
Construction of an Immune Escape-Related Signature in Clear Cell Renal Cell Carcinoma and Identification of the Relationship between IFNAR1 and Immune Infiltration by Multiple Immunohistochemistry

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Normalization
2.2. Exploring the Genomic Alterations of Core Cancer Immune Escape Genes
2.3. Identification of the Immune Escape-Related Subgroups in ccRCC
2.4. Further Optimization of the IER Cluster
2.5. Calculating the Immune Escape Score and Construction of Immune-Related Nomogram
2.6. Exploring the Potential Ability for Predicting Immunotherapy Response of the Biomarkers and TME Associations
2.7. Using Multiplex Immunohistochemistry to Explore IFNAR1 Expression and TME
2.8. Cell Culture and Transfection
2.9. CCK-8 Assays
2.10. Transwell Assays
3. Results
3.1. Copy Number Variation of Core Cancer Innate Immune Escape Genes in ccRCC
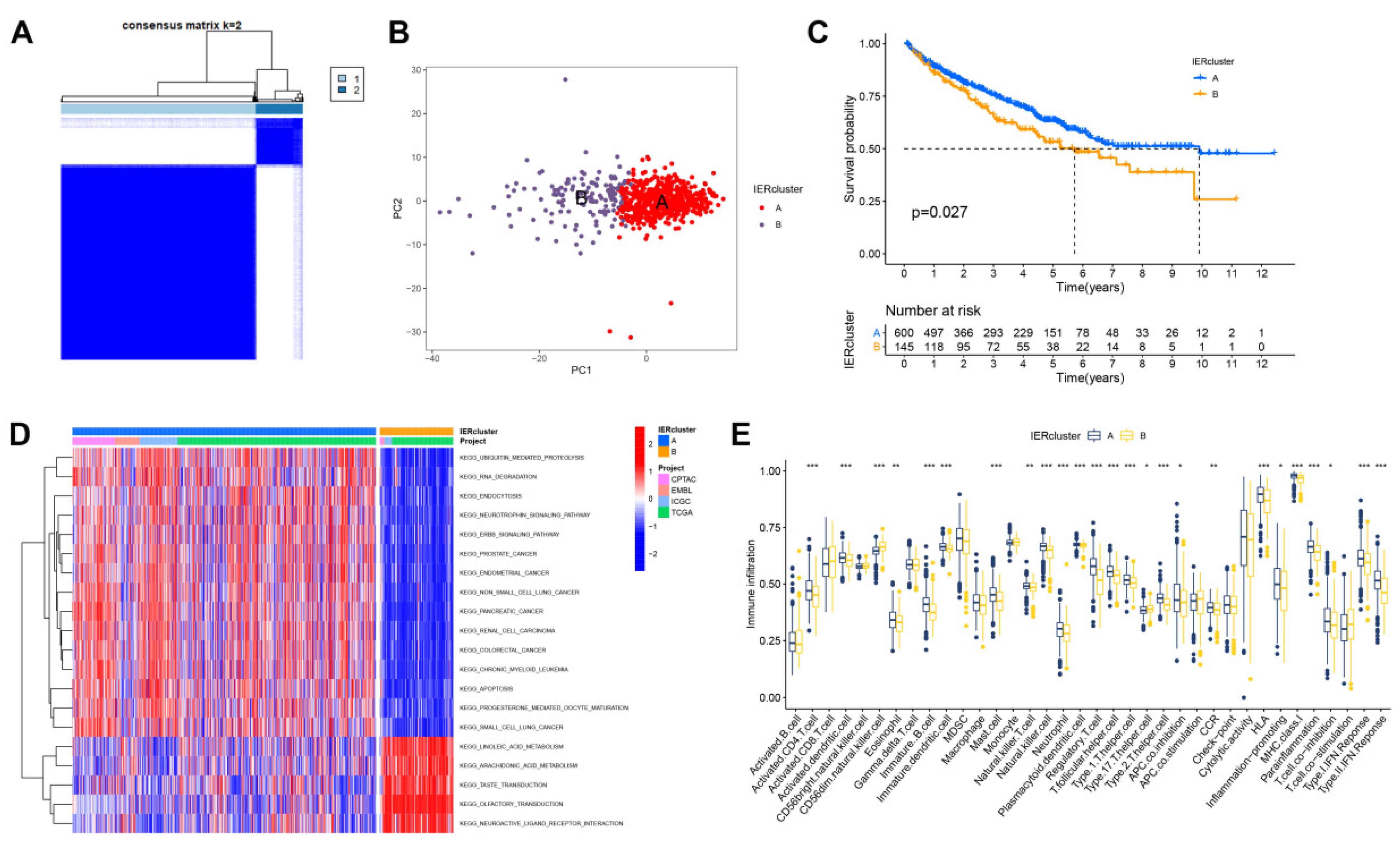
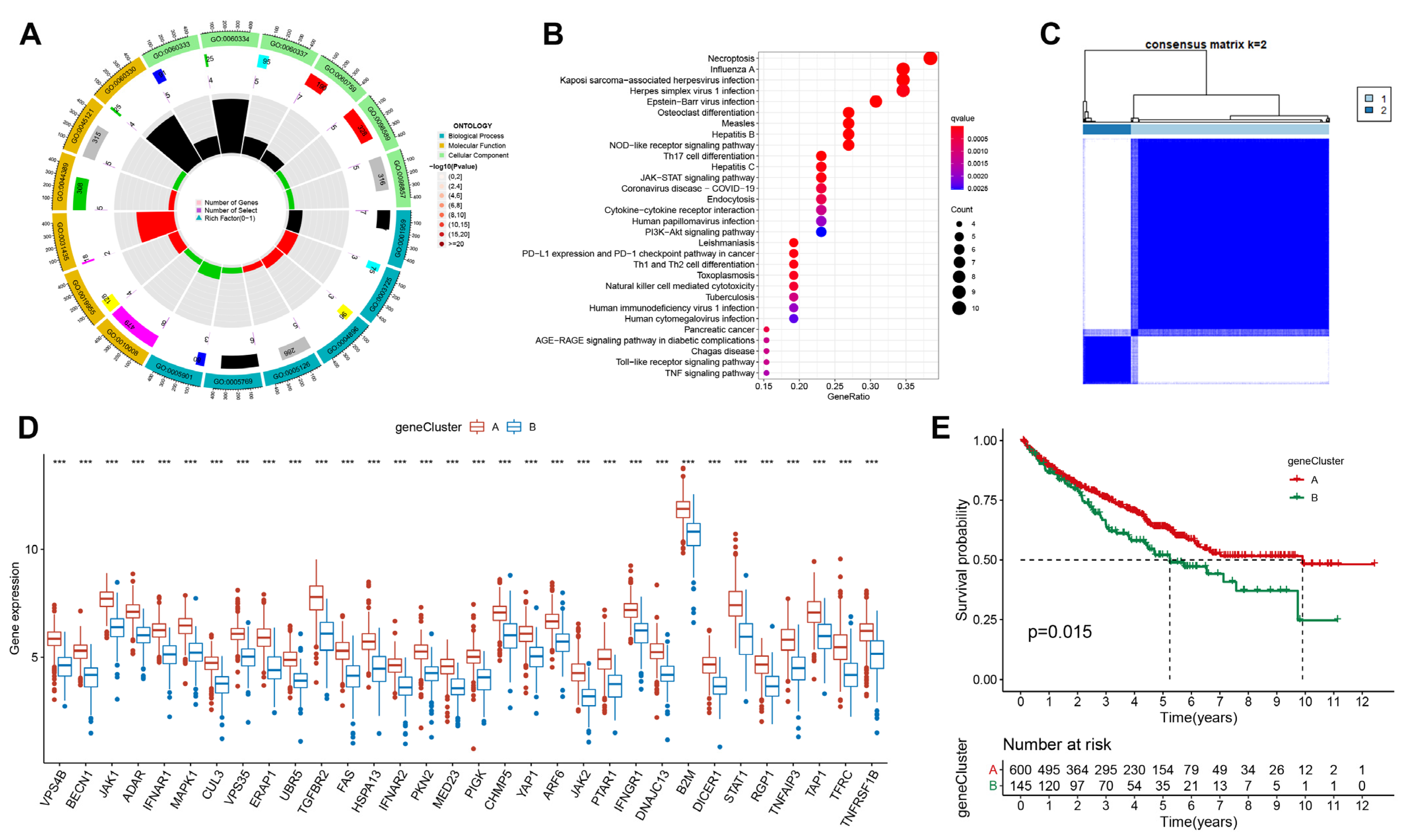
3.2. Identification of Immune Escape Clusters
3.3. Exploring Potential Biological Function of Immune Escape-Related Genes in ccRCC
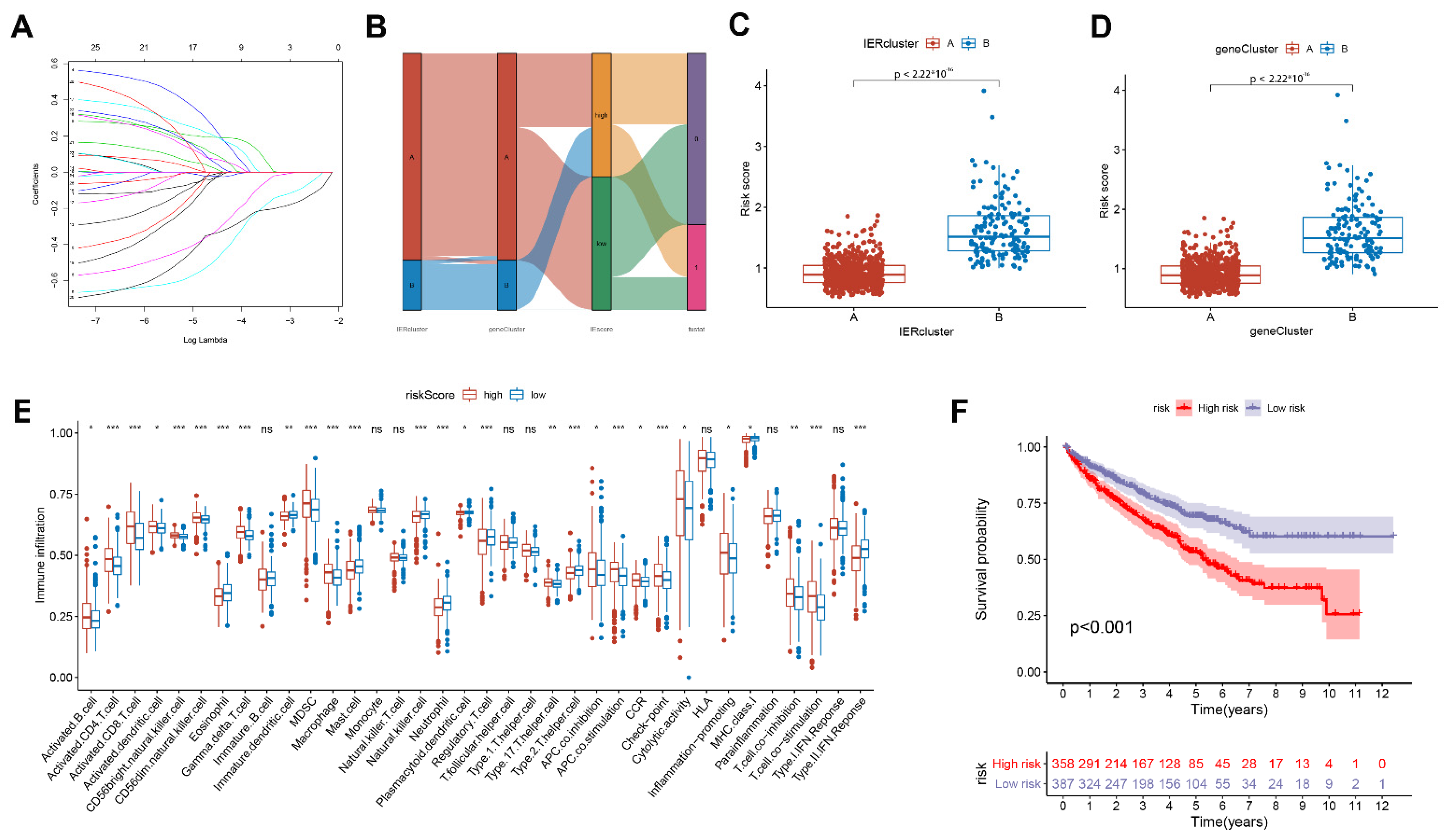
3.4. Calculate the Immune Escape Score and Explore Potential Clinical Implications
3.5. Construction of Related Immune Escape Nomogram
3.6. The Expression Level of IFNAR1 Is Closely Related to Immunotherapy Response and Immune Microenvironment
3.7. IFNAR1 Promotes Immune Activation by Facilitating CD4+ T Cell and CD8+ T Cell Infiltration
3.8. IFNAR1 Regulates the Proliferation and Migration Properties of ccRCC
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Xie, Y.; Xie, F.; Zhang, L.; Zhou, X.; Huang, J.; Wang, F.; Jin, J.; Zhang, L.; Zeng, L.; Zhou, F. Targeted Anti-Tumor Immunotherapy Using Tumor Infiltrating Cells. Adv. Sci. 2021, 8, e2101672. [Google Scholar] [CrossRef]
- Lawson, K.A.; Sousa, C.M.; Zhang, X.; Kim, E.; Akthar, R.; Caumanns, J.J.; Yao, Y.; Mikolajewicz, N.; Ross, C.; Brown, K.R.; et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 2020, 586, 120–126. [Google Scholar] [CrossRef]
- Deleuze, A.; Saout, J.; Dugay, F.; Peyronnet, B.; Mathieu, R.; Verhoest, G.; Bensalah, K.; Crouzet, L.; Laguerre, B.; Belaud-Rotureau, M.-A.; et al. Immunotherapy in Renal Cell Carcinoma: The Future Is Now. Int. J. Mol. Sci. 2020, 21, 2532. [Google Scholar] [CrossRef] [Green Version]
- Ljungberg, B.; Campbell, S.C.; Cho, H.Y.; Jacqmin, D.; Lee, J.E.; Weikert, S.; Kiemeney, L.A. The epidemiology of renal cell carcinoma. Eur. Urol. 2011, 60, 615–621. [Google Scholar] [CrossRef]
- Wagstaff, J.; Jones, R.; Hawkins, R.; Porfiri, E.; Pickering, L.; Bahl, A.; Brown, J.; Buchan, S. Treatment patterns and clinical outcomes in patients with renal cell carcinoma in the UK: Insights from the RECCORD registry. Ann. Oncol. 2016, 27, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, H.; Kamai, T. Recent advances in the treatment of metastatic renal cell carcinoma. Int. J. Urol. 2013, 20, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Mathew, L.K.; Lee, S.S.; Skuli, N.; Rao, S.; Keith, B.; Nathanson, K.L.; Lal, P.; Simon, M.C. Restricted expression of miR-30c-2-3p and miR-30a-3p in clear cell renal cell carcinomas enhances HIF2α activity. Cancer Discov. 2014, 4, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Eng, L.; Ding, D.; Chen, Y.; Dai, H.; Liu, G.; Qiao, Z.; An, R. Anti-tumor effect of ribavirin in combination with interferon-α on renal cell carcinoma cell lines in vitro. Cancer Cell Int. 2014, 14, 63. [Google Scholar]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Deweerd, N.A.; Stifter, S.A.; Liu, L.; Zhou, B.; Wang, W.; Zhou, Y.; Ying, B.; Hu, X.; Matthews, A.Y.; et al. A proline deletion in IFNAR1 impairs IFN-signaling and underlies increased resistance to tuberculosis in humans. Nat. Commun. 2018, 9, 85. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.-J.; Hu, F.-F.; Xia, M.-X.; Han, L.; Zhang, Q.; Guo, A.-Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Braun, D.A.; Hou, Y.; Bakouny, Z.; Ficial, M.; Angelo, M.S.; Forman, J.; Ross-Macdonald, P.; Berger, A.C.; Jegede, O.A.; Elagina, L.; et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 2020, 26, 909–918. [Google Scholar] [CrossRef]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor–immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Immunosuppressive cells in tumor immune escape and metastasis. J. Mol. Med. 2016, 94, 509–522. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Perdiguero, E.G.; Geissmann, F. Identifying the infiltrators. Science 2014, 344, 801–802. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [Green Version]
- Jago, C.B.; Yates, J.; Câmara, N.O.S.; Lechler, R.I.; Lombardi, G. Differential expression of CTLA-4 among T cell subsets. Clin. Exp. Immunol. 2004, 136, 463–471. [Google Scholar] [PubMed]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2008, 10, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar]
- Oleinika, K.; Nibbs, R.J.; Graham, G.J.; Fraser, A.R. Suppression, subversion and escape: The role of regulatory T cells in cancer progression. Clin. Exp. Immunol. 2013, 171, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Klement, J.D.; Ibrahim, M.L.; Xiao, W.; Redd, P.S.; Nayak-Kapoor, A.; Zhou, G.; Liu, K. Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. J. Immunother. Cancer 2019, 7, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Chen, M.; Da Xu, D.; Li, T.-E.; Zhang, Z.; Li, J.-H.; Wang, X.-Y.; Yang, X.; Lu, L.; Jia, H.-L.; et al. The combination of PD-1 blockade with interferon-α has a synergistic effect on hepatocellular carcinoma. Cell. Mol. Immunol. 2022, 19, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, X.; Huang, X.; Dong, X.; Jing, S.; Zhang, Y.; Zhao, B.; Wang, Z.; Qu, H. Correlation between IFNAR1 expression in peripheral blood T lymphocytes and inflammatory cytokines, tumor-infiltrating lymphocytes, and chemosensitivity in patients with colorectal cancer. Cytokine 2022, 159, 156008. [Google Scholar] [CrossRef]
- Isaacs, A.; Lindenmann, J. Virus interference: I. The interferon. Proc. R. Soc. Lond. Ser. B-Biol. Sci. 1957, 147, 258–267. [Google Scholar]
- Von Locquenghien, M.; Rozalén, C.; Celià-Terrassa, T. Interferons in cancer immunoediting: Sculpting metastasis and immunotherapy response. J. Clin. Investig. 2021, 131, e143296. [Google Scholar] [CrossRef]
- Adámková, L.; Součkova, K.; Kovarík, J. Transcription protein STAT1: Biology and relation to cancer. Folia Biol. 2007, 53, 1–6. [Google Scholar]
- Widschwendter, A.; Tonko-Geymayer, S.; Welte, T.; Daxenbichler, G.; Marth, C.; Doppler, W. Prognostic significance of signal transducer and activator of transcription 1 activation in breast cancer. Clin. Cancer Res. 2002, 8, 3065–3074. [Google Scholar]
- Simpson, J.A.D.; Al-Attar, A.; Watson, N.F.S.; Scholefield, J.H.; Ilyas, M.; Durrant, L.G. Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut 2010, 59, 926–933. [Google Scholar] [CrossRef]
- Bottos, A.; Gotthardt, D.; Gill, J.W.; Gattelli, A.; Frei, A.; Tzankov, A.; Sexl, V.; Wodnar-Filipowicz, A.; Hynes, N.E. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat. Commun. 2016, 7, 12258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rautela, J.; Baschuk, N.; Slaney, C.Y.; Jayatilleke, K.M.; Xiao, K.; Bidwell, B.N.; Lucas, E.C.; Hawkins, E.D.; Lock, P.; Wong, C.S.; et al. Loss of Host Type-I IFN Signaling Accelerates Metastasis and Impairs NK-cell Antitumor Function in Multiple Models of Breast Cancer. Cancer Immunol. Res. 2015, 3, 1207–1217. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, K.; Xu, F.; Zhang, X.; Zeng, B.; Zhang, W.; Shi, G.; Ye, D. Construction of an Immune Escape-Related Signature in Clear Cell Renal Cell Carcinoma and Identification of the Relationship between IFNAR1 and Immune Infiltration by Multiple Immunohistochemistry. Cancers 2023, 15, 169. https://doi.org/10.3390/cancers15010169
Chang K, Xu F, Zhang X, Zeng B, Zhang W, Shi G, Ye D. Construction of an Immune Escape-Related Signature in Clear Cell Renal Cell Carcinoma and Identification of the Relationship between IFNAR1 and Immune Infiltration by Multiple Immunohistochemistry. Cancers. 2023; 15(1):169. https://doi.org/10.3390/cancers15010169
Chicago/Turabian StyleChang, Kun, Fujiang Xu, Xuanzhi Zhang, Bohan Zeng, Wei Zhang, Guohai Shi, and Dingwei Ye. 2023. "Construction of an Immune Escape-Related Signature in Clear Cell Renal Cell Carcinoma and Identification of the Relationship between IFNAR1 and Immune Infiltration by Multiple Immunohistochemistry" Cancers 15, no. 1: 169. https://doi.org/10.3390/cancers15010169
APA StyleChang, K., Xu, F., Zhang, X., Zeng, B., Zhang, W., Shi, G., & Ye, D. (2023). Construction of an Immune Escape-Related Signature in Clear Cell Renal Cell Carcinoma and Identification of the Relationship between IFNAR1 and Immune Infiltration by Multiple Immunohistochemistry. Cancers, 15(1), 169. https://doi.org/10.3390/cancers15010169