
Ex Vivo High-Resolution Magic Angle Spinning (HRMAS) 1H NMR Spectroscopy for Early Prostate Cancer Detection

 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
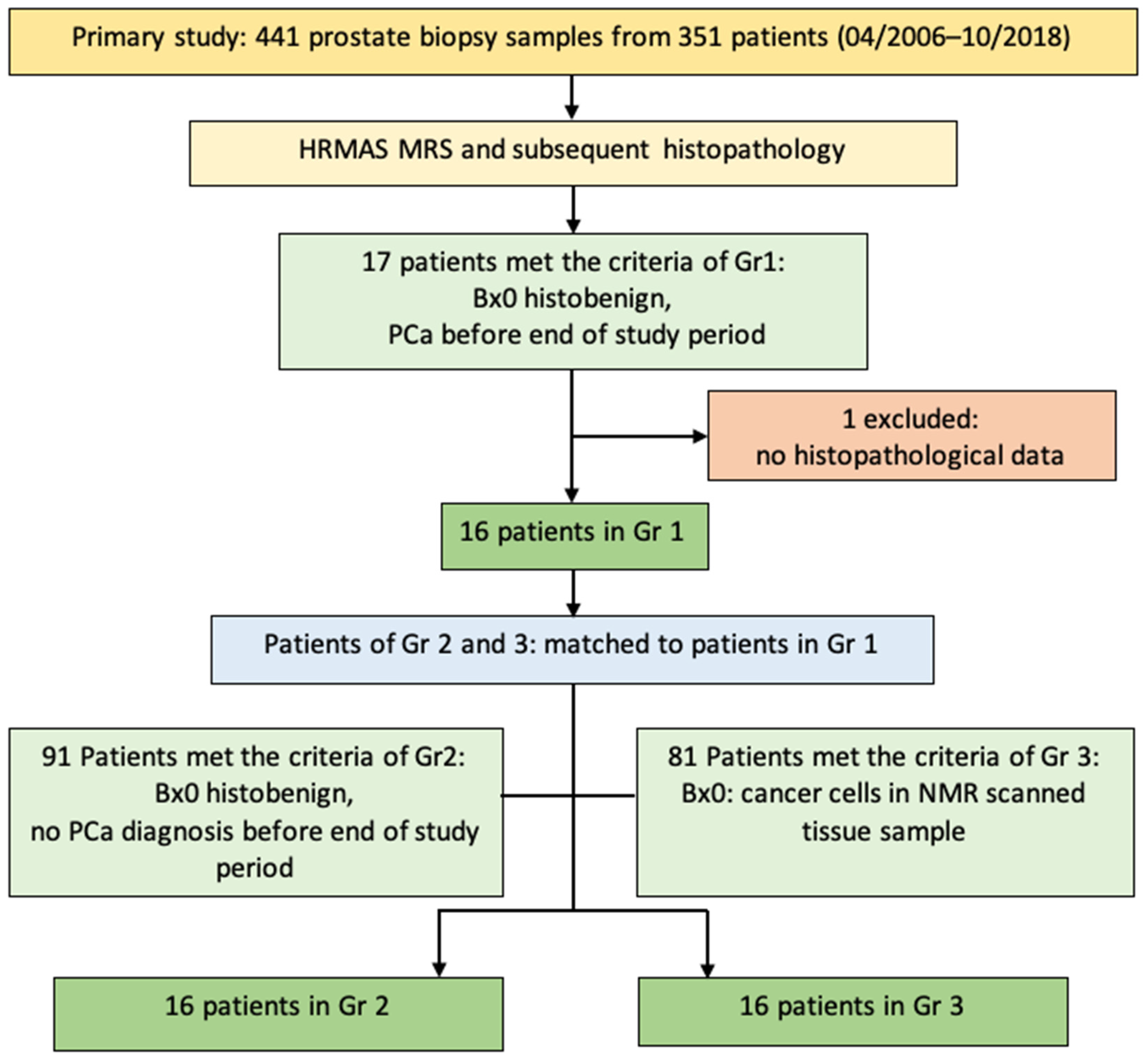
2.1. Patients
2.2. Intact Tissue Magnetic Resonance Spectroscopy (MRS)
2.3. Quantitative Histopathology
2.4. Statistical Analysis
3. Results
3.1. Clinical and (Histo)Pathological Patient Data
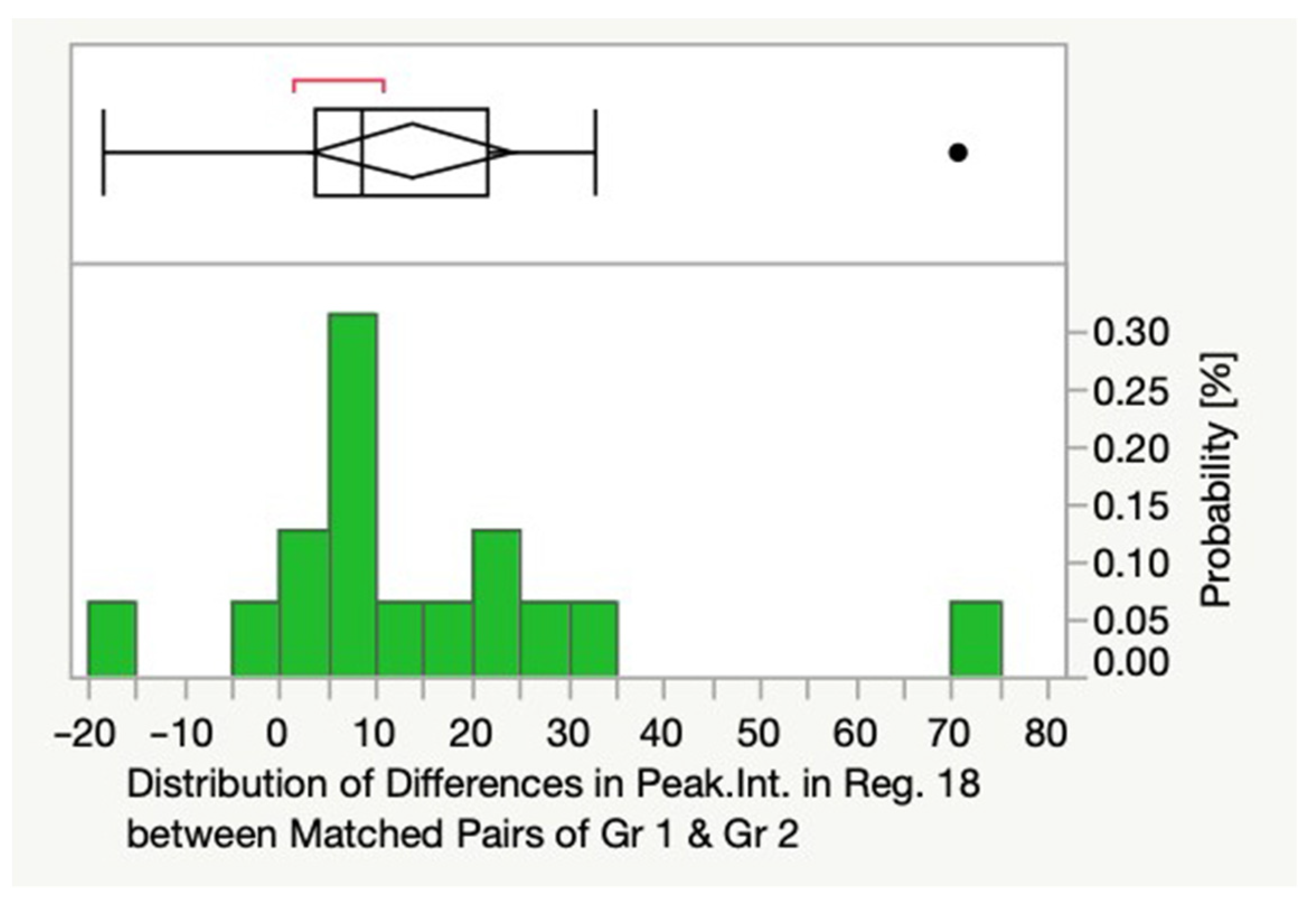
3.2. Differences between Histobenign and Malignant Prostate Tissue
3.3. Differences between Histobenign and Premalignant Prostate Tissue
3.4. Differences between Premalignant and Malignant Prostate Tissue
3.5. Differences between Gleason Score Categories GS 3 + 3 = 6 and 3 + 4 = 7
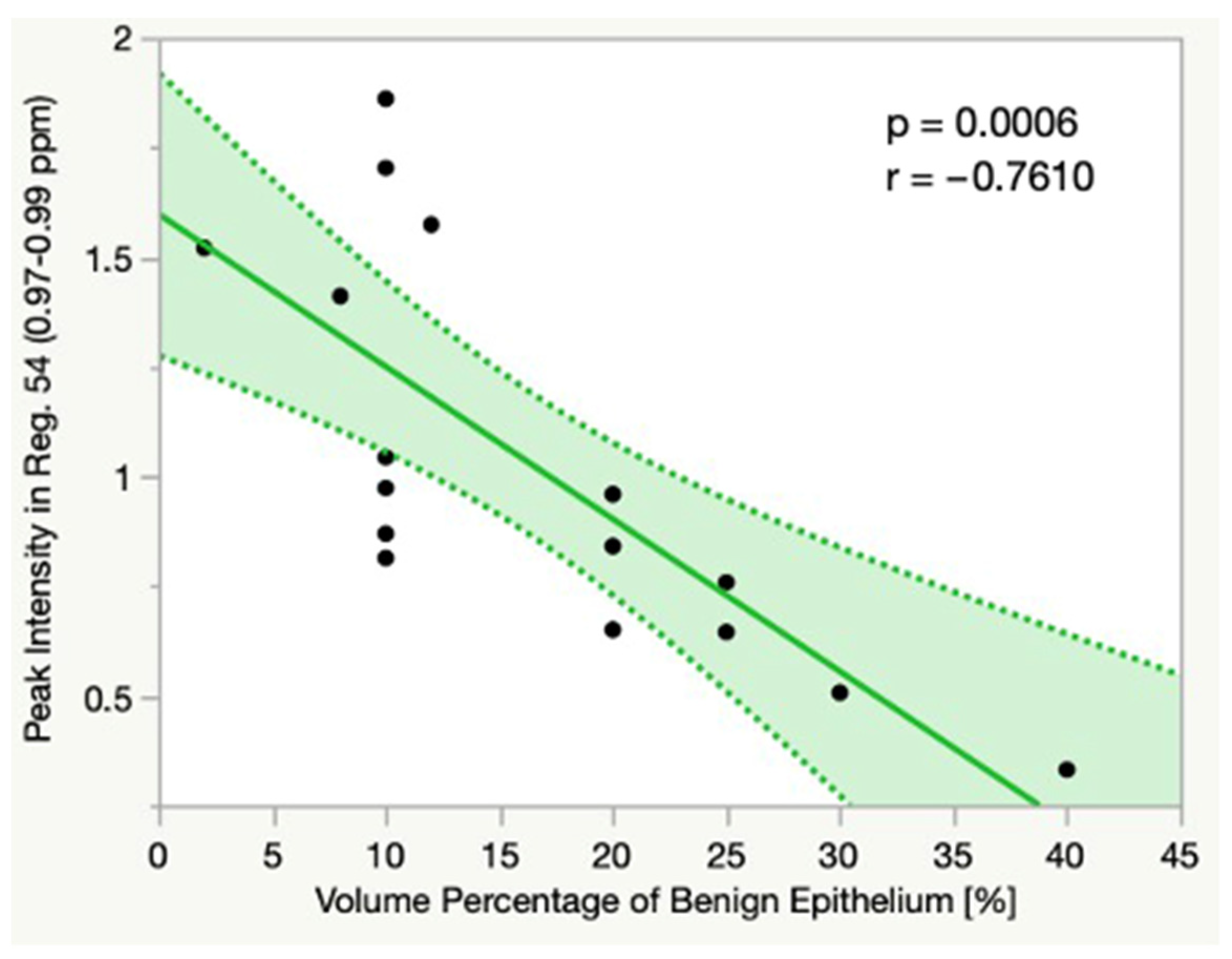
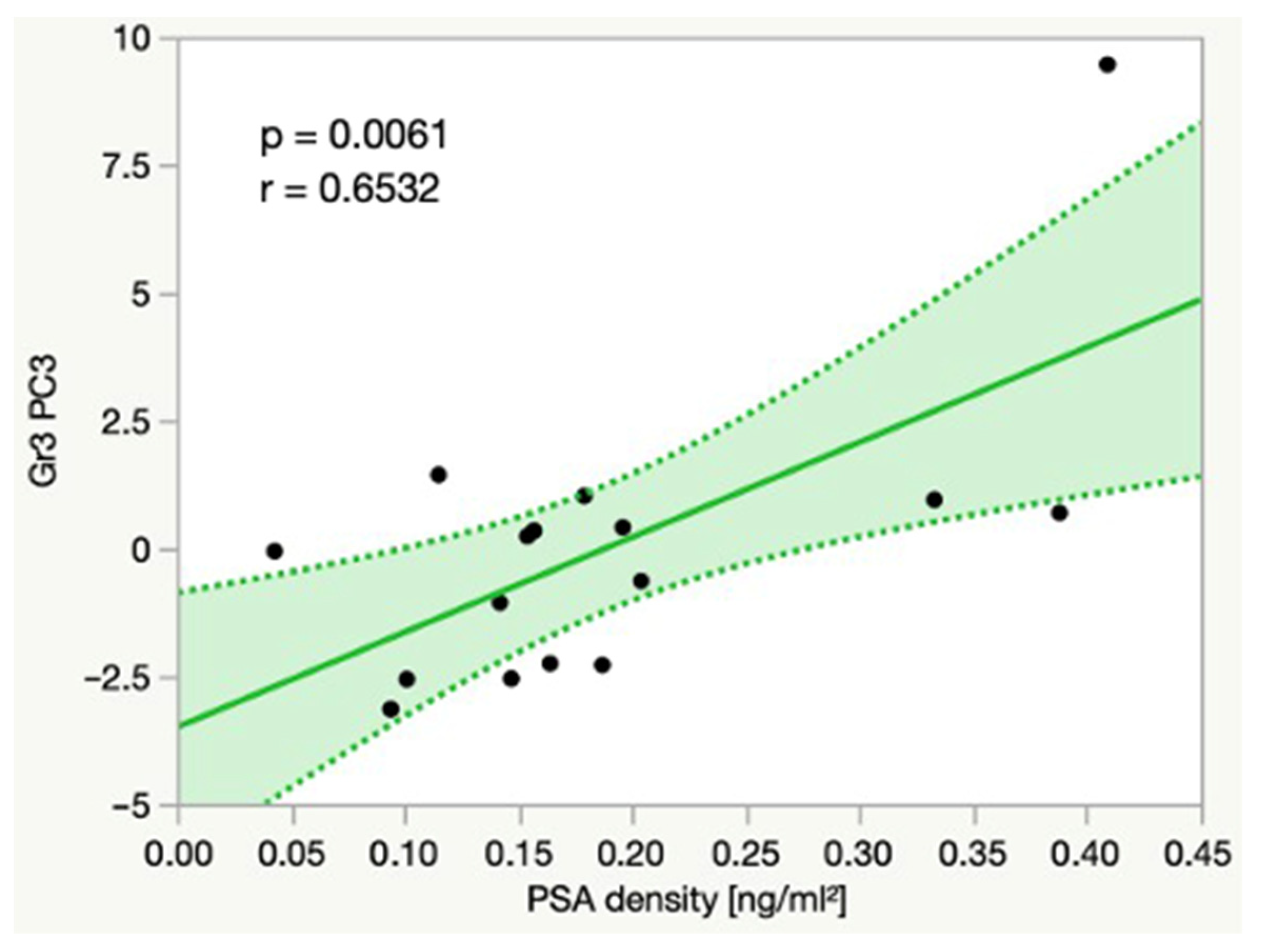
3.6. Linear Correlations
4. Discussion
4.1. Differentiation between Histobenign, Premalignant and Malignant Prostate Tissue
4.2. Correlations between Metabolite Intensities and Histopathology
4.3. Correlations between Metabolite Intensities and PSA Density
4.4. Metabolite Concentrations for the Estimation of Tumor Aggressiveness
4.5. Metabolomic Profiles for Early PCa Detection
4.6. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | ppm Range | Region | ppm Range | Region | ppm Range |
|---|---|---|---|---|---|
| 1 | 4.41–4.50 | 21 | 3.13–3.17 | 41 | 1.99–2.05 |
| 2 | 4.36–4.40 | 22 | 3.09–3.12 | 42 | 1.91–1.96 |
| 3 | 4.28–4.35 | 23 | 3.05–3.08 | 43 | 1.82–1.90 |
| 4 | 4.19–4.27 | 24 | 3.00–3.04 | 44 | 1.74–1.80 |
| 5 | 4.10–4.18 | 25 | 2.96–2.99 | 45 | 1.65–1.73 |
| 6 | 4.03–4.06 | 26 | 2.87–2.95 | 46 | 1.58–1.61 |
| 7 | 3.95–3.99 | 27 | 2.80–2.86 | 47 | 1.51–1.56 |
| 8 | 3.92–3.94 | 28 | 2.75–2.79 | 48 | 1.45–1.48 |
| 9 | 3.9–3.91 | 29 | 2.69–2.74 | 49 | 1.40–1.44 |
| 10 | 3.85–3.89 | 30 | 2.64–2.68 | 50 | 1.35–1.39 |
| 11 | 3.80–3.84 | 31 | 2.58–2.63 | 51 | 1.27–1.34 |
| 12 | 3.76–3.79 | 32 | 2.50–2.57 | 52 | 1.17–1.26 |
| 13 | 3.73–3.75 | 33 | 2.46–2.49 | 53 | 1.00–1.06 |
| 14 | 3.68–3.72 | 34 | 2.39–2.45 | 54 | 0.97–0.99 |
| 15 | 3.66–3.67 | 35 | 2.30–2.38 | 55 | 0.93–0.96 |
| 16 | 3.63–3.65 | 36 | 2.22–2.29 | 56 | 0.77–0.92 |
| 17 | 3.59–3.61 | 37 | 2.19–2.20 | 57 | 0.68–0.74 |
| 18 | 3.30–3.35 | 38 | 2.12–2.17 | 58 | 0.51–0.53 |
| 19 | 3.26–3.29 | 39 | 2.09–2.11 | ||
| 20 | 3.20–3.25 | 40 | 2.06–2.08 |
Appendix B
| Regions | ppm Range | Metabolites | ppm Values | References |
|---|---|---|---|---|
| 1 | 4.41–4.5 | |||
| 2 | 4.36–4.4 | |||
| 3 | 4.28–4.35 | Phosphocholine | 4.28 | Govindaraju 2000 |
| ATP | 4.295 | Govindaraju 2000 | ||
| 4 | 4.19–4.27 | Threonine | 4.24 | Govindaraju 2000 |
| 4.26 | Swindle 2008 | |||
| 5 | 4.1–4.18 | Lactate | 4.10 | Govindaraju 2000 |
| 4.10 | Swindle 2008 | |||
| 4.10–4.14 | Jordan 2007 | |||
| Fructose | 4.10–4.11 | Mickiewicz 2014 | ||
| Proline | 4.12 | Mickiewicz 2014 | ||
| 6 | 4.03–4.06 | Choline | 4.05 | Govindaraju 2000 |
| Tryptophan | 4.05 | Govindaraju 2000 | ||
| 7 | 3.95–3.99 | Serine | 3.97 | Govindaraju 2000 |
| Phenylalanine | 3.98 | Govindaraju 2000 | ||
| Phosphoethanolamine | 3.98 | Govindaraju 2000 | ||
| Histidine | 3.99 | Govindaraju 2000 | ||
| Fructose | 3.99 | Mickiewicz 2014 | ||
| 8 | 3.92–3.94 | Phosphocreatine | 3.92 | Govindaraju 2000 |
| Serine | 3.93 | Govindaraju 2000 | ||
| Tyrosine | 3.93 | Govindaraju 2000 | ||
| Creatine | 3.94 | Stenman 2011 | ||
| 9 | 3.9–3.91 | Creatine | 3.90 | Swindle 2008 |
| 3.91 | Govindaraju 2000 | |||
| 10 | 3.85–3.89 | Glucose | 3.88 | Govindaraju 2000 |
| Aspartate | 3.89 | Govindaraju 2000 | ||
| Fructose | 3.89 | Mickiewicz 2014 | ||
| 11 | 3.8–3.84 | Fructose | 3.82 | Mickiewicz 2014 |
| Glucose | 3.82–3.83 | Govindaraju 2000 | ||
| Serine | 3.83 | Govindaraju 2000 | ||
| 12 | 3.76–3.79 | Glutamine | 3.76 | Govindaraju 2000 |
| Alanine | 3.76 | Govindaraju 2000 | ||
| 3.78 | Stenman 2011 | |||
| Fructose | 3.79 | Mickiewicz 2014 | ||
| 13 | 3.73–3.75 | Glutamate | 3.74 | Govindaraju 2000 |
| Glucose | 3.75 | Govindaraju 2000 | ||
| 14 | 3.68–3.72 | Glycerophosphocholine | 3.68 | Zektzer 2005 |
| 3.69 | Swindle 2008 | |||
| Fructose | 3.70 | Mickiewicz 2014 | ||
| Glucose | 3.70–3.71 | Govindaraju 2000 | ||
| 15 | 3.66–3.67 | Fructose | 3.67 | Mickiewicz 2014 |
| Fructose | 3.63–3.65 | Phosphocholine | 3.62 | Govindaraju 2000 |
| Myo-inositol | 3.63 | Swanson 2006 | ||
| 17 | 3.59–3.61 | Myo-inositol | 3.51–3.61 | Govindaraju 2000 |
| 3.52–3.62 | Stenman 2011 | |||
| Valine | 3.60 | Govindaraju 2000 | ||
| Phosphocholine | 3.61 | Swindle 2008 | ||
| 3.62 | Zektzer 2005 | |||
| 18 | 3.3–3.35 | Glycerophosphoethanolamine | 3.30 | Swanson 2006 |
| Scyllo-inositol | 3.30 | Zektzer 2005 | ||
| 3.33 | Govindaraju 2000 | |||
| 3.35 | Stenman 2010, Stenman 2011 | |||
| 3.35 | Swanson 2006 | |||
| 19 | 3.26–3.29 | Histidine | 3.26 | Govindaraju 2000 |
| Taurine | 3.26 | Swanson 2006 | ||
| 3.26 | Zektzer 2005 | |||
| 3.28 | Swindle 2008 | |||
| Myo-inositol | 3.27 | Govindaraju 2000 | ||
| 3.28 | Swanson 2006 | |||
| 3.28 | Zektzer 2005 | |||
| 3.29 | Stenman 2010, Stenman 2011 | |||
| Phenylalanine | 3.28 | Govindaraju 2000 | ||
| 20 | 3.2–3.25 | Choline | 3.19 | Van Asten 2008 |
| 3.20 | Stenman 2010, Stenman 2011 | |||
| 3.21 | Swanson 2006 | |||
| 3.21 | Swindle 2008 | |||
| 3.21 | Tessem 2008 | |||
| Phosphoethanolamine | 3.21 | Govindaraju 2000 | ||
| 3.22 | Zektzer 2005 | |||
| Glycerophosphocholine | 3.21 | Van Asten 2008 | ||
| 3.21 | Swindle 2008 | |||
| 3.22 | Stenman 2010, Stenman 2011 | |||
| 3.24 | Swanson 2006 | |||
| 3.24 | Tessem 2008 | |||
| Phosphocholine | 3.21 | Van Asten 2008 | ||
| 3.21 | Swindle 2008 | |||
| 3.22 | Stenman 2010, Stenman2011 | |||
| 3.23 | Swanson 2006 | |||
| 3.23 | Tessem 2008 | |||
| Taurine | 3.24 | Govindaraju 2000 | ||
| 3.25 | Stenman 2010, Stenman 2011 | |||
| 3.25 | Swindle 2008 | |||
| Inositol | 3.25 | Swindle 2008 | ||
| 21 | 3.13–3.17 | Polyamines | 3.05–3.15 | Stenman 2010, Stenman 2011 |
| 3.10–3.14 | Tessem 2008 | |||
| Spermine | 3.1–3.2 | Swindle 2008 | ||
| 3.14 | Van Asten 2008 | |||
| Ethanolamine | 3.15 | Zektzer 2005 | ||
| 22 | 3.09–3.12 | Polyamines | 3.05–3.15 | Stenman 2010, Stenman 2011 |
| 3.10–3.14 | Swanson 2006 | |||
| Polyamines (Spermine, Spermidine, Putrescine) | 3.11 | Tessem 2008 | ||
| Spermine | 3.09–3.13 | Tessem 2008 | ||
| Phenylalanine | 3.11 | Govindaraju 2000 | ||
| 23 | 3.05–3.08 | Lysine | 3.05 | Swindle 2008 |
| Polyamine | 3.05–3.15 | Stenman 2010, Stenman 2011 | ||
| 24 | 3–3.04 | Creatine | 3.02 | Stenman 2010, Stenman 2011 |
| 3.026 | Govindaraju 2000 | |||
| 3.03 | Van Asten 2008 | |||
| 3.03 | Swindle 2008 | |||
| 3.04 | Swanson 2006 | |||
| Tyrosine | 3.04 | Govindaraju 2000 | ||
| 25 | 2.96–2.99 | |||
| 26 | 2.87–2.95 | |||
| 27 | 2.8–2.86 | PUFA n6 species | 2.80 | Stenman 2009 |
| Diallylic protons (Omega 6.20) | 2.80 | Stenman 2011 | ||
| Aspartate | 2.80 | Govindaraju 2000 | ||
| Lipid | 2.82 | Swindle 2008 | ||
| 28 | 2.75–2.79 | |||
| 29 | 2.69–2.74 | Citrate | 2.70 | Van Asten 2008 |
| 2.70 | Dittrich 2012 | |||
| 2.72 | Swanson 2006 | |||
| 30 | 2.64–2.68 | Aspartate | 2.65 | Govindaraju 2000 |
| Citrate | 2.65 | Stenman 2010, Stenman 2011 | ||
| 2.66 | Swindle 2008 | |||
| 2.67 | Van Asten 2008 | |||
| 2.67 | Dittrich 2012 | |||
| 31 | 2.58–2.63 | Citrate | 2.62 | Tessem 2008 |
| 32 | 2.5–2.57 | Citrate | 2.51, 2.54 | Dittrich 2012 |
| 2.52 | Swindle 2008 | |||
| 2.54 | Swanson 2006 | |||
| 2.55 | Stenman 2010, Stenman 2011 | |||
| 33 | 2.46–2.49 | Taurine | 2.46 | Stenman 2010, Stenman 2011 |
| Glutamine | 2.47 | Stenman 2010, Stenman 2011 | ||
| 34 | 2.39–2.45 | Succinate | 2.39 | Govindaraju 2000 |
| Glutamine | 2.43, 2.45 | Govindaraju 2000 | ||
| 35 | 2.3–2.38 | Lipid | 2.3 | Swindle 2008 |
| 2.33, 2.35 | Govindaraju 2000 | |||
| Glutamate | 2.35 | Stenman 2010, Stenman 2011 | ||
| Pyruvate | 2.36 | Govindaraju 2000 | ||
| 36 | 2.22–2.29 | Valine | 2.26 | Govindaraju 2000 |
| Lipid | 2.27 | Giskeødegård 2013 | ||
| 37 | 2.19–2.2 | |||
| 38 | 2.12–2.17 | Glutamate | 2.12 | Govindaraju 2000 |
| 2.15 | Stenman 2010, Stenman 2011 | |||
| Glutamine | 2.13 | Govindaraju 2000 | ||
| 2.14 | Stenman 2010, Stenman 2011 | |||
| 39 | 2.09–2.11 | Spermine & Spermidine | 2.10 | Swanson 2006 |
| Spermine | 2.10 | Swindle 2008 | ||
| Polyamines (Spermine, Spermidine, Putrescine) | 2.10 | Tessem 2008 | ||
| 40 | 2.06–2.08 | |||
| 41 | 1.99–2.05 | Proline | 2.02 | Mickiewicz 2014 |
| Lipid | 2.02 | Swindle 2008 | ||
| 2.05 | Giskeødegård 2013 | |||
| Glutamate | 2.04 | Govindaraju 2000 | ||
| 2.05 | Stenman 2010, Stenman 2011 | |||
| 42 | 1.91–1.96 | Acetate | 1.90 | Govindaraju 2000 |
| 43 | 1.82–1.9 | |||
| 44 | 1.74–1.8 | Polyamines (Spermine, Spermidine, Putrescine) Spermine | 1.78 1.78 1.8 | Swanson 2006 Tessem 2008 Swindle 2008 |
| 45 | 1.65–1.73 | Lysine | 1.72 | Swindle 2008 |
| 46 | 1.58–1.61 | Lipid | 1.60 | Giskeødegård 2013 |
| 1.6 | Swindle 2008 | |||
| 47 | 1.51–1.56 | |||
| 48 | 1.45–1.48 | Alanine | 1.47 | Van Asten 2008 |
| 1.47 | Govindaraju 2000 | |||
| 1.47 | Swindle 2008 | |||
| 1.48 | Stenman 2010, Stenman 2011 | |||
| 1.49 | Swanson 2006 | |||
| 1.49 | Tessem 2008 | |||
| 49 | 1.4–1.44 | Lysine | 1.44 | Swindle 2008 |
| 50 | 1.35–1.39 | |||
| 51 | 1.27–1.34 | Lactate | 1.30 | Swindle 2008 |
| 1.31 | Govindaraju 2000 | |||
| 1.33 | Van Asten 2008 | |||
| 1.33 | Stenman 2010, Stenman 2011 | |||
| 1.33 | Tessem 2008 | |||
| 1.34 | Swanson 2006 | |||
| Threonine | 1.31 | Govindaraju 2000 | ||
| 1.31 | Swindle 2008 | |||
| Lipid | 1.33 | Swindle 2008 | ||
| 52 | 1.17–1.26 | |||
| 53 | 1–1.06 | Valine | 1.03 | Govindaraju 2000 |
| 1.03 | Swindle 2008 | |||
| 54 | 0.97–0.99 | (Iso)Leucine | 0.97 | Swindle 2008 |
| Valine | 0.98 | Govindaraju 2000 | ||
| 55 | 0.93–0.96 | |||
| 56 | 0.77–0.92 | Lipid | 0.9 | Swindle 2008 |
| 57 | 0.68–0.74 | |||
| 58 | 0.51–0.53 |
Appendix C. Tables of all Significant Results
| Groups | Principal Component | p |
|---|---|---|
| Gr1 & Gr2 | PC11 | 0.037 |
| Gr1 & 2 PC11 | 0.037 | |
| Gr1 & Gr3 | PC 1 | 0.021 |
| Gr1 & 3 PC1 | 0.011 | |
| Gr2 & Gr3 | Gr2 & 3 PC6 | 0.033 |
| Groups | Region | p |
|---|---|---|
| Gr1 & Gr2 | R17 | 0.021 |
| R18 | 0.003 | |
| R20 | 0.029 | |
| R23 | 0.018 | |
| R27 | 0.026 | |
| R40 | 0.044 | |
| R49 | 0.040 | |
| Gr1 & Gr3 | R17 | 0.029 |
| R18 | 0.013 | |
| R24 | 0.036 | |
| R35 | 0.009 | |
| Gr2 & Gr3 | R23 | 0.005 |
| R36 | 0.023 | |
| R44 | 0.029 | |
| R46 | 0.016 | |
| R52 | 0.032 |
| Gleason Scores | Region | p |
|---|---|---|
| 3 + 3 = 6 & 3 + 4 = 7 | R23 | 0.048 |
| R27 | 0.021 | |
| R28 | 0.013 |
| Groups | Region/PC | p | r |
|---|---|---|---|
| All | R23 | 0.0399 | 0.2976 |
| R30 | 0.0008 | 0.4670 | |
| Gr1 | R1 | 0.0015 | 0.7251 |
| R8 | 0.0219 | 0.5675 | |
| R16 | 0.0095 | 0.6257 | |
| P11 | 0.0414 | −0.5146 | |
| Gr1 PC10 | 0.0304 | −0.5411 | |
| Gr2 | R30 | 0.0487 | 0.4999 |
| Gr3 | R54 | 0.0006 | −0.7610 |
| Groups | Region/PC | p | r |
|---|---|---|---|
| All | R9 | 0.0253 | −0.3227 |
| R50 | 0.0213 | −0.3317 | |
| PC9 | 0.0158 | −0.3466 | |
| Gr1 | R9 | 0.0232 | −0.5631 |
| Gr1 PC8 | 0.0195 | 0.5762 | |
| Gr2 | R9 | 0.0487 | −0.4999 |
| Gr3 | R3 | 0.0326 | 0.5354 |
| R4 | 0.0260 | 0.5539 | |
| R53 | 0.0047 | 0.6682 | |
| R58 | 0.0002 | 0.7984 | |
| PC4 | 0.0238 | −0.5608 | |
| Gr3 PC3 | 0.0061 | 0.6532 |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Pienta, K.J.; Esper, P.S. Risk Factors for Prostate Cancer. Ann. Intern. Med. 1993, 118, 793–803. [Google Scholar] [CrossRef]
- Albertsen, P.C.; Hanley, J.A.; Fine, J. 20-Year Outcomes Following Conservative Management of Clinically Localized Prostate Cancer. JAMA 2005, 293, 2095–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeb, S.; Bjurlin, M.A.; Nicholson, J.; Tammela, T.L.; Penson, D.F.; Carter, H.B.; Carroll, P.; Etzioni, R. Overdiagnosis and Overtreatment of Prostate Cancer. Eur. Urol. 2014, 65, 1046–1055. [Google Scholar] [CrossRef] [Green Version]
- Heidenreich, A. Guidelines and Counselling for Treatment Options in the Management of Prostate Cancer. In Prostate Cancer: Recent Results in Cancer Research; Ramon, J., Denis, L.J., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 131–162. [Google Scholar]
- Mottet, N.; Bellmunt, J.; Bolla, M.; Briers, E.; Cumberbatch, M.G.; De Santis, M.; Fossati, N.; Gross, T.; Henry, A.M.; Joniau, S.; et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part 1: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2017, 71, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Gleason, D.F. Classification of prostatic carcinomas. Cancer Chemother. Rep. 1966, 50, 125–128. [Google Scholar] [PubMed]
- Epstein, J.I.; Allsbrook, W.C., Jr.; Amin, M.B.; Egevad, L.L. The 2005 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma. Am. J. Surg. Pathol. 2005, 29, 1228–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Master, V.A.; Chi, T.; Simko, J.P.; Weinberg, V.; Carroll, P.R. The Independent Impact of Extended Pattern Biopsy on Prostate Cancer Stage Migration. J. Urol. 2005, 174, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Catalona, W.J.; Smith, D.S.; Ratliff, T.L.; Basler, J.W. Detection of organ-confined prostate cancer is increased through prostate-specific antigen-based screening. JAMA 1993, 270, 948–954. [Google Scholar] [CrossRef]
- Cheng, L.L.; Burns, M.A.; Taylor, J.L.; He, W.; Halpern, E.F.; McDougal, W.S.; Wu, C.-L. Metabolic Characterization of Human Prostate Cancer with Tissue Magnetic Resonance Spectroscopy. Cancer Res. 2005, 65, 3030–3034. [Google Scholar] [CrossRef] [Green Version]
- Stamey, T.A.; Yang, N.; Hay, A.R.; McNeal, J.E.; Freiha, F.S.; Redwine, E. Prostate-Specific Antigen as a Serum Marker for Adenocarcinoma of the Prostate. N. Engl. J. Med. 1987, 317, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Robles, J.M.; Morell, A.R.; Redorta, J.P.; de Torres Mateos, J.; Roselló, A.S. Clinical Behavior of Prostatic Specific Antigen and Prostatic Acid Phosphatase: A Comparative Study. Eur. Urol. 1988, 14, 360–366. [Google Scholar] [CrossRef]
- Sakr, W.; Brawer, M.; Moul, J.; Donohue, R.; Schulman, C.G.; Sakr, D. Pathology and bio markers of prostate cancer. Prostate Cancer Prostatic Dis. 1999, 2, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presti, J.C.; O’Dowd, G.J.; Miller, M.C.; Mattu, R.; Veltri, R.W. Extended peripheral zone biopsy schemes increase cancer detection rates and minimize variance in prostate specific antigen and age related cancer rates: Results of a community multi-practice study. J. Urol. 2003, 169, 125–129. [Google Scholar] [CrossRef]
- Cheng, L.L.; Lean, C.L.; Bogdanova, A.; Wright, S.C., Jr.; Ackerman, J.L.; Brady, T.J.; Garrido, L. Enhanced resolution of proton NMR spectra of malignant lymph nodes using magic-angle spinning. Magn. Reson. Med. 1996, 36, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Bertilsson, H.; Tessem, M.-B.; Flatberg, A.; Viset, T.; Gribbestad, I.; Angelsen, A.; Halgunset, J. Changes in Gene Transcription Underlying the Aberrant Citrate and Choline Metabolism in Human Prostate Cancer Samples. Clin. Cancer Res. 2012, 18, 3261–3269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.L.; Wu, C.-L.; Smith, M.R.; Gonzalez, R. Non-destructive quantitation of spermine in human prostate tissue samples using HRMAS 1H NMR spectroscopy at 9.4 T. FEBS Lett. 2001, 494, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Jordan, K.W.; Cheng, L.L. NMR-based metabolomics approach to target biomarkers for human prostate cancer. Expert Rev. Proteom. 2007, 4, 389–400. [Google Scholar] [CrossRef]
- Fuss, T.L.; Cheng, L.L. Evaluation of Cancer Metabolomics Using ex vivo High Resolution Magic Angle Spinning (HRMAS) Magnetic Resonance Spectroscopy (MRS). Metabolites 2016, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Liao, G.-Q.; Wen, X.-F.; Chen, W.-H.; Cheng, S.; Stolzenburg, J.-U.; Ganzer, R.; Neuhaus, J. Nuclear magnetic resonance spectroscopy as a new approach for improvement of early diagnosis and risk stratification of prostate cancer. J. Zhejiang Univ. Sci. B 2017, 18, 921–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurth, J.; Defeo, E.; Cheng, L.L. Magnetic resonance spectroscopy: A promising tool for the diagnostics of human prostate cancer? Urol. Oncol. 2011, 29, 562–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trock, B.J. Application of metabolomics to prostate cancer. Urol. Oncol. 2011, 29, 572–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-L.; Jordan, K.W.; Ratai, E.M.; Sheng, J.; Adkins, C.B.; DeFeo, E.M.; Jenkins, B.G.; Ying, L.; McDougal, W.S.; Cheng, L.L. Metabolomic Imaging for Human Prostate Cancer Detection. Sci. Transl. Med. 2010, 2, 16ra8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandergrift, L.A.; Decelle, E.A.; Kurth, J.; Wu, S.; Fuss, T.L.; Defeo, E.M.; Halpern, E.F.; Taupitz, M.; McDougal, W.S.; Olumi, A.F.; et al. Metabolomic Prediction of Human Prostate Cancer Aggressiveness: Magnetic Resonance Spectroscopy of Histologically Benign Tissue. Sci. Rep. 2018, 8, 4997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilgner, M.; Vater, T.S.; Habbel, P.; Cheng, L.L. High-Resolution Magic Angle Spinning (HRMAS) NMR Methods in Metabolomics. Methods Mol. Biol. 2019, 2037, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.R.; De Bastos, M.L.; Carvalho, M.; de Pinho, P.G. Biomarker Discovery in Human Prostate Cancer: An Update in Metabolomics Studies. Transl. Oncol. 2016, 9, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Swanson, M.G.; Vigneron, D.B.; Tabatabai, Z.L.; Males, R.G.; Schmitt, L.; Carroll, P.R.; James, J.K.; Hurd, R.E.; Kurhanewicz, J. Proton HR-MAS spectroscopy and quantitative pathologic analysis of MRI/3D-MRSI-targeted postsurgical prostate tissues. Magn. Reson. Med. 2003, 50, 944–954. [Google Scholar] [CrossRef]
- Swanson, M.G.; Zektzer, A.S.; Tabatabai, Z.L.; Simko, J.; Jarso, S.; Keshari, K.R.; Schmitt, L.; Carroll, P.R.; Shinohara, K.; Vigneron, D.B.; et al. Quantitative analysis of prostate metabolites using1H HR-MAS spectroscopy. Magn. Reson. Med. 2006, 55, 1257–1264. [Google Scholar] [CrossRef]
- Lima, A.R.; Pinto, J.; Bastos, M.D.L.; Carvalho, M.; De Pinho, P.G. NMR-based metabolomics studies of human prostate cancer tissue. Metabolomics 2018, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Stenman, K.; Stattin, P.; Stenlund, H.; Riklund, K.; Gröbner, G.; Bergh, A. 1H HRMAS NMR Derived Bio-markers Related to Tumor Grade, Tumor Cell Fraction, and Cell Proliferation in Prostate Tissue Samples. Biomark. Insights 2011, 6, BMI-S6794. [Google Scholar] [CrossRef] [Green Version]
- Swanson, M.G.; Keshari, K.R.; Tabatabai, Z.L.; Simko, J.P.; Shinohara, K.; Carroll, P.R.; Zektzer, A.S.; Kurhanewicz, J. Quantification of choline- and ethanolamine-containing metabolites in human prostate tissues using 1H HR-MAS total correlation spectroscopy. Magn. Reson. Med. 2008, 60, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell. Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Madhu, B.; Shaw, G.L.; Warren, A.Y.; Neal, D.E.; Griffiths, J.R. Absolute quantitation of metabolites in human prostate cancer biopsies by HR-MAS 1H NMR spectroscopy. In Proceedings of the International Society for Magnetic Resonance in Medicine 2014, Milan, Italy, 10–16 May 2014; p. 4096. [Google Scholar]
- Costello, L.C.; Franklin, R.B. Citrate metabolism of normal and malignant prostate epithelial cells. Urology 1997, 50, 3–12. [Google Scholar] [CrossRef]
- Burns, M.A.; Taylor, J.L.; Wu, C.-L.; Zepeda, A.G.; Bielecki, A.; Cory, D.; Cheng, L.L. Reduction of spinning sidebands in proton NMR of human prostate tissue with slow high-resolution magic angle spinning. Magn. Reson. Med. 2005, 54, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Teahan, O.; Bevan, C.L.; Waxman, J.; Keun, H.C. Metabolic signatures of malignant progression in prostate epithelial cells. Int. J. Biochem. Cell Biol. 2011, 43, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, R.; Kurth, J.; Decelle, E.A.; Defeo, E.M.; Taupitz, M.; Wu, S.; Wu, C.-L.; McDougal, W.S.; Cheng, L.L. Assessing prostate cancer growth with citrate measured by intact tissue proton magnetic resonance spectroscopy. Prostate Cancer Prostatic Dis. 2012, 15, 278–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenman, K.; Hauksson, J.B.; Gröbner, G.; Stattin, P.; Bergh, A.; Riklund, K. Detection of polyunsaturated omega-6 fatty acid in human malignant prostate tissue by 1D and 2D high-resolution magic angle spinning NMR spectroscopy. Magn. Reson. Mater. Phys. Biol. Med. 2009, 22, 327. [Google Scholar] [CrossRef] [PubMed]
- Van Asten, J.J.A.; Cuijpers, V.; De Kaa, C.H.-V.; Soede-Huijbregts, C.; Witjes, J.A.; Verhofstad, A.; Heerschap, A. High resolution magic angle spinning NMR spectroscopy for metabolic assessment of cancer presence and Gleason score in human prostate needle biopsies. Magn. Reson. Mater. Phys. Biol. Med. 2008, 21, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Giskeødegård, G.F.; Bertilsson, H.; Selnæs, K.M.; Wright, A.J.; Bathen, T.F.; Viset, T.; Halgunset, J.; Angelsen, A.; Gribbestad, I.S.; Tessem, M.-B. Spermine and Citrate as Metabolic Biomarkers for Assessing Prostate Cancer Aggressiveness. PLoS ONE 2013, 8, e62375. [Google Scholar] [CrossRef] [PubMed] [Green Version]





| Clinical Parameter | Group | Mean | Standard Deviation | Minimum | Maximum | Unit |
|---|---|---|---|---|---|---|
| Age at Bx0 | All Gr | 62.29 | 7.23 | 44 | 77 | years |
| Gr1 | 60.25 | 6.28 | 46 | 71 | ||
| Gr2 | 62.13 | 6.52 | 44 | 73 | ||
| Gr3 | 64.50 | 8.49 | 46 | 77 | ||
| Pre-Bx0 PSA | All Gr | 7.74 | 3.62 | 2.33 | 18.14 | ng/mL |
| Gr1 | 6.75 | 2.46 | 2.70 | 12.56 | ||
| Gr2 | 8.84 | 3.43 | 3.50 | 18.14 | ||
| Gr3 | 7.63 | 4.56 | 2.33 | 18.00 | ||
| Prostate Vol. | All Gr | 46.58 | 28.84 | 18.14 | 182.00 | mL |
| Gr1 | 40.77 | 26.51 | 18.14 | 126.00 | ||
| Gr2 | 57.00 | 39.35 | 24.00 | 182.00 | ||
| Gr3 | 41.98 | 13.47 | 22.90 | 71.00 | ||
| PSAd | All Gr | 0.20 | 0.11 | 0.04 | 0.50 | ng/mL2 |
| Gr1 | 0.21 | 0.12 | 0.05 | 0.43 | ||
| Gr2 | 0.19 | 0.11 | 0.06 | 0.50 | ||
| Gr3 | 0.19 | 0.10 | 0.04 | 0.41 | ||
| Biopsy characteristics | Number of Patients | |||||
| Biopsy type: | ||||||
| Fusion bx with 2 samples | 14 | |||||
| Regular bx with 1 sample | 34 | |||||
| Bx0 as 1st, 2nd or 3rd biopsy: | ||||||
| 1st | 23 | |||||
| 2nd | 13 | |||||
| 3rd | 12 | |||||
| Prostate region of Bx sample at regular biopsies: | ||||||
| Right mid | 27 | |||||
| Right apex | 1 | |||||
| Right base | 1 | |||||
| No details provided | 5 | |||||
| Target region at fusion biopsies: | ||||||
| Right target | 4 | |||||
| Left target | 10 | |||||
| Parameter | Number of Patients |
|---|---|
| Highest Bx GS until end of study period in Gr1 and Gr3: | |
| 3 + 3 = 6 | 12 |
| 3 + 4 = 7 | 16 |
| 4 + 3 = 7 | 4 |
| Pi-RADS all groups: | |
| 2 | 1 |
| 3 | 5 |
| 4 | 4 |
| 5 | 5 |
| Date of first PCa diagnosis in relation to date of Bx0 in Gr1: | |
| >2 y after Bx0 (Max: 5 y 6 m) | 6 |
| 1–2 y after Bx0 | 6 |
| <1 y after Bx0 (Min: 0 y 7 m) | 4 |
| Date of first PCa diagnosis in relation to date of Bx0 in Gr3 | |
| At Bx0 | 12 |
| <1 y before Bx0 | 1 |
| 1–2 y before Bx0 | 1 |
| >2 y. before Bx0 (Max: 5 y 4 m) | 2 |
| Prostatectomy before end of study period in Gr1 and Gr3 | |
| Yes | 20 |
| No | 12 |
| GS Prostatectomy | |
| 3 + 3 = 6 | 2 |
| 3 + 4 = 7 | 13 |
| 4 + 3 = 7 | 4 |
| 4 + 5 = 9 | 1 |
| Comparison of GS at Bx0 vs. GS at prostatectomy | |
| Same | 10 |
| Higher at PE | 8 |
| Higher at Bx0 | 2 |
| pTNM | |
| T1c | 1 |
| T2a | 1 |
| T2c | 5 |
| T3a | 12 |
| N+ | 3 |
| M+ | 3 |
| Histopathological Parameter | Group | Mean | Standard Deviation | Minimum | Maximum | Unit |
|---|---|---|---|---|---|---|
| Vol.%Epi | All groups | 18.77 | 12.36 | 0 | 55 | % |
| Gr1 | 21.38 | 16.72 | 0 | 55 | ||
| Gr2 | 18.56 | 9.37 | 2 | 35 | ||
| Gr3 | 16.38 | 9.91 | 2 | 40 | ||
| Vol.%Ca | Gr3 | 20.06 | 18.37 | 5 | 60 | % |
| Vol.% Stroma | All groups | 74.54 | 16.16 | 30 | 100 | % |
| Gr1 | 78.63 | 16.72 | 45 | 100 | ||
| Gr2 | 81.44 | 9.37 | 65 | 98 | ||
| Gr3 | 63.56 | 15.95 | 30 | 85 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steiner, A.; Schmidt, S.A.; Fellmann, C.S.; Nowak, J.; Wu, C.-L.; Feldman, A.S.; Beer, M.; Cheng, L.L. Ex Vivo High-Resolution Magic Angle Spinning (HRMAS) 1H NMR Spectroscopy for Early Prostate Cancer Detection. Cancers 2022, 14, 2162. https://doi.org/10.3390/cancers14092162
Steiner A, Schmidt SA, Fellmann CS, Nowak J, Wu C-L, Feldman AS, Beer M, Cheng LL. Ex Vivo High-Resolution Magic Angle Spinning (HRMAS) 1H NMR Spectroscopy for Early Prostate Cancer Detection. Cancers. 2022; 14(9):2162. https://doi.org/10.3390/cancers14092162
Chicago/Turabian StyleSteiner, Annabel, Stefan Andreas Schmidt, Cara Sophie Fellmann, Johannes Nowak, Chin-Lee Wu, Adam Scott Feldman, Meinrad Beer, and Leo L. Cheng. 2022. "Ex Vivo High-Resolution Magic Angle Spinning (HRMAS) 1H NMR Spectroscopy for Early Prostate Cancer Detection" Cancers 14, no. 9: 2162. https://doi.org/10.3390/cancers14092162
APA StyleSteiner, A., Schmidt, S. A., Fellmann, C. S., Nowak, J., Wu, C. -L., Feldman, A. S., Beer, M., & Cheng, L. L. (2022). Ex Vivo High-Resolution Magic Angle Spinning (HRMAS) 1H NMR Spectroscopy for Early Prostate Cancer Detection. Cancers, 14(9), 2162. https://doi.org/10.3390/cancers14092162