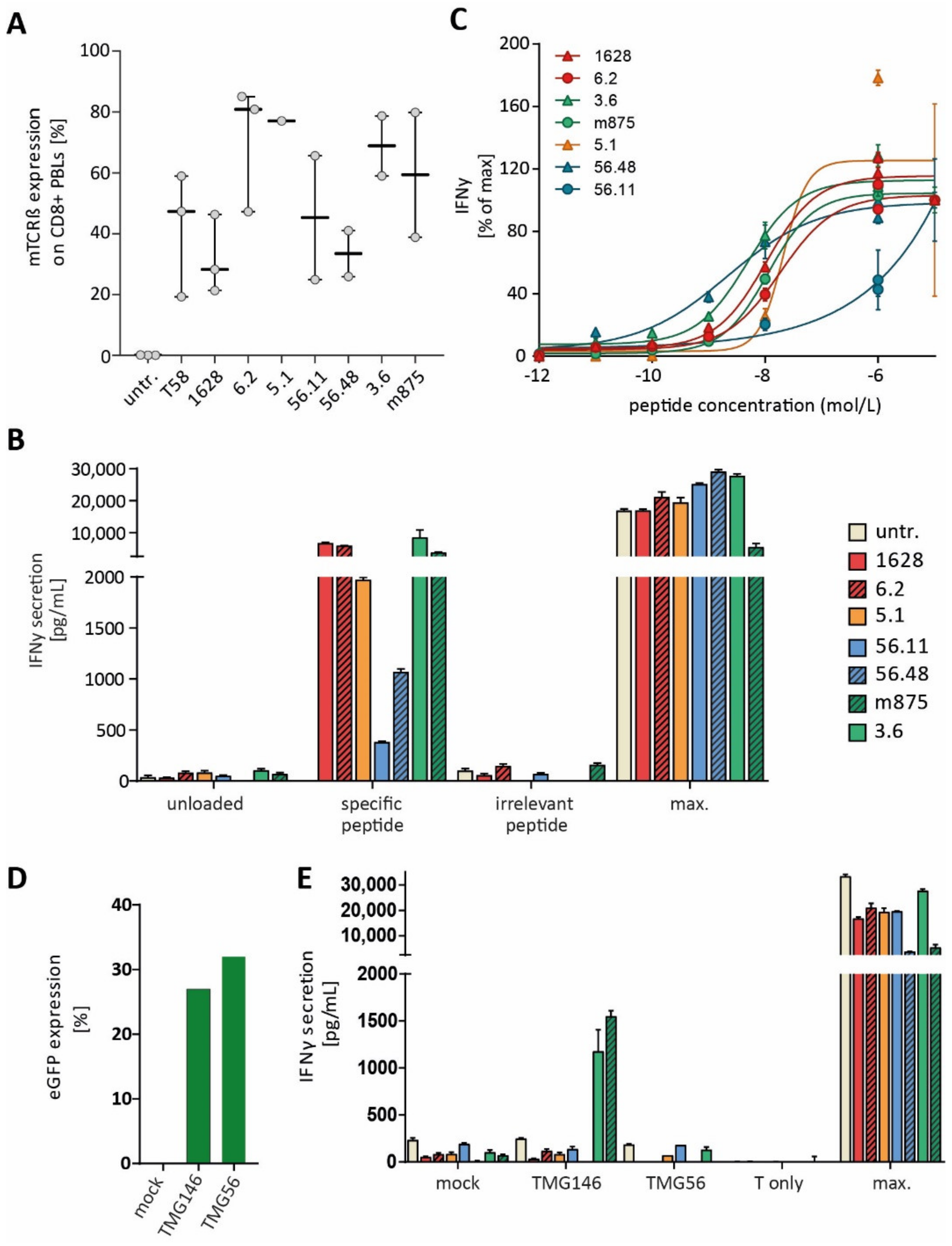
3. Results
3.1. Identification of Candidate Neoepitopes for an Ovarian Cancer Patient and Colon Cancer Patient
3.2. TCRs Identified from TIL or Patients’ PB Repertoire Did Not Show Reactivity against Selected Neoantigens
3.3. Detection of Neoantigen-Reactive CTLs after Stimulation of Healthy HLA-Matched Donor CD8+ T Cells
3.4. Identified TCRs from the Human Repertoire Exhibit Reactivity against Neoantigen Candidates
3.5. Immunization of ABabDII Transgenic Mice Led to the Isolation of a High-Affinity TCR
3.6. Endogenous Processing of Predicted Neoepitope Candidates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anandakrishnan, R.; Varghese, R.T.; Kinney, N.A.; Garner, H.R. Estimating the Number of Genetic Mutations (Hits) Required for Carcinogenesis Based on the Distribution of Somatic Mutations. PLoS Comput. Biol. 2019, 15, e1006881. [Google Scholar] [CrossRef] [PubMed]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.-C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune Recognition of Somatic Mutations Leading to Complete Durable Regression in Metastatic Breast Cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Leisegang, M.; Engels, B.; Schreiber, K.; Yew, P.Y.; Kiyotani, K.; Idel, C.; Arina, A.; Duraiswamy, J.; Rweichselbaum, R.; Uckert, W.; et al. Eradication of Large Solid Tumors by Gene Therapy with a T Cell Receptor Targeting a Single Cancer-Specific Point Mutation. Clin. Cancer Res. 2016, 22, 2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merhi, M.; Zhang, X.; Wang, S.; Hu, Y.; Liu, L.; Wu, D.; Liu, Y.; Li, X.; Liu, Y.; Yang, Q.; et al. Identification of Clonal Neoantigens Derived From Driver Mutations in an EGFR-Mutated Lung Cancer Patient Benefitting From Anti-PD-1. Front. Immunol. 2020, 1, 1366. [Google Scholar] [CrossRef]
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; Shamalov, K.; Trebska-McGowan, K.; Bliskovsky, V.V.; Parkhurst, M.R.; Ankri, C.; Prickett, T.D.; Crystal, J.S.; et al. Isolation of Neoantigen-Specific T Cells from Tumor and Peripheral Lymphocytes. J. Clin. Investig. 2015, 125, 3981–3991. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining Exomic Sequencing Data to Identify Mutated Antigens Recognized by Adoptively Transferred Tumor-Reactive T Cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of Tumor-Infiltrating Lymphocyte Cultures for Use in Adoptive Transfer Therapy for Melanoma Patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef]
- Stronen, E.; Toebes, M.; Kelderman, S.; Van Buuren, M.M.; Yang, W.; Van Rooij, N.N.; Donia, M.; Boeschen, M.-L.; Lund-Johansen, F.; Olweus, J.; et al. Targeting of Cancer Neoantigens with Donor-Derived T Cell Receptor Repertoires. Sci. Rep. 2016, 352, 1337–1341. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective Identification of Neoantigen-Specific Lymphocytes in the Peripheral Blood of Melanoma Patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.-C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of Somatic Mutations in Human Gastrointestinal Cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Scheper, W.; Kelderman, S.; Fanchi, L.F.; Linnemann, C.; Bendle, G.; J Rooij, M.A.; Hirt, C.; Mezzadra, R.; Slagter, M.; Dijkstra, K.; et al. Low and Variable Tumor Reactivity of the Intratumoral TCR Repertoire in Human Cancers. Nat. Med. 2018, 25, 89–94. [Google Scholar] [CrossRef]
- Penter, L.; Dietze, K.; Ritter, J.; Lammoglia Cobo, M.F.; Garmshausen, J.; Aigner, F.; Bullinger, L.; Hackstein, H.; Wienzek-Lischka, S.; Blankenstein, T.; et al. Localization-Associated Immune Phenotypes of Clonally Expanded Tumor-Infiltrating T Cells and Distribution of Their Target Antigens in Rectal Cancer. Oncoimmunology 2019, 8, e1586409. [Google Scholar] [CrossRef]
- Oliveira, G.; Stromhaug, K.; Klaeger, S.; Kula, T.; Frederick, D.T.; Le, P.M.; Forman, J.; Huang, T.; Li, S.; Zhang, W.; et al. Phenotype, Specificity and Avidity of Antitumour CD8 + T Cells in Melanoma Cell State of CD8 + TIL-TCR Clonotypes. Nature 2021, 596, 119. [Google Scholar] [CrossRef]
- Cafri, G.; Yossef, R.; Pasetto, A.; Deniger, D.C.; Lu, Y.C.; Parkhurst, M.; Gartner, J.J.; Jia, L.; Ray, S.; Ngo, L.T.; et al. Memory T Cells Targeting Oncogenic Mutations Detected in Peripheral Blood of Epithelial Cancer Patients. Nat. Commun. 2019, 10, 449. [Google Scholar] [CrossRef] [PubMed]
- Çınar, Ö.; Brzezicha, B.; Grunert, C.; Kloetzel, P.M.; Beier, C.; Peuker, C.A.; Keller, U.; Pezzutto, A.; Busse, A. High-Affinity T-Cell Receptor Specific for MyD88 L265P Mutation for Adoptive T-Cell Therapy of B-Cell Malignancies. J. Immunother. Cancer 2021, 9, 2410. [Google Scholar] [CrossRef]
- Ali, M.; Foldvari, Z.; Giannakopoulou, E.; Böschen, M.-L.; Strønen, E.; Yang, W.; Toebes, M.; Schubert, B.; Kohlbacher, O.; Schumacher, T.N.; et al. Induction of Neoantigen-Reactive T Cells from Healthy Donors. Nat. Protoc. 2019, 14, 1926–1943. [Google Scholar] [CrossRef]
- Kato, T.; Matsuda, T.; Ikeda, Y.; Park, J.-H.; Leisegang, M.; Yoshimura, S.; Hikichi, T.; Harada, M.; Zewde, M.; Sato, S.; et al. Effective Screening of T Cells Recognizing Neoantigens and Construction of T-Cell Receptor-Engineered T Cells. Oncotarget 2018, 9, 11009. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Lampert, J.C.; Chen, X.; Leitão, C.; Popović, J.; Müller, W.; Blankenstein, T. Transgenic Mice with a Diverse Human T Cell Antigen Receptor Repertoire. Nat. Med. 2010, 16, 1029–1034. [Google Scholar] [CrossRef]
- Çakmak-Görür, N.; Radke, J.; Rhein, S.; Schumann, E.; Willimsky, G.; Heppner, F.L.; Blankenstein, T.; Pezzutto, A. Intracellular Expression of FLT3 in Purkinje Cells: Implications for Adoptive T-Cell Therapies. Leukemia 2019, 33, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Obenaus, M.; Leitão, C.; Leisegang, M.; Chen, X.; Gavvovidis, I.; van der Bruggen, P.; Uckert, W.; Schendel, D.J.; Blankenstein, T. Identification of Human T-Cell Receptors with Optimal Affinity to Cancer Antigens Using Antigen-Negative Humanized Mice. Nat. Biotechnol. 2015, 33, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Poncette, L.; Chen, X.; Lorenz, F.K.M.; Blankenstein, T. Effective NY-ESO-1-Specific MHC II-Restricted T Cell Receptors from Antigen-Negative Hosts Enhance Tumor Regression. J. Clin. Investig. 2019, 129, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-B.; Lee, I.-G.; Joo, Y.-H.; Hong, S.-H.; Seo, Y.-J. Molecular Sciences TCR Transgenic Mice: A Valuable Tool for Studying Viral Immunopathogenesis Mechanisms. Int. J. Mol. Sci. 2020, 21, 9690. [Google Scholar] [CrossRef] [PubMed]
- Karosiene, E.; Lundegaard, C.; Lund, O.; Nielsen, M. NetMHCcons: A Consensus Method for the Major Histocompatibility Complex Class i Predictions. Immunogenetics 2012, 64, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Spiotto, M.T.; Yu, P.; Rowley, D.A.; Nishimura, M.I.; Meredith, S.C.; Gajewski, T.F.; Fu, Y.X.; Schreiber, H. Increasing Tumor Antigen Expression Overcomes “Ignorance” to Solid Tumors via Crosspresentation by Bone Marrow-Derived Stromal Cells. Immunity 2002, 17, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Dauer, M.; Schad, K.; Herten, J.; Junkmann, J.; Bauer, C.; Kiefl, R.; Endres, S.; Eigler, A. FastDC Derived from Human Monocytes within 48 h Effectively Prime Tumor Antigen-Specific Cytotoxic T Cells. J. Immunol. Methods 2005, 302, 145–155. [Google Scholar] [CrossRef]
- Wölfl, M.; Greenberg, P.D. Antigen-Specific Activation and Cytokine-Facilitated Expansion of Naive, Human CD8+ T Cells. Nat. Protoc. 2014, 9, 950–966. [Google Scholar] [CrossRef] [Green Version]
- Han, A.; Glanville, J.; Hansmann, L.; Davis, M.M. Linking T-Cell Receptor Sequence to Functional Phenotype at the Single-Cell Level. Nat. Biotechnol. 2014, 4, 684–692. [Google Scholar] [CrossRef]
- Penter, L.; Dietze, K.; Bullinger, L.; Westermann, J.; Rahn, H.-P.; Hansmann, L. FACS Single Cell Indexsorting Is Highlyreliable Anddetermines Immunephenotypes of Clonallyexpanded T Cells. Eur. J. Immunol. 2018, 8, 1248–1250. [Google Scholar] [CrossRef]
- Campillo-Davo, D.; Versteven, M.; Roex, G.; De Reu, H.; Van Der Heijden, S.; Anguille, S.; Berneman, Z.N.; Van Tendeloo, V.F.I.; Lion, E. Rapid Assessment of Functional Avidity of Tumor-Specific T Cell Receptors Using an Antigen-Presenting Tumor Cell Line Electroporated with Full-Length Tumor Antigen MRNA. Cancers 2020, 12, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobisse, S.; Genolet, R.; Roberti, A.; Tanyi, J.L.; Racle, J.; Stevenson, B.J.; Iseli, C.; Michel, A.; Le Bitoux, M.A.; Guillaume, P.; et al. Sensitive and Frequent Identification of High Avidity Neo-Epitope Specific CD8+ T Cells in Immunotherapy-Naive Ovarian Cancer. Nat. Commun. 2018, 9, 1092. [Google Scholar] [CrossRef] [PubMed]
- Bhaduri-McIntosh, S.; Rotenberg, M.J.; Gardner, B.; Robert, M.; Miller, G. Repertoire and Frequency of Immune Cells Reactive to Epstein-Barr Virus–Derived Autologous Lymphoblastoid Cell Lines. Blood 2008, 111, 1334–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasetto, A.; Gros, A.; Robbins, P.F.; Deniger, D.C.; Prickett, T.D.; Matus-Nicodemos, R.; Douek, D.C.; Howie, B.; Robins, H.; Parkhurst, M.R.; et al. Tumor- and Neoantigen-Reactive T-Cell Receptors Can Be Identified Based on Their Frequency in Fresh Tumor. Cancer Immunol. Res. 2016, 4, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Yossef, R.; Tran, E.; Deniger, D.C.; Gros, A.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Cafri, G.; Robbins, P.F.; et al. Enhanced Detection of Neoantigen-Reactive T Cells Targeting Unique and Shared Oncogenes for Personalized Cancer Immunotherapy. JCI Insight 2018, 3, e122467. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient with Epithelial Cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Martin, S.D.; Wick, D.A.; Nielsen, J.S.; Little, N.; Holt, R.A.; Nelson, B.H. A Library-Based Screening Method Identifies Neoantigen-Reactive T Cells in Peripheral Blood Prior to Relapse of Ovarian Cancer. Oncoimmunology 2018, 7, e1371895. [Google Scholar] [CrossRef] [Green Version]
- Popović, J.; Li, A.P.; Kloetzel, P.M.; Leisegang, M.; Uckert, W.; Blankenstein, T. The Only Proposed T-Cell Epitope Derived from the TEL-AML1 Translocation Is Not Naturally Processed. Blood 2011, 118, 946–954. [Google Scholar] [CrossRef]
- Sgourakis, N.G.; Bradley, P.; Riemer, A.B.; Riley, T.P.; Baker, B.M.; J Keller, G.L.; Smith, A.R.; Davancaze, L.M.; Arbuiso, A.G.; Devlin, J.R. Structure Based Prediction of Neoantigen Immunogenicity. Front. Immunol. 2019, 10, 2047. [Google Scholar] [CrossRef]
- Fritsch, E.F.; Rajasagi, M.; Ott, P.A.; Brusic, V.; Hacohen, N.; Wu, C.J. HLA-Binding Properties of Tumor Neoepitopes in Humans. Cancer Immunol. Res. 2014, 2, 522–529. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Kishton, R.J.; Angel, M.; Conn, C.S.; Dalla-Venezia, N.; Marcel, V.; Vincent, A.; Catez, F.; Ferré, S.; Ayadi, L.; et al. Ribosomal Proteins Regulate MHC Class I Peptide Generation for Immunosurveillance. Mol. Cell 2019, 73, 1162–1173.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, C.; Chowell, D.; Gönen, M.; Elhanati, Y.; Chan, T.A. Genetic and Environmental Determinants of Human TCR Repertoire Diversity. Immun. Ageing 2020, 17, 26. [Google Scholar] [CrossRef] [PubMed]
- Britanova, O.V.; Putintseva, E.V.; Shugay, M.; Merzlyak, E.M.; Turchaninova, M.A.; Staroverov, D.B.; Bolotin, D.A.; Lukyanov, S.; Bogdanova, E.A.; Mamedov, I.Z.; et al. Age-Related Decrease in TCR Repertoire Diversity Measured with Deep and Normalized Sequence Profiling. J. Immunol. 2014, 192, 2689–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA Class I Genotype Influences Cancer Response to Checkpoint Blockade Immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Patient | TMG Name | Epitope | Gene/ Mutation | HLA Restriction | % Rank | IC50 (nM) |
|---|---|---|---|---|---|---|
| 146 | TMG146 | ILISYIGMV | SLC35D1T324S | A2:01 | 0.12 | 9.76 |
| RLNGYFAQL | CDH2D660Y | A2:01 | 0.6 | 44.92 | ||
| FLTEGERSPYL | PIGMV62L | A2:01 | 0.12 | 10.64 | ||
| MLFTIGQSKV | TBC1D1E299K | A2:01 | 0.6 | 43.62 | ||
| SLFPEILLRV | ATP11CI1108R | A2:01 | 0.15 | 12.01 | ||
| FLGTSTLLVSV | MCOLN2W390S | A2:01 | 0.5 | 36.71 | ||
| QLMEHSENGAV | CADPS2E695Q | A2:01 | 3.5 | 653.37 | ||
| 56 | TMG56 | HILSFVYPI | SLC27A4R408H | A2:01 | 0.8 | 62.41 |
| SPSRPPGPT | PLCB3R918S | B7:02 | 0.25 | 50.2 | ||
| LAVDTDEIEKY | NBPFM674T | B35:01 | 2.00 | 1356.1 | ||
| HSHELNGPY | CRYBB2C38Y | B35:01 | 0.2 | 36.43 | ||
| WLDGKHVVF | PPIAL4GA128V | C4:01 | 0.03 | 1679.01 | ||
| TKFDVQVLK | INF2E670Q | C4:01 | 0.4 | 4893.37 | ||
| YRQQAGRELL | IGSF9BE63Q | C7:02 | 0.01 | 48.03 | ||
| RRNPTGSVVM | PDXKA305T | C7:02 | 1.2 | 3589.15 | ||
| YRRDVHHVACY | KLHL22Q281H | C7:02 | 1.9 | 5719.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grunert, C.; Willimsky, G.; Peuker, C.A.; Rhein, S.; Hansmann, L.; Blankenstein, T.; Blanc, E.; Beule, D.; Keller, U.; Pezzutto, A.; et al. Isolation of Neoantigen-Specific Human T Cell Receptors from Different Human and Murine Repertoires. Cancers 2022, 14, 1842. https://doi.org/10.3390/cancers14071842
Grunert C, Willimsky G, Peuker CA, Rhein S, Hansmann L, Blankenstein T, Blanc E, Beule D, Keller U, Pezzutto A, et al. Isolation of Neoantigen-Specific Human T Cell Receptors from Different Human and Murine Repertoires. Cancers. 2022; 14(7):1842. https://doi.org/10.3390/cancers14071842
Chicago/Turabian StyleGrunert, Corinna, Gerald Willimsky, Caroline Anna Peuker, Simone Rhein, Leo Hansmann, Thomas Blankenstein, Eric Blanc, Dieter Beule, Ulrich Keller, Antonio Pezzutto, and et al. 2022. "Isolation of Neoantigen-Specific Human T Cell Receptors from Different Human and Murine Repertoires" Cancers 14, no. 7: 1842. https://doi.org/10.3390/cancers14071842
APA StyleGrunert, C., Willimsky, G., Peuker, C. A., Rhein, S., Hansmann, L., Blankenstein, T., Blanc, E., Beule, D., Keller, U., Pezzutto, A., & Busse, A. (2022). Isolation of Neoantigen-Specific Human T Cell Receptors from Different Human and Murine Repertoires. Cancers, 14(7), 1842. https://doi.org/10.3390/cancers14071842