
Genetic and Drug Inhibition of LDH-A: Effects on Murine Gliomas


 and
and 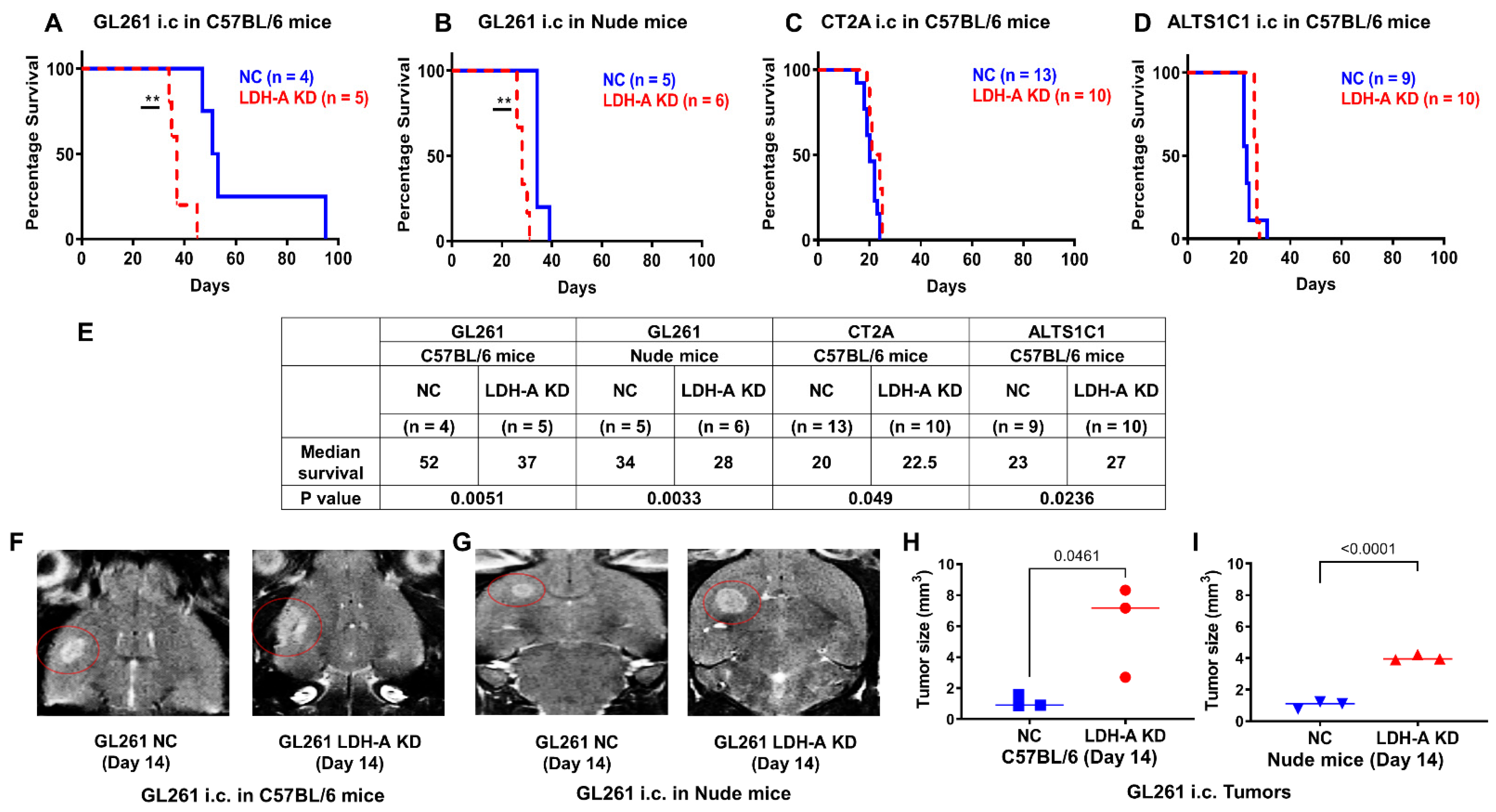{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. Generation of LDH-A Knockdown and Control Cell Lines
2.3. Western Blotting
2.4. LDH Enzyme Activity and Lactate-Glo Assay
2.5. LDH Zymography
2.6. Metabolic Extracellular Flux Analysis
2.7. mRNA Gene Expression Profile Analysis
2.8. Proliferation Assay In Vitro
2.9. Animal Models
2.10. Alzet Pump
2.11. MR Imaging
2.12. Histological Staining and Image Analysis
2.13. Statistical Analysis
3. Results
3.1. LDH-A Knockdown: Effects on Animal Survival and Tumor Growth
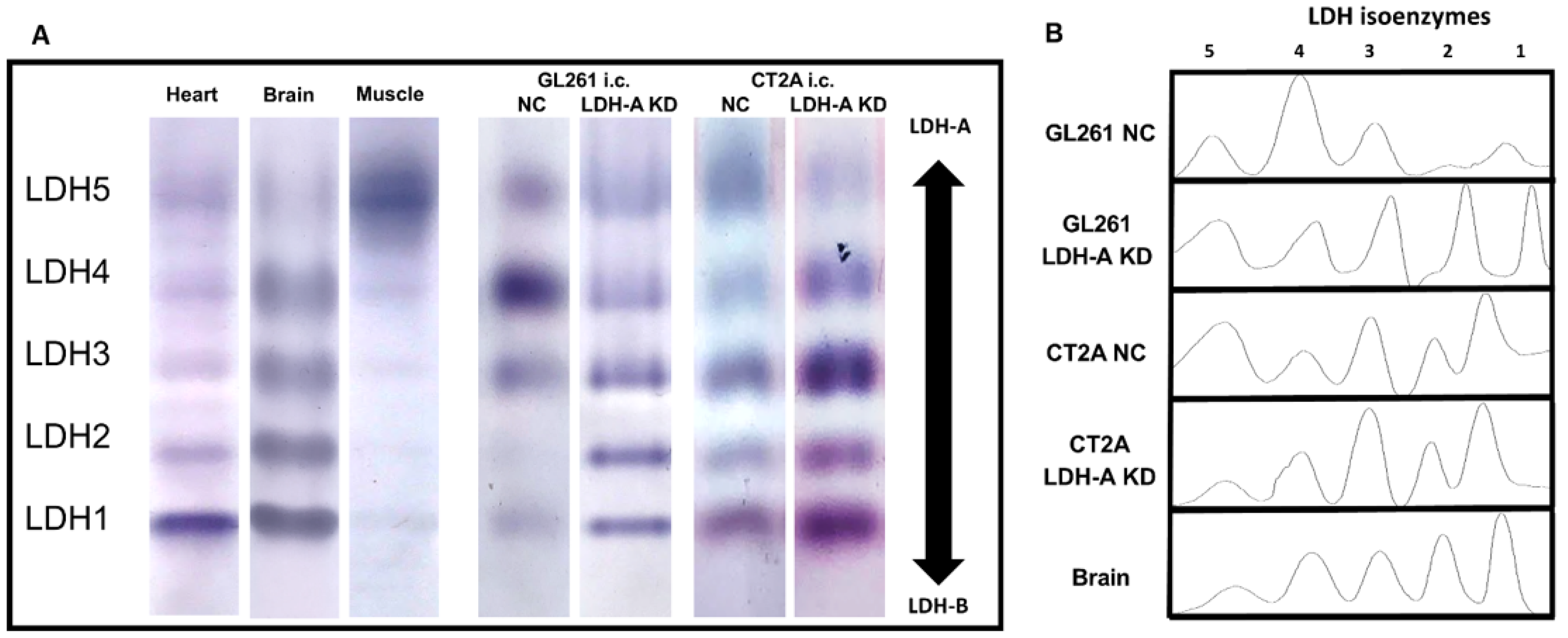
3.2. LDH Isoenzyme Pattern of i.c. Murine Gliomas
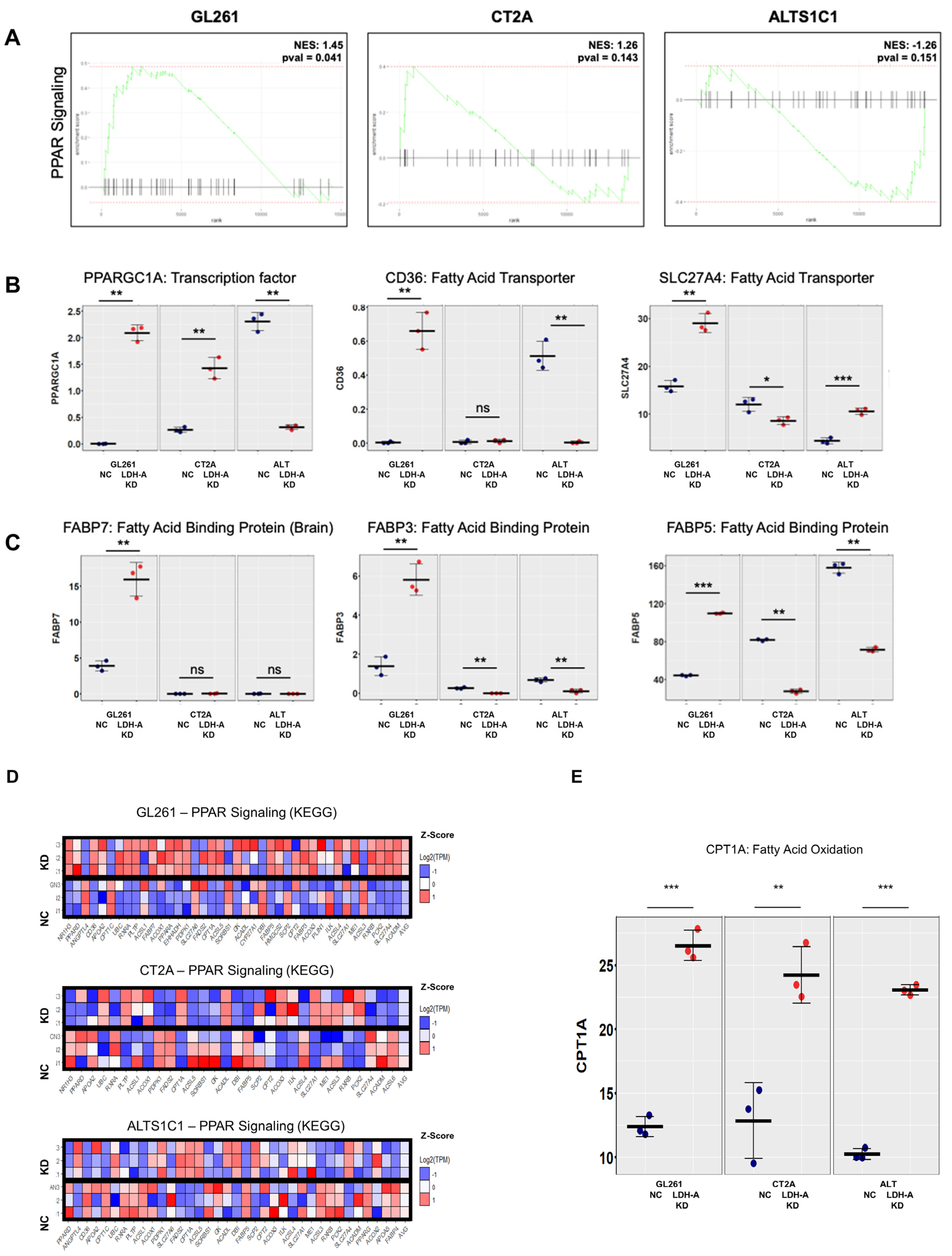
3.3. Overexpression of Lipid Metabolism Genes in LDHA KD Glioma Cells
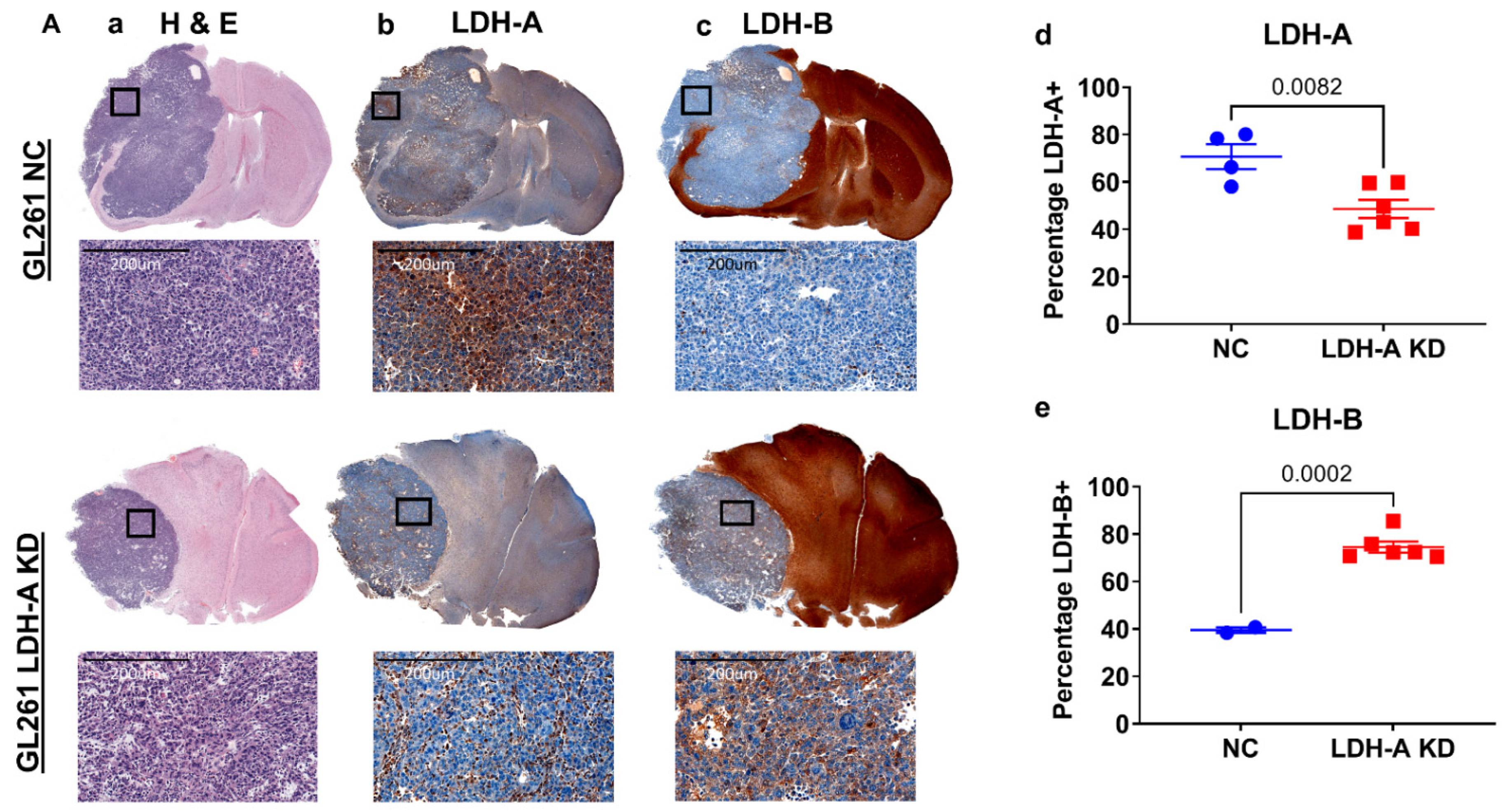
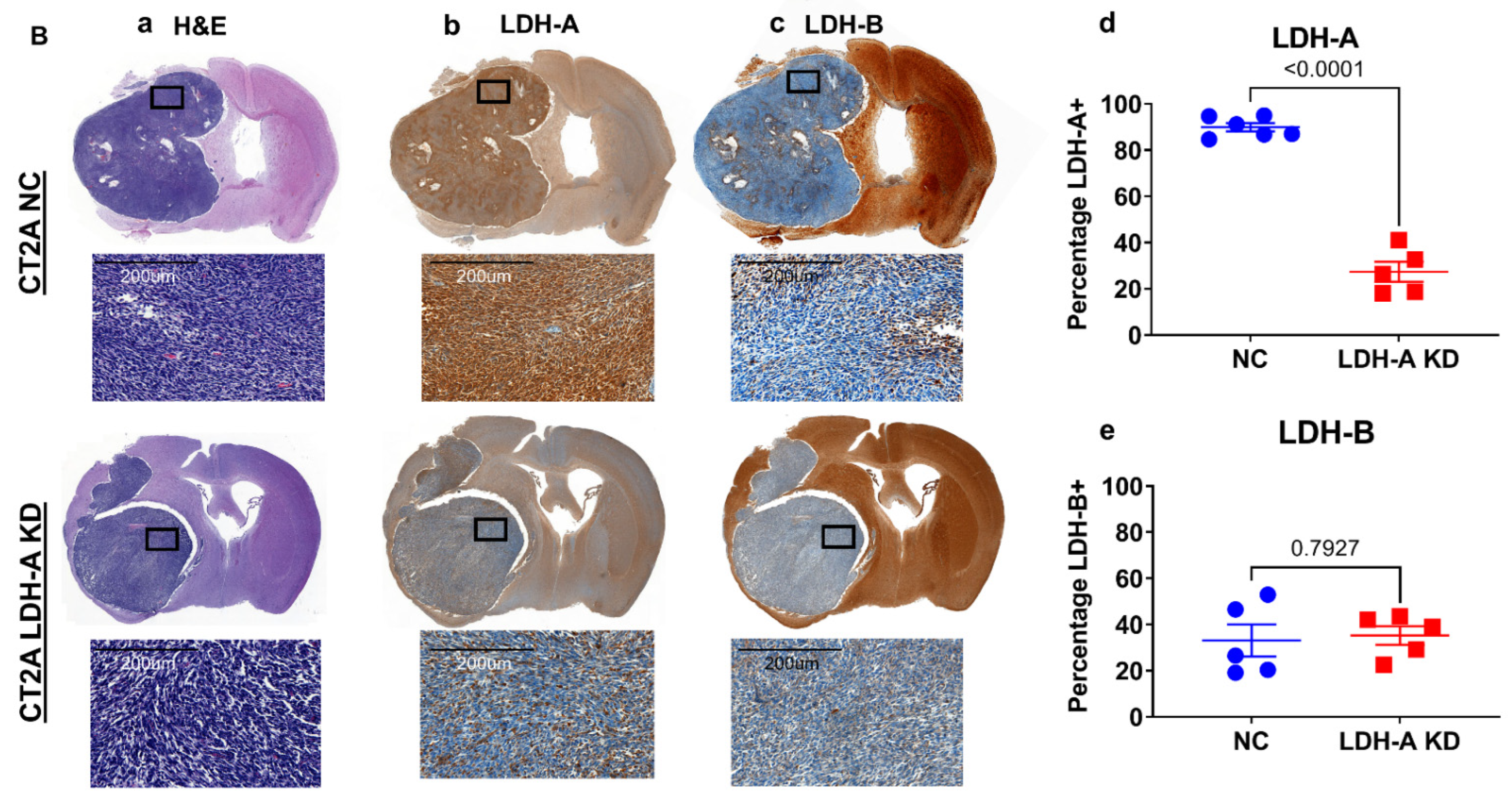
3.4. LDH-A and LDH-B Staining Pattern GL261 and CT2A the Intracranial Tumors
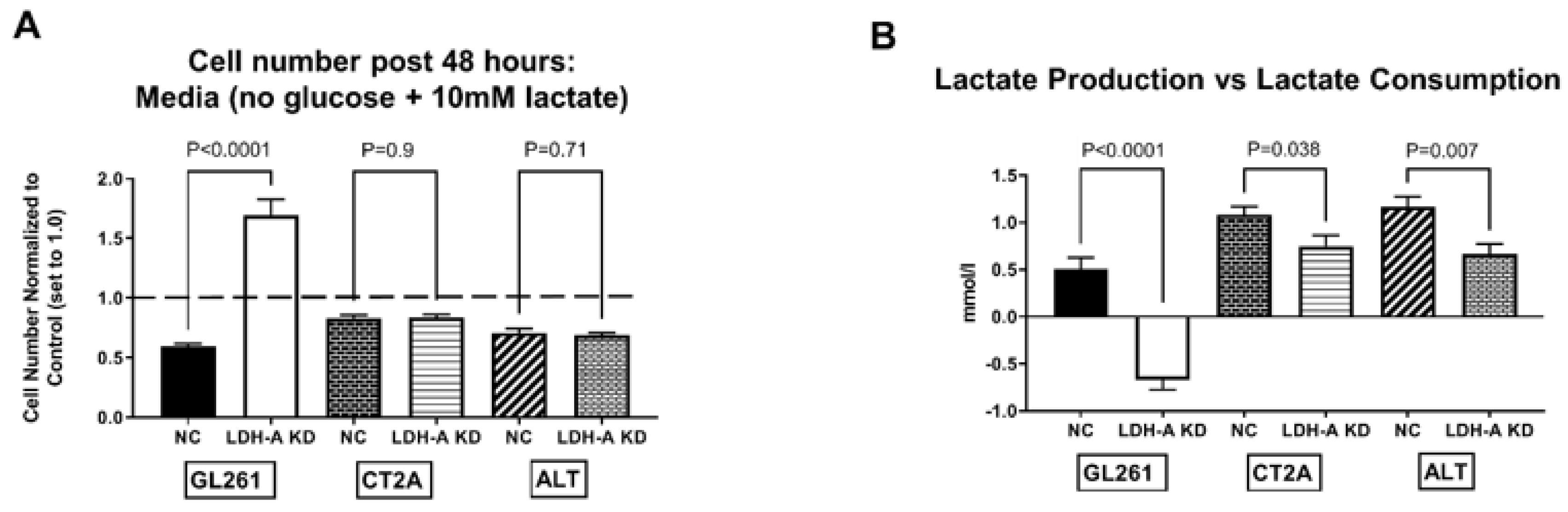
3.5. Impact of Nutrients on Tumor-Cell Metabolism and Proliferation
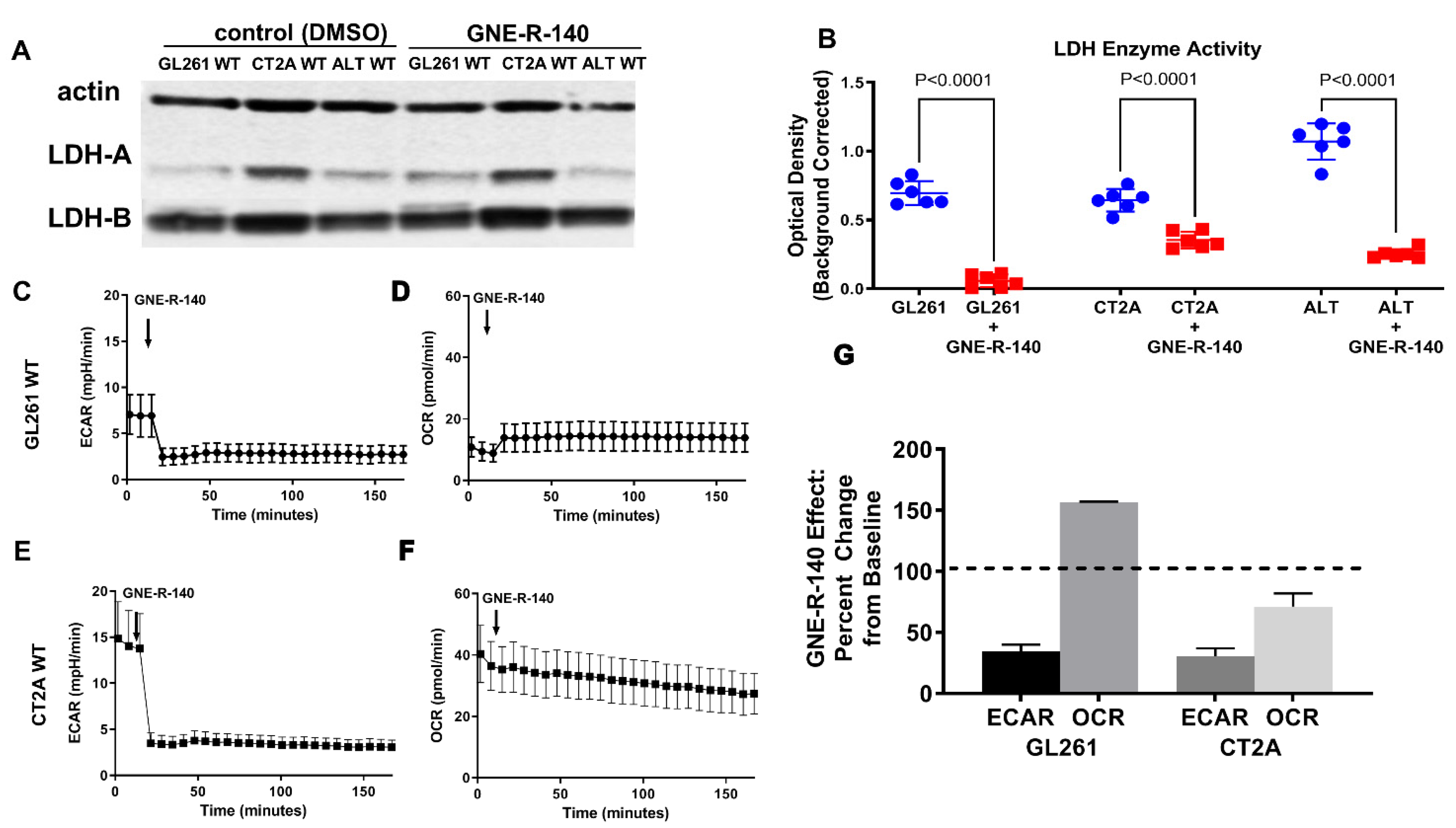
3.6. GNE-R-140: Effects on GL261, CT2A Glioma Cells in Culture
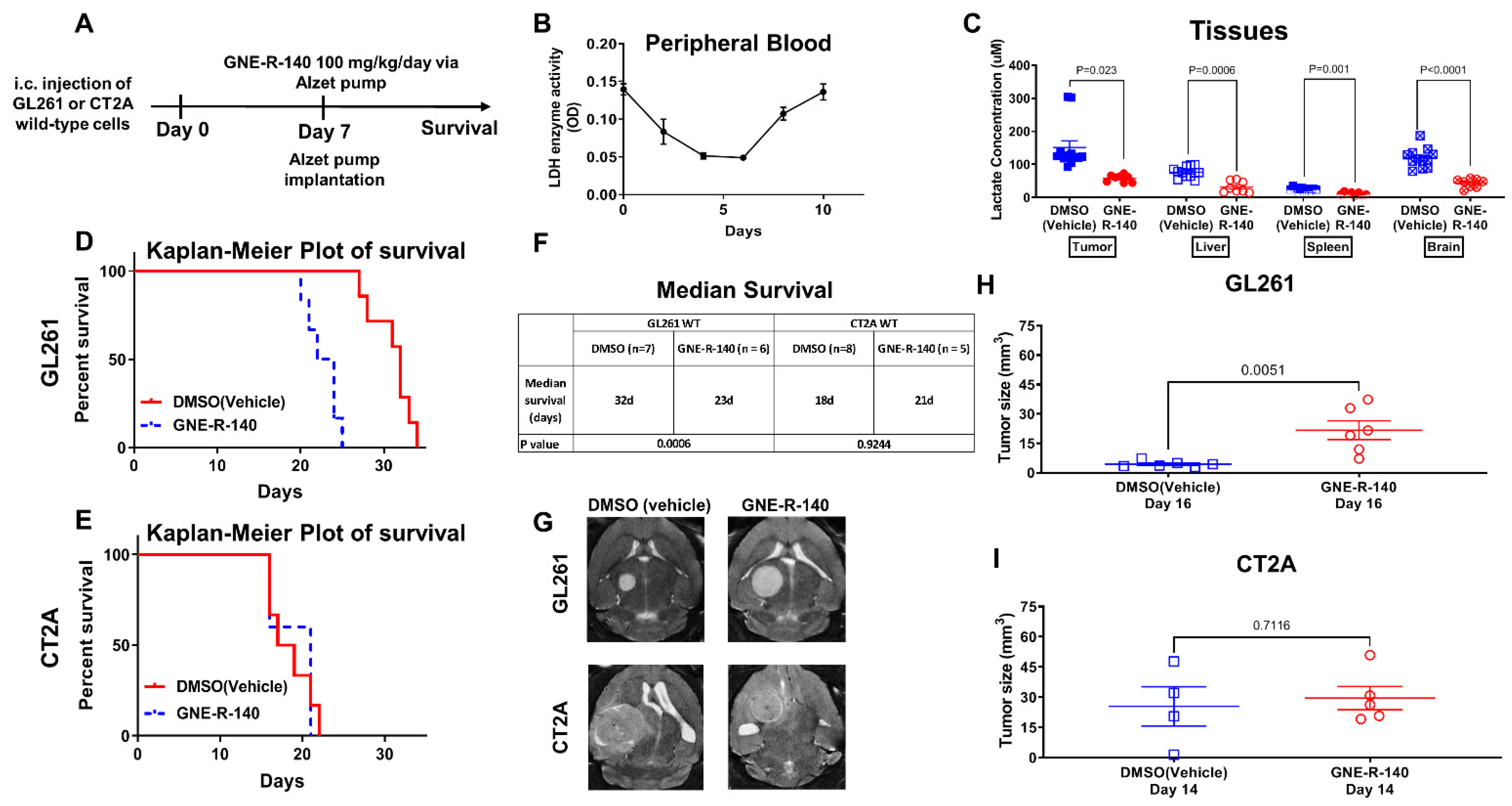
3.7. GNE-R-140: Effects on i.c. GL261, CT2A Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robertson, F.L.; Marqués-Torrejón, M.-A.; Morrison, G.M.; Pollard, S.M. Experimental models and tools to tackle glioblastoma. Dis. Models Mech. 2019, 12, dmm040386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Jagt, D.L.V.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Long, D.M.; Frame, A.K.; Reardon, P.N.; Cumming, R.C.; Hendrix, D.A.; Kretzschmar, D.; Giebultowicz, J.M. Lactate dehydrogenase expression modulates longevity and neurodegeneration in Drosophila melanogaster. Aging 2020, 12, 10041–10058. [Google Scholar] [CrossRef]
- Cahn, R.D.; Zwilling, E.; Kaplan, N.O.; Levine, L. Nature and Development of Lactic Dehydrogenases: The two major types of this enzyme form molecular hybrids which change in makeup during development. Science 1962, 136, 962–969. [Google Scholar] [CrossRef]
- Plagemann, P.G.; Gregory, K.F.; Wroblewski, F. The electrophoretically distinct forms of mammalian lactic dehydrogenase. 1. Distribution of lactic dehydrogenase. 1. Distribution of lactic dehydrogenases in rabbit and human tissue. J. Biol. Chem. 1960, 235, 2282–2287. [Google Scholar] [CrossRef]
- Li, J.; Zhu, S.; Tong, J.; Hao, H.; Yang, J.; Liu, Z.; Wang, Y. Suppression of lactate dehydrogenase A compromises tumor progression by downregulation of the Warburg effect in glioblastoma. Neuroreport 2016, 27, 110–115. [Google Scholar] [CrossRef] [Green Version]
- Leiblich, A.; Cross, S.S.; Catto, J.W.F.; Phillips, J.T.; Leung, H.Y.; Hamdy, F.C.; Rehman, I. Lactate dehydrogenase-B is silenced by promoter hypermethylation in human prostate cancer. Oncogene 2006, 25, 2953–2960. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, M.; Taniguchi, T.; Ishikawa, J.; Sugimura, H.; Sugano, K.; Kanno, T. Promoter hypermethylation in cancer silences LDHB, eliminating lactate dehydrogenase isoenzymes 1-4. Clin. Chem. 2003, 49, 1518–1520. [Google Scholar] [CrossRef] [PubMed]
- McCleland, M.L.; Adler, A.S.; Deming, L.; Cosino, E.; Lee, L.; Blackwood, E.M.; Solon, M.; Tao, J.; Li, L.; Shames, D.; et al. Lactate dehydrogenase B is required for the growth of KRAS-dependent lung adenocarcinomas. Clin. Cancer Res. 2013, 19, 773–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogatzki, M.J.; Ferguson, B.S.; Goodwin, M.L.; Gladden, L.B. Lactate is always the end product of glycolysis. Front. Neurosci. 2015, 9, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurr, A. Lactate: The ultimate cerebral oxidative energy substrate? J. Cereb. Blood Flow Metab. 2006, 26, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Rizwan, A.; Serganova, I.; Khanin, R.; Karabeber, H.; Ni, X.; Thakur, S.; Zakian, K.L.; Blasberg, R.; Koutcher, J.A. Relationships between LDH-A, lactate, and metastases in 4T1 breast tumors. Clin. Cancer Res. 2013, 19, 5158–5169. [Google Scholar] [CrossRef] [Green Version]
- Moroz, E.; Carlin, S.; Dyomina, K.; Burke, S.; Thaler, H.T.; Blasberg, R.; Serganova, I. Real-Time Imaging of HIF-1α Stabilization and Degradation. PLoS ONE 2009, 4, e5077. [Google Scholar] [CrossRef] [Green Version]
- Shindo, M.; Maeda, M.; Myat, K.; Mane, M.; Cohen, I.J.; Vemuri, K.; Albeg, A.S.; Serganova, I.; Blasberg, R.G. LDH-A Knockdown: Changes in the LDH isoenzyme profile and variability of glioma responses in different tumor microenvironments. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Mane, M.M.; Cohen, I.J.; Ackerstaff, E.; Shalaby, K.; Ijoma, J.N.; Ko, M.; Maeda, M.; Albeg, A.S.; Vemuri, K.; Satagopan, J.; et al. Lactate Dehydrogenase A Depletion Alters MyC-CaP Tumor Metabolism, Microenvironment, and CAR T Cell Therapy. Mol. Ther. Oncol. 2020, 18, 382–395. [Google Scholar] [CrossRef]
- Serganova, I.; Cohen, I.J.; Vemuri, K.; Shindo, M.; Maeda, M.; Mane, M.; Moroz, E.; Khanin, R.; Satagopan, J.; Koutcher, J.A.; et al. LDH-A regulates the tumor microenvironment via HIF-signaling and modulates the immune response. PLoS ONE 2018, 13, e0203965. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.L.; Chen, T.B.; Yang, H.; Lv, K. Lactate induced up-regulation of KLHDC8A (Kelch domain-containing 8A) contributes to the proliferation, migration and apoptosis of human glioma cells. J. Cell Mol. Med. 2020, 24, 11691–11702. [Google Scholar] [CrossRef]
- Mishra, D.; Banerjee, D. Lactate Dehydrogenases as Metabolic Links between Tumor and Stroma in the Tumor Microenvironment. Cancers 2019, 11, 750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Chen, Y.J.; Mahieu, N.G.; Huang, X.J.; Singh, M.; Crawford, P.A.; Johnson, S.L.; Gross, R.W.; Schaefer, J.; Patti, G.J. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol. 2016, 12, 937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, A.; Purkey, H.E.; Hitz, A.; Robarge, K.; Peterson, D.; Labadie, S.; Kwong, M.; Hong, R.; Gao, M.; Del Nagro, C.; et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat. Chem. Biol. 2016, 12, 779. [Google Scholar] [CrossRef] [PubMed]
- Purkey, H.E.; Robarge, K.; Chen, J.; Chen, Z.; Corson, L.B.; Ding, C.Z.; DiPasquale, A.G.; Dragovich, P.S.; Eigenbrot, C.; Evangelista, M.; et al. Cell Active Hydroxylactam Inhibitors of Human Lactate Dehydrogenase with Oral Bioavailability in Mice. ACS Med. Chem. Lett. 2016, 7, 896–901. [Google Scholar] [CrossRef] [Green Version]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Branco, M.; Linhares, P.; Carvalho, B.; Santos, P.; Costa, B.M.; Vaz, R. Serum lactate levels are associated with glioma malignancy grade. Clin. Neurol. Neurosurg. 2019, 186, 105546. [Google Scholar] [CrossRef]
- Hu, S.; Jiang, Q.; Luo, D.D.; Zhao, L.; Fu, X.; Chen, Y.Q.; Song, X.; Li, L.H.; Zhao, H.L.; He, Y.F.; et al. miR-200b is a key regulator of tumor progression and metabolism targeting lactate dehydrogenase A in human malignant glioma. Oncotarget 2016, 7, 48423–48431. [Google Scholar] [CrossRef]
- Chesnelong, C.; Chaumeil, M.M.; Blough, M.D.; Al-Najjar, M.; Stechishin, O.D.; Chan, J.A.; Pieper, R.O.; Ronen, S.M.; Weiss, S.; Luchman, H.A.; et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro Oncol. 2014, 16, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Griguer, C.E.; Oliva, C.R.; Gillespie, G.Y. Glucose metabolism heterogeneity in human and mouse malignant glioma cell lines. J. Neurooncol. 2005, 74, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Luan, Y.; Cai, J.; Wu, S.; Mai, J.; Gu, J.; Zhang, H.; Li, K.; Lin, Y.; Xiao, X.; et al. The Anti-Warburg Effect Elicited by the cAMP-PGC1α Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep. 2017, 18, 468–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, K.; Cluntun, A.; Schubert, H.; Hackett, S.; Berg, J.; Leonard, P.; Ajalla Aleixo, M.; Blevins, A.; Barta, P.; Tilley, S.; et al. Protein-Metabolite Interactomics Reveals Novel Regulation of Carbohydrate Metabolism. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Tumor metabolism: Cancer cells give and take lactate. J. Clin. Investig. 2008, 118, 3835–3837. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Ghergurovich, J.M.; Lang, J.D.; Levin, M.K.; Briones, N.; Facista, S.J.; Mueller, C.; Cowan, A.J.; McBride, M.J.; San Roman Rodriguez, E.; Killian, A.; et al. Local production of lactate, ribose phosphate, and amino acids by human triple-negative breast cancer. Medicine 2021, 6, 736–754. [Google Scholar] [CrossRef]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e359. [Google Scholar] [CrossRef] [Green Version]
- Tanner, L.B.; Goglia, A.G.; Wei, M.H.; Sehgal, T.; Parsons, L.R.; Park, J.O.; White, E.; Toettcher, J.E.; Rabinowitz, J.D. Four Key Steps Control Glycolytic Flux in Mammalian Cells. Cell Syst. 2018, 7, 49–62.e48. [Google Scholar] [CrossRef]
- Zdralevic, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.M.; et al. Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadal-Bufi, F.; Mason, J.M.; Chan, L.Y.; Craik, D.J.; Kaas, Q.; Troeira Henriques, S. Designed beta-Hairpins Inhibit LDH5 Oligomerization and Enzymatic Activity. J. Med. Chem 2021, 64, 3767–3779. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Thunborg, K.; Azimzadeh, O.; von Toerne, C.; Werner, C.; Shevtsov, M.; Di Genio, T.; Zdralevic, M.; Pouyssegur, J.; Renner, K.; et al. Targeting Cancer Metabolism Breaks Radioresistance by Impairing the Stress Response. Cancers 2021, 13, 3762. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maeda, M.; Ko, M.; Mane, M.M.; Cohen, I.J.; Shindo, M.; Vemuri, K.; Serganova, I.; Blasberg, R. Genetic and Drug Inhibition of LDH-A: Effects on Murine Gliomas. Cancers 2022, 14, 2306. https://doi.org/10.3390/cancers14092306
Maeda M, Ko M, Mane MM, Cohen IJ, Shindo M, Vemuri K, Serganova I, Blasberg R. Genetic and Drug Inhibition of LDH-A: Effects on Murine Gliomas. Cancers. 2022; 14(9):2306. https://doi.org/10.3390/cancers14092306
Chicago/Turabian StyleMaeda, Masatomo, Myat Ko, Mayuresh M. Mane, Ivan J. Cohen, Masahiro Shindo, Kiranmayi Vemuri, Inna Serganova, and Ronald Blasberg. 2022. "Genetic and Drug Inhibition of LDH-A: Effects on Murine Gliomas" Cancers 14, no. 9: 2306. https://doi.org/10.3390/cancers14092306
APA StyleMaeda, M., Ko, M., Mane, M. M., Cohen, I. J., Shindo, M., Vemuri, K., Serganova, I., & Blasberg, R. (2022). Genetic and Drug Inhibition of LDH-A: Effects on Murine Gliomas. Cancers, 14(9), 2306. https://doi.org/10.3390/cancers14092306