
Senescence Is the Main Trait Induced by Temozolomide in Glioblastoma Cells


 ,
,  , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Drugs and Treatments
2.3. Quantification of Apoptosis and Necrosis
2.4. Quantification of Cellular Senescence
2.5. Quantification of Polyploidy
2.6. Quantification of γH2AX, 53BP1, and Trf1 Foci
2.7. Neutral and FPG-Alkaline Comet Assays
2.8. Protein Extraction and Western Blot Analysis
2.9. Immunohistochemistry
2.10. Patients and Ethic Statement
2.11. Statistical Analysis and Mathematical Assessments
3. Results
3.1. Kinetics of CSEN
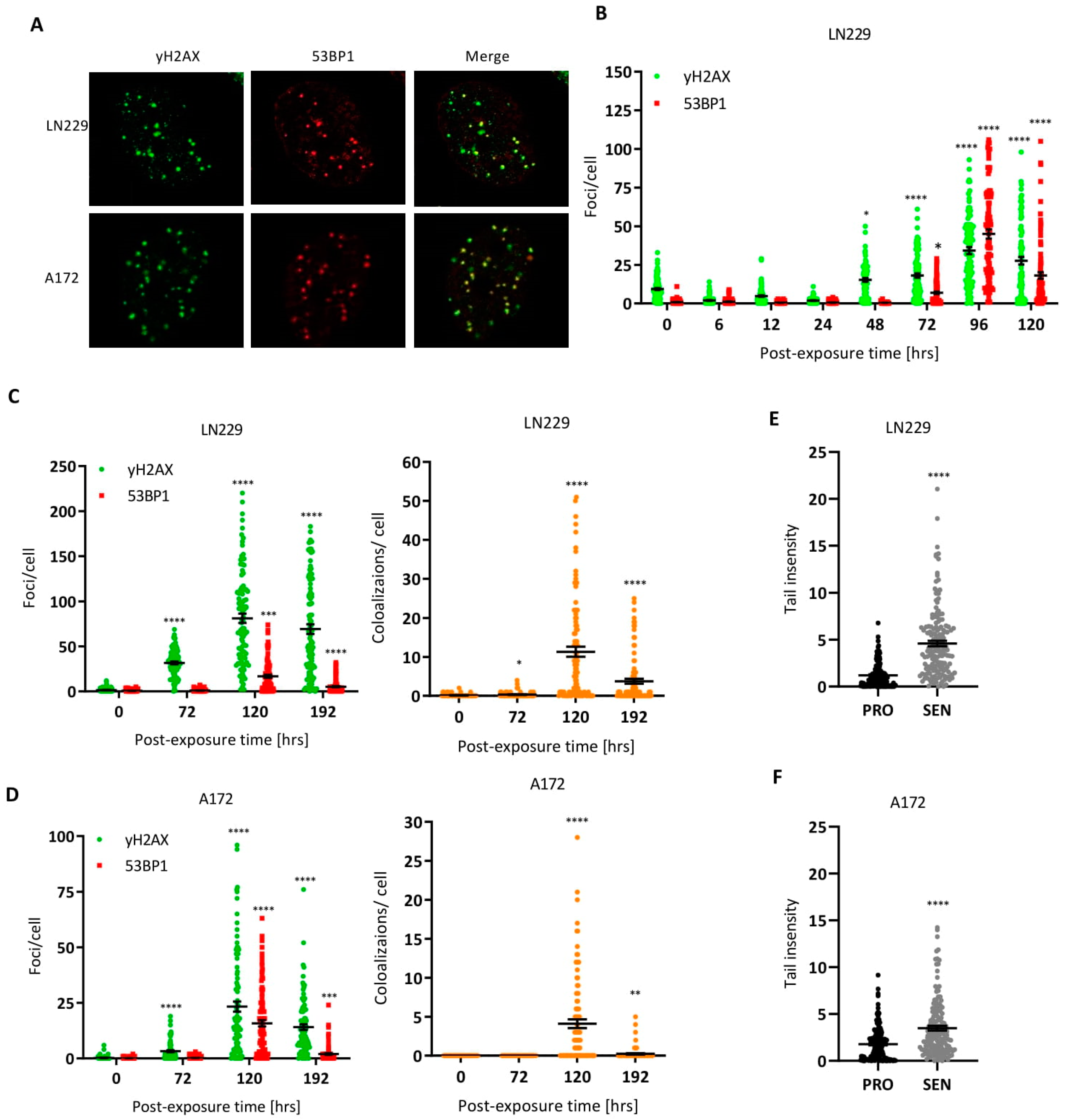
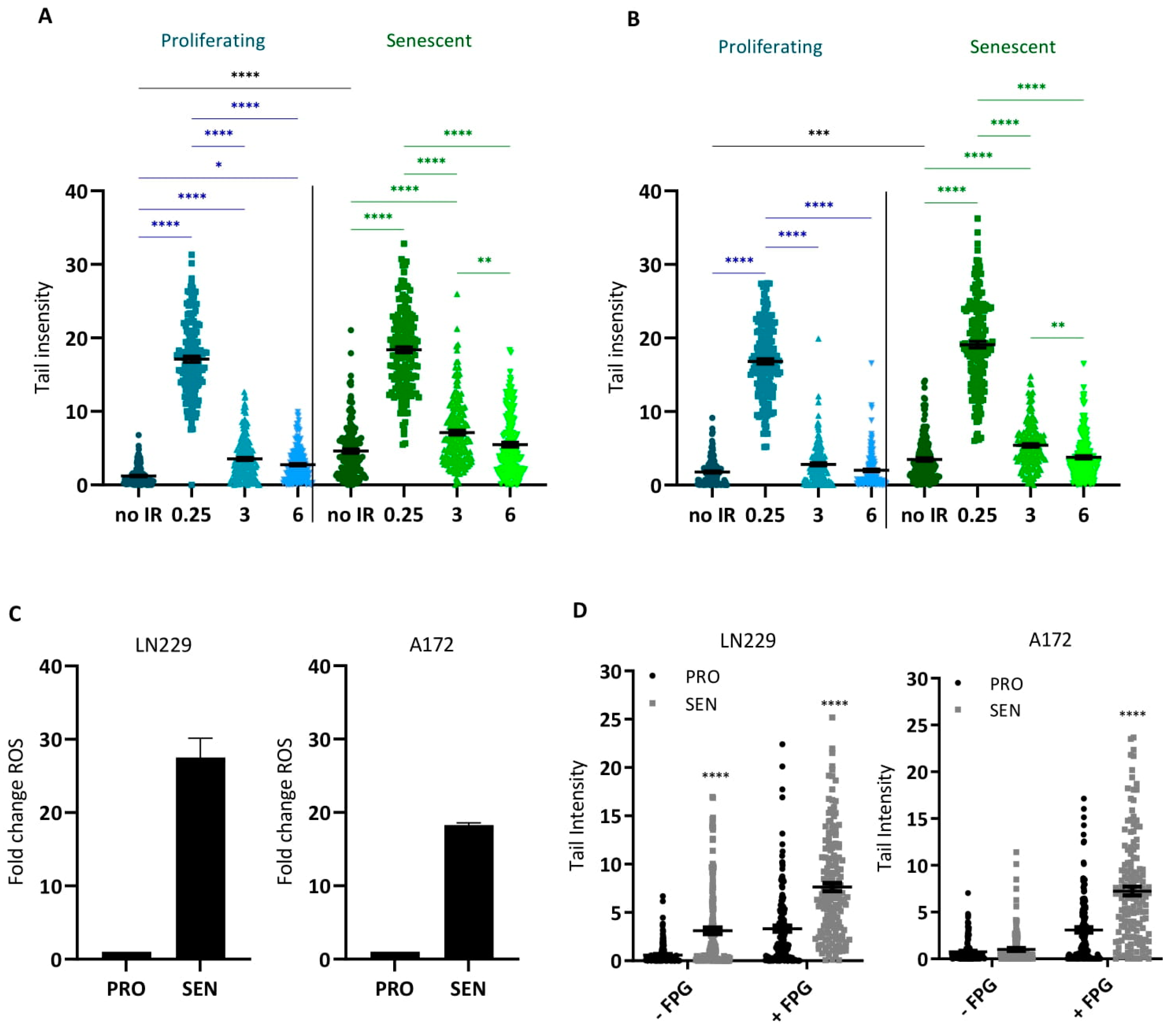
3.2. DNA Double-Strand Breaks in Senescent Cells
3.3. Are TMZ-Induced DSBs in CSEN Cells Localized in Telomeres?
3.4. Are DSBs Subject to Repair in Senescent Cells?
3.5. ROS and Oxidative DNA Damage in Senescent Cells
3.6. Is O6MeG Required for Maintaining the Senescent State?
3.7. Sustained Activation of the DNA Damage Response in Senescent Cells
3.8. Is Acquired TMZ Resistance Associated with Resistance to Induction of CSEN?
3.9. TMZ-Induced CSEN Displays the Inflammatory Phenotype
3.10. Senescence in Glioblastoma In Situ Following TMZ-Based Therapy
4. Discussion
5. Conclusions and Hypotheses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newlands, E.; Stevens, M.; Wedge, S.; Wheelhouse, R.; Brock, C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Fritz, G.; Mitra, S.; Coquerelle, T. Transfection and expression of human O6-methylguanine-DNA methyltransferase (MGMT) cDNA in Chinese hamster cells: The role of MGMT in protection against the genotoxic effects of alkylating agents. Carcinogenesis 1991, 12, 1857–1867. [Google Scholar] [PubMed]
- Margison, G.P.; Santibanez Koref, M.F.; Povey, A.C. Mechanisms of carcinogenicity/chemotherapy by O6-methylguanine. Mutagenesis 2002, 17, 483–487. [Google Scholar] [CrossRef] [Green Version]
- Beranek, D.T. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat. Res. 1990, 231, 11–30. [Google Scholar] [CrossRef]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE 2013, 8, e55665. [Google Scholar] [CrossRef] [Green Version]
- Pegg, A.E. Repair of O6-alkylguanine by alkyltransferases. Mutat. Res. 2000, 462, 83–100. [Google Scholar] [CrossRef]
- Christmann, M.; Nagel, G.; Horn, S.; Krahn, U.; Wiewrodt, D.; Sommer, C.; Kaina, B. MGMT activity, promoter methylation and immunohistochemistry of pretreatment and recurrent malignant gliomas: A comparative study on astrocytoma and glioblastoma. Int. J. Cancer 2010, 127, 2106–2118. [Google Scholar] [CrossRef]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of. gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Switzeny, O.J.; Christmann, M.; Renovanz, M.; Giese, A.; Sommer, C.; Kaina, B. MGMT promoter methylation determined by HRM in comparison to MSP and pyrosequencing for predicting high-grade glioma response. Clin. Epigenet. 2016, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaina, B.; Christmann, M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair 2019, 78, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Ochs, K.; Kaina, B. Apoptosis induced by DNA damage O6-methylguanine is Bcl-2 and caspase-9/3 regulated and Fas/caspase-8 independent. Cancer Res. 2000, 60, 5815–5824. [Google Scholar]
- Quiros, S.; Roos, W.P.; Kaina, B. Processing of O6-methylguanine into DNA double-strand breaks requires two rounds of replication whereas apoptosis is also induced in subsequent cell cycles. Cell Cycle 2010, 9, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Eich, M.; Roos, W.P.; Nikolova, T.; Kaina, B. Contribution of ATM and ATR to the resistance of glioblastoma and malignant melanoma cells to the methylating anticancer drug temozolomide. Mol. Cancer Ther. 2013, 12, 2529–2540. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Roos, W.P.; Wu, Q.; Hofmann, T.G.; Kaina, B. The SIAH1-HIPK2-p53ser46 Damage Response Pathway is Involved in Temozolomide-Induced Glioblastoma Cell Death. Mol. Cancer Res. 2019, 17, 1129–1141. [Google Scholar] [CrossRef] [Green Version]
- Aasland, D.; Gotzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR-CHK1, p21, and NF-kappaB. Cancer Res. 2019, 79, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Kaina, B. Temozolomide in Glioblastoma Therapy: Role of Apoptosis, Senescence and Autophagy. Comment on Strobel et al., Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. [Google Scholar] [CrossRef] [Green Version]
- Ensminger, M.; Iloff, L.; Ebel, C.; Nikolova, T.; Kaina, B.; Löbrich, M. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 2014, 206, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Anton, K.; Baehring, J.M.; Mayer, T. Glioblastoma multiforme: Overview of current treatment and future perspectives. Hematol. Oncol. Clin. N. Am. 2012, 26, 825–853. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Winkler, F. Regimen of procarbazine, lomustine, and vincristine versus temozolomide for gliomas. Cancer 2018, 124, 2674–2676. [Google Scholar] [CrossRef] [PubMed]
- Stratenwerth, B.; Geisen, S.M.; He, Y.; Beltzig, L.; Sturla, S.J.; Kaina, B. Molecular Dosimetry of Temozolomide: Quantification of Critical Lesions, Correlation to Cell Death Responses, and Threshold Doses. Mol. Cancer Ther. 2021, 20, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Tano, K.; Shiota, S.; Collier, J.; Foote, R.S.; Mitra, S. Isolation and structural characterization of a cDNA clone encoding the human DNA repair protein for O6-alkylguanine. Proc. Natl. Acad. Sci. USA 1990, 87, 686–690. [Google Scholar] [CrossRef] [Green Version]
- Glousker, G.; Briod, A.S.; Quadroni, M.; Lingner, J. Human shelterin protein POT1 prevents severe telomere instability induced by homology-directed DNA repair. EMBO J. 2020, 39, e104500. [Google Scholar] [CrossRef]
- He, Y.; Kaina, B. Are There Thresholds in Glioblastoma Cell Death Responses Triggered by Temozolomide? Int. J. Mol. Sci. 2019, 20, 1562. [Google Scholar] [CrossRef] [Green Version]
- Kaina, B.; Beltzig, L.; Piee-Staffa, A.; Haas, B. Cytotoxic and Senolytic Effects of Methadone in Combination with Temozolomide in Glioblastoma Cells. Int. J. Mol. Sci. 2020, 21, 7006. [Google Scholar] [CrossRef]
- Schwarzenbach, C.; Tatsch, L.; Brandstetter Vilar, J.; Rasenberger, B.; Beltzig, L.; Kaina, B.; Tomicic, M.T.; Christmann, M. Targeting c-IAP1, c-IAP2, and Bcl-2 Eliminates Senescent Glioblastoma Cells Following Temozolomide Treatment. Cancers 2021, 13, 3585. [Google Scholar] [CrossRef]
- Roos, W.P.; Batista, L.F.Z.; Naumann, S.; Wick, W.; Weller, M.; Menck, C.F.M.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Galli, M.; Frigerio, C.; Longhese, M.P.; Clerici, M. The regulation of the DNA damage response at telomeres: Focus on kinases. Biochem. Soc. Trans. 2021, 49, 933–943. [Google Scholar] [CrossRef]
- Bianchi, A.; Smith, S.; Chong, L.; Elias, P.; de Lange, T. TRF1 is a dimer and bends telomeric DNA. EMBO J. 1997, 16, 1785–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muruzabal, D.; Collins, A.; Azqueta, A. The enzyme-modified comet assay: Past, present and future. Food Chem. Toxicol. 2021, 147, 111865. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.L.; Yang, C.C.; Tseng, H.C.; Hsiao, L.D.; Lin, C.C.; Yang, C.M. Haem oxygenase-1 up-regulation by rosiglitazone via ROS-dependent Nrf2-antioxidant response elements axis or PPARgamma attenuates LPS-mediated lung inflammation. Br. J. Pharmacol. 2018, 175, 3928–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agger, K.; Cloos, P.A.; Rudkjaer, L.; Williams, K.; Andersen, G.; Christensen, J.; Helin, K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 2009, 23, 1171–1176. [Google Scholar] [CrossRef] [Green Version]
- Wiewrodt, D.; Nagel, G.; Dreimuller, N.; Hundsberger, T.; Perneczky, A.; Kaina, B. MGMT in primary and recurrent human glioblastomas after radiation and chemotherapy and comparison with p53 status and clinical outcome. Int. J. Cancer 2008, 122, 1391–1399. [Google Scholar] [CrossRef]
- Esteller, M.; Hamilton, S.R.; Burger, P.C.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999, 59, 793–797. [Google Scholar]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.D.; Krex, D.; Grauer, O.; et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): A randomised, open-label, phase 3 trial. Lancet 2019, 393, 678–688. [Google Scholar] [CrossRef]
- Stepanenko, A.A.; Chekhonin, V.P. Recent Advances in Oncolytic Virotherapy and Immunotherapy for Glioblastoma: A Glimmer of Hope in the Search for an Effective Therapy? Cancers 2018, 10, 492. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.; Steinbach, J.P.; Wick, W. Temozolomide: A milestone in the pharmacotherapy of brain tumors. Future Oncol. 2005, 1, 747–754. [Google Scholar] [CrossRef] [PubMed]
- van Steensel, B.; Smogorzewska, A.; de Lange, T. TRF2 protects human telomeres from end-to-end fusions. Cell 1998, 92, 401–413. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, Z.D.; Kitange, G.J.; Gupta, S.K.; Joughin, B.A.; Chaim, I.A.; Mazzucato, P.; Lauffenburger, D.A.; Sarkaria, J.N.; Samson, L.D. DNA Repair Capacity in Multiple Pathways Predicts Chemoresistance in Glioblastoma Multiforme. Cancer Res. 2017, 77, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Batchelor, T. Temozolomide for malignant brain tumours. Lancet 2000, 355, 1115–1116. [Google Scholar] [CrossRef]
- Danson, S.J.; Middleton, M.R. Temozolomide: A novel oral alkylating agent. Expert Rev. Anticancer Ther. 2001, 1, 13–19. [Google Scholar] [CrossRef]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar]
- Marzolini, C.; Decosterd, L.A.; Shen, F.; Gander, M.; Leyvraz, S.; Bauer, J.; Buclin, T.; Biollaz, J.; Lejeune, F. Pharmacokinetics of temozolomide in association with fotemustine in malignant melanoma and malignant glioma patients: Comparison of oral, intravenous, and hepatic intra-arterial administration. Cancer Chemother. Pharmacol. 1998, 42, 433–440. [Google Scholar] [CrossRef]
- Portnow, J.; Badie, B.; Chen, M.; Liu, A.; Blanchard, S.; Synold, T.W. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: Potential implications for the current approach to chemoradiation. Clin. Cancer Res. 2009, 15, 7092–7098. [Google Scholar] [CrossRef] [Green Version]
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. 2004, 10, 3728–3736. [Google Scholar] [CrossRef] [Green Version]
- Beltzig, L.; Stratenwerth, B.; Kaina, B. Accumulation of Temozolomide-Induced Apoptosis, Senescence and DNA Damage by Metronomic Dose Schedule: A Proof-of-Principle Study with Glioblastoma Cells. Cancers 2021, 13, 6287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. FEBS J. 2022. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beltzig, L.; Schwarzenbach, C.; Leukel, P.; Frauenknecht, K.B.M.; Sommer, C.; Tancredi, A.; Hegi, M.E.; Christmann, M.; Kaina, B. Senescence Is the Main Trait Induced by Temozolomide in Glioblastoma Cells. Cancers 2022, 14, 2233. https://doi.org/10.3390/cancers14092233
Beltzig L, Schwarzenbach C, Leukel P, Frauenknecht KBM, Sommer C, Tancredi A, Hegi ME, Christmann M, Kaina B. Senescence Is the Main Trait Induced by Temozolomide in Glioblastoma Cells. Cancers. 2022; 14(9):2233. https://doi.org/10.3390/cancers14092233
Chicago/Turabian StyleBeltzig, Lea, Christian Schwarzenbach, Petra Leukel, Katrin B. M. Frauenknecht, Clemens Sommer, Alessandro Tancredi, Monika E. Hegi, Markus Christmann, and Bernd Kaina. 2022. "Senescence Is the Main Trait Induced by Temozolomide in Glioblastoma Cells" Cancers 14, no. 9: 2233. https://doi.org/10.3390/cancers14092233
APA StyleBeltzig, L., Schwarzenbach, C., Leukel, P., Frauenknecht, K. B. M., Sommer, C., Tancredi, A., Hegi, M. E., Christmann, M., & Kaina, B. (2022). Senescence Is the Main Trait Induced by Temozolomide in Glioblastoma Cells. Cancers, 14(9), 2233. https://doi.org/10.3390/cancers14092233