

Implementation of Comprehensive Genomic Profiling in Ovarian Cancer Patients: A Retrospective Analysis

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Protocol and Population
2.2. Comprehensive Genomic Profiling
2.3. Study Measures
2.4. Statistical Analysis
3. Results
3.1. Baseline Clinical and Demographic Characteristics of the Study Population and Comparison to the Control Group
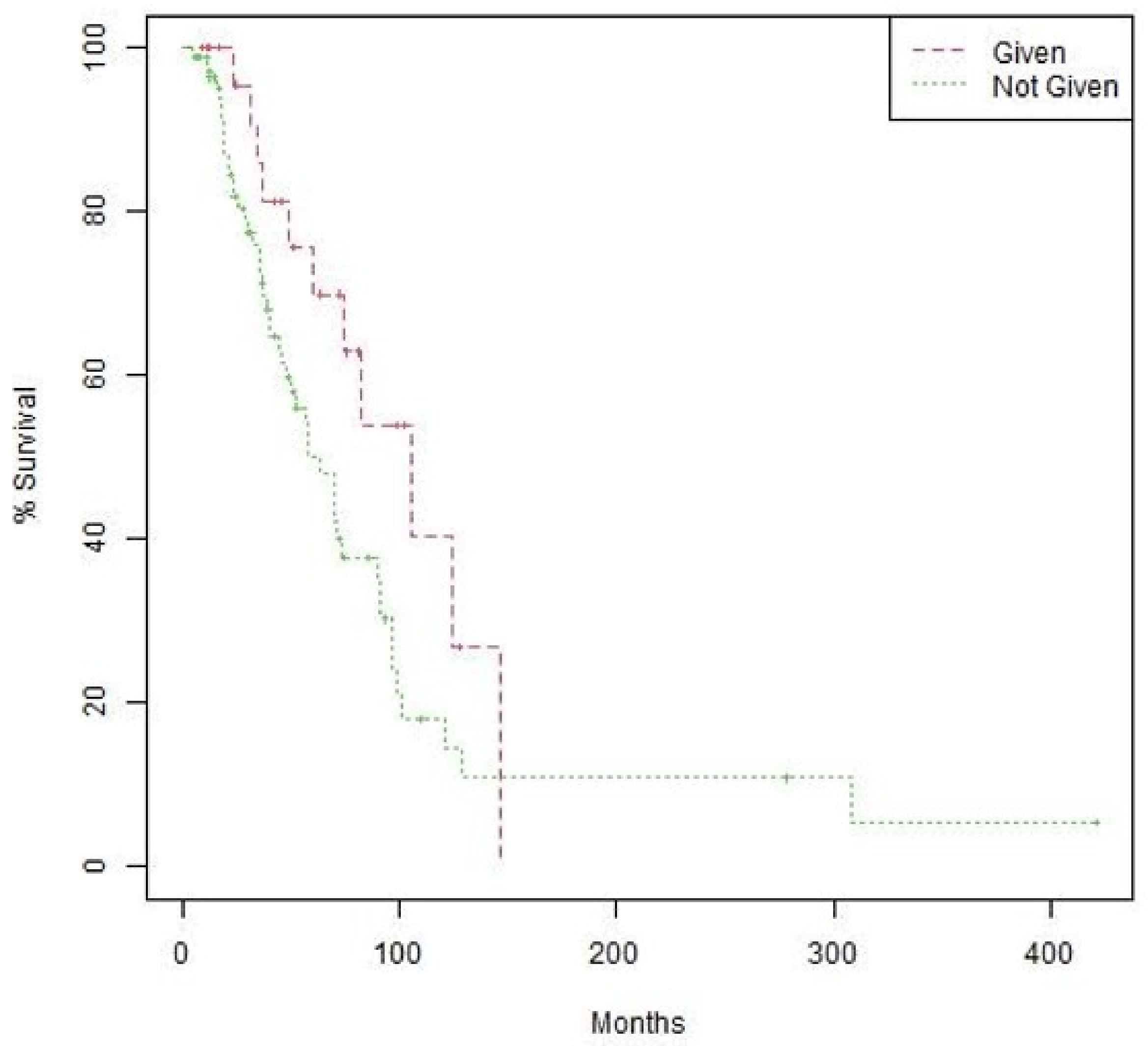
3.2. Comparison of PFS and OS in the CGP and Historical Control Groups
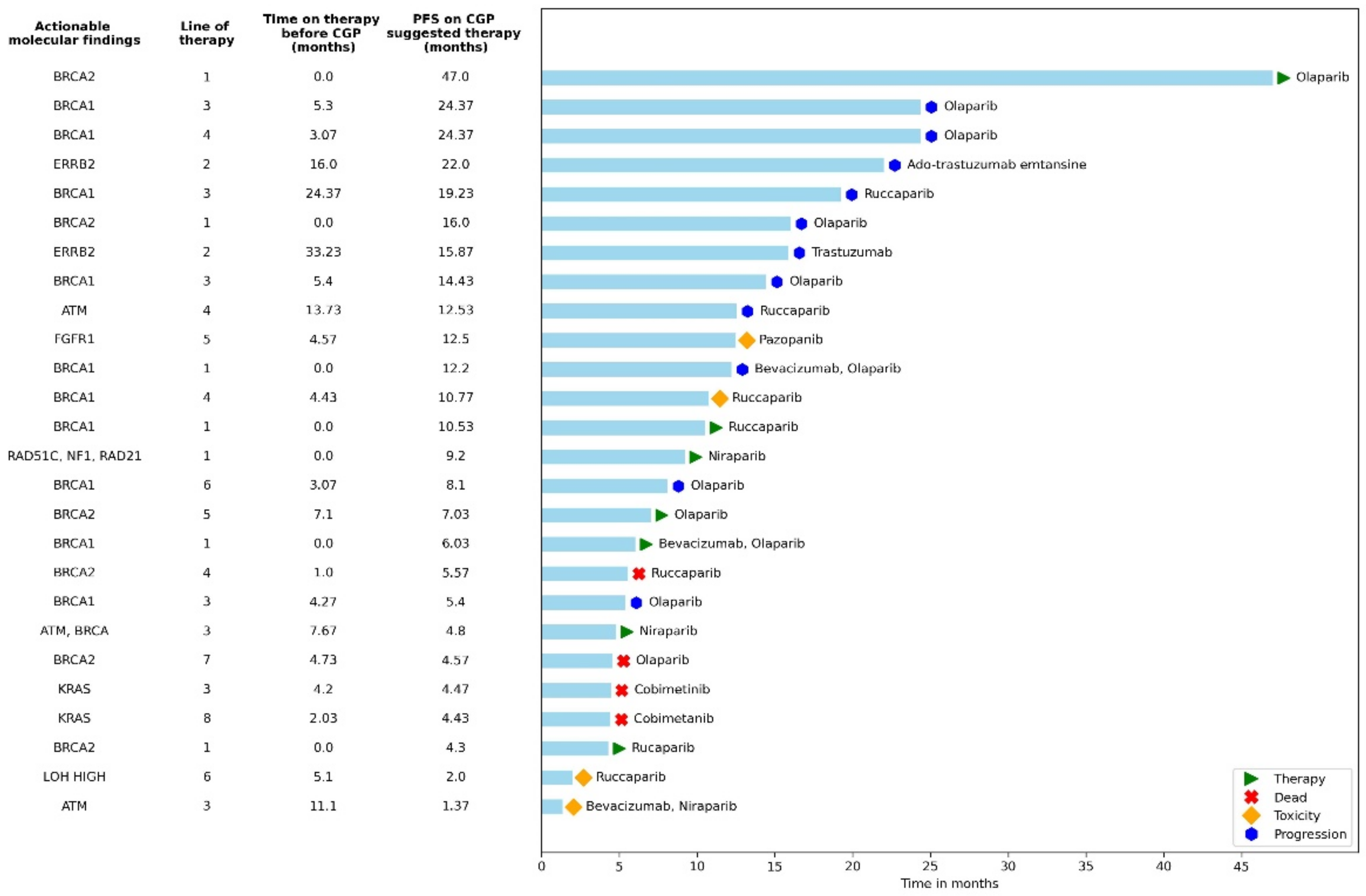
3.3. Analysis of the CGP Group
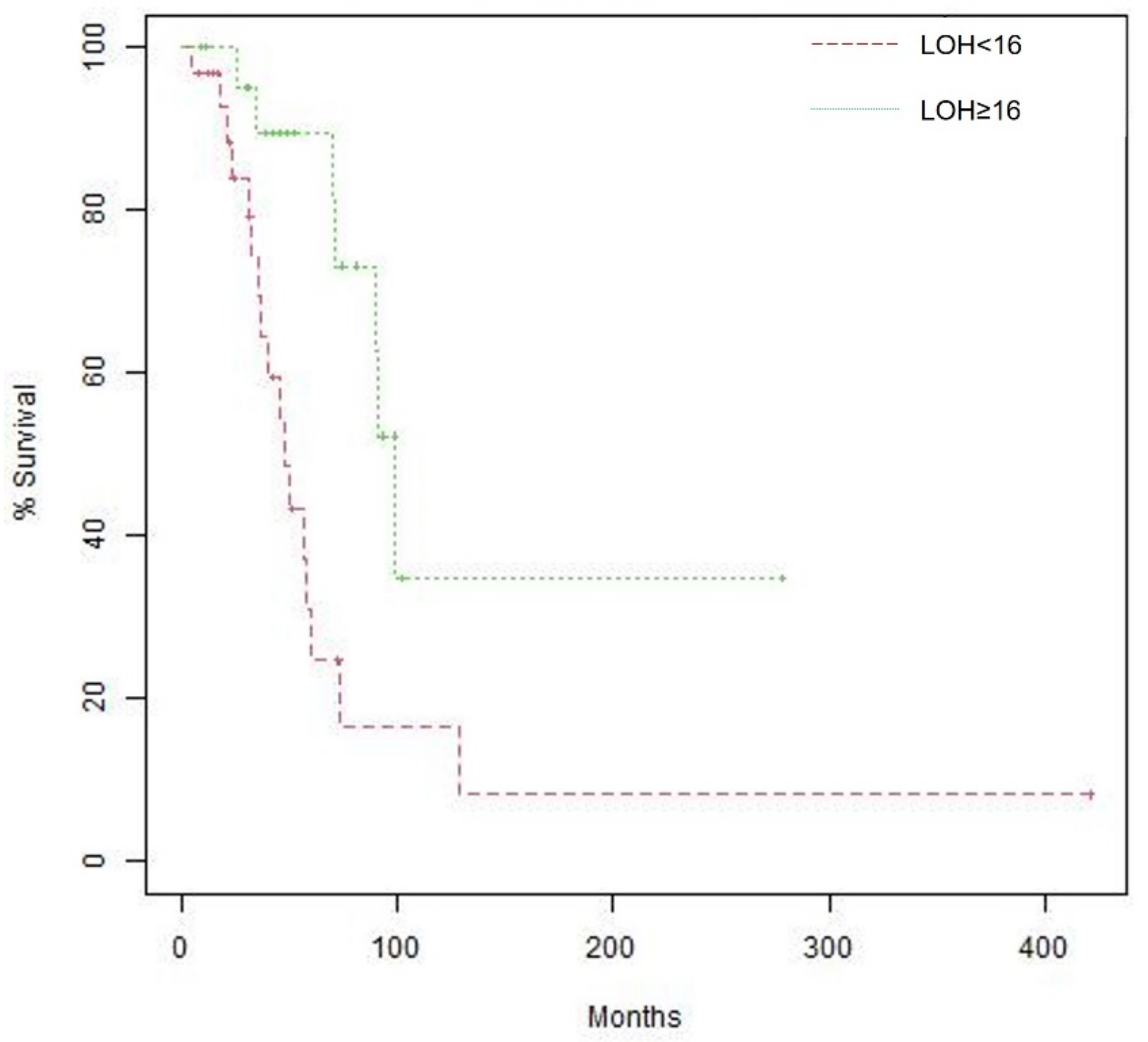
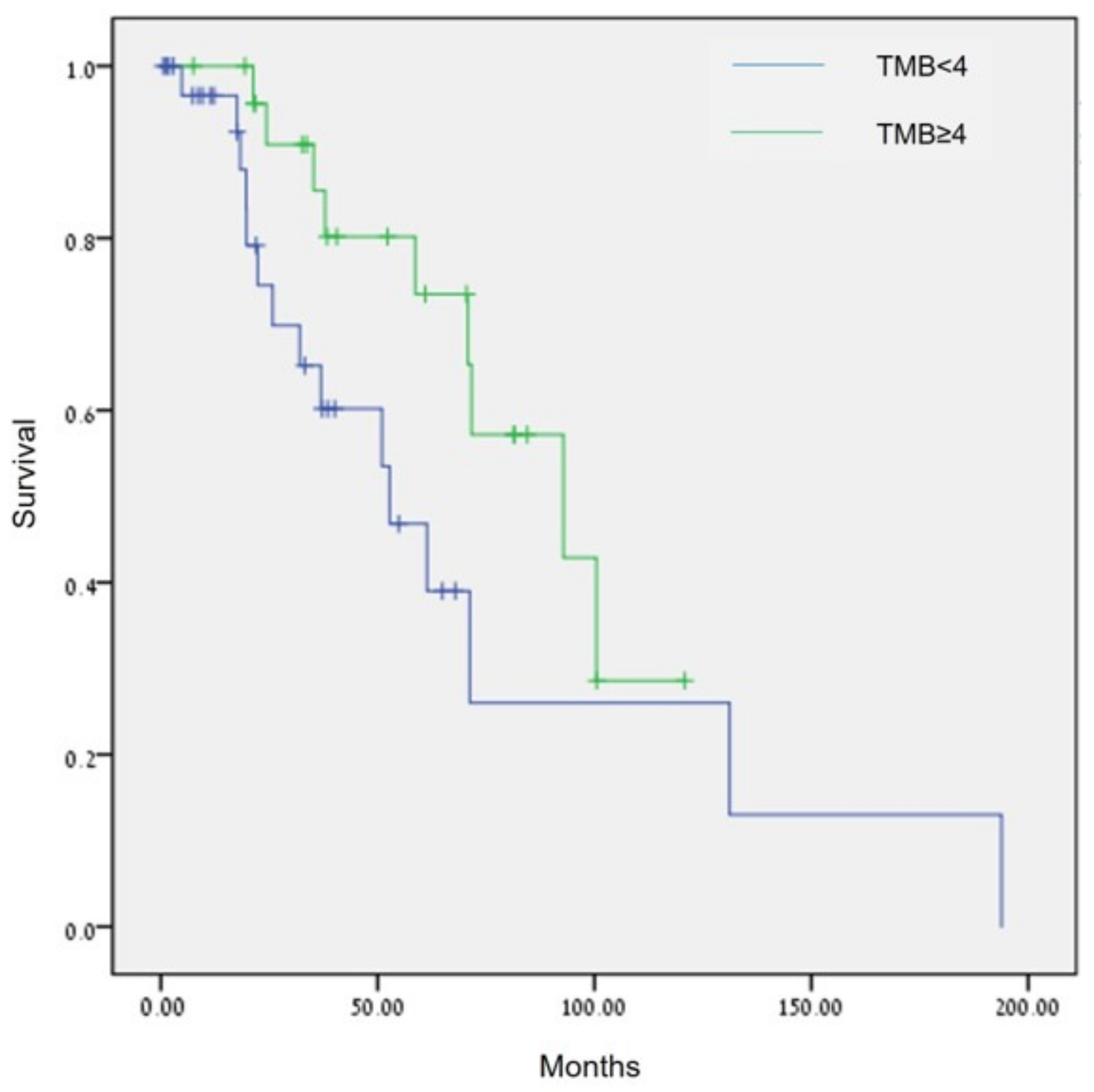
3.4. Effect of Biomarkers on Overall Survival
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Estimated Number of Incident Cases and Deaths Worldwide, Females, All Ages. Available online: https://gco.iarc.fr/today/online-analysis-multi-bars?v=2020&mode=cancer&mode_population=countries&population=900&populations=900&key=total&sex=2&cancer=39&type=0&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&nb_items=10&group_cancer=1&include_nmsc=1&include_nmsc_other=1&type_multiple=%257B%2522inc%2522%253Atrue%252C%2522mort%2522%253Atrue%252C%2522prev%2522%253Afalse%257D&orientation=horizontal&type_sort=0&type_nb_items=%257B%2522top%2522%253Atrue%252C%2522bottom%2522%253Afalse%257D&population_group_globocan_id (accessed on 20 September 2022).
- Reid, F. World Ovarian Cancer Coalition Atlas 2020: Global Trends in Incidence, Mortality, and Survival; World Ovarian Cancer Coalition: Toronto, ON, USA, 2020. [Google Scholar]
- National Institutes of Health; National Cancer Institute, Surveillance, Epidemiology, and End Results Program. Cancer Stat Facts: Female Breast Cancer; National Cancer Institute, NIH: Bethesda, MD, USA, 2020. Available online: https://seer.cancer.gov/statfacts/html/ovary.html (accessed on 20 September 2022).
- Kossaï, M.; Leary, A.; Scoazec, J.-Y.; Genestie, C. Ovarian Cancer: A Heterogeneous Disease. Pathobiology 2017, 85, 41–49. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domchek, S.M.; Aghajanian, C.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol. Oncol. 2015, 140, 199–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients with Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-Ribose Polymerase Inhibition: Frequent Durable Responses in BRCA Carrier Ovarian Cancer Correlating With Platinum-Free Interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Tinker, A.V.; Gelmon, K. The Role of PARP Inhibitors in the Treatment of Ovarian Carcinomas. Curr. Pharm. Des. 2012, 18, 3770–3774. [Google Scholar] [CrossRef]
- Verhoeven, Y.; Quatannens, D.; Trinh, X.; Wouters, A.; Smits, E.; Lardon, F.; De Waele, J.; van Dam, P. Targeting the PD-1 Axis with Pembrolizumab for Recurrent or Metastatic Cancer of the Uterine Cervix: A Brief Update. Int. J. Mol. Sci. 2021, 22, 1807. [Google Scholar] [CrossRef]
- The AACR Project GENIE Consortium; André, F.; Arnedos, M.; Baras, A.S.; Baselga, J.; Bedard, P.L.; Berger, M.F.; Bierkens, M.; Calvo, F.; Cerami, E.; et al. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Horak, P.; Fröhling, S.; Glimm, H. Integrating next-generation sequencing into clinical oncology: Strategies, promises and pitfalls. ESMO Open 2016, 1, e000094. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.T.; Kim, K.; Lee, H.; Kozarewa, I.; Mortimer, P.G.; Odegaard, J.I.; Harrington, E.A.; Lee, J.; Lee, T.; et al. Tumor Genomic Profiling Guides Patients with Metastatic Gastric Cancer to Targeted Treatment: The VIKTORY Umbrella Trial. Cancer Discov. 2019, 9, 1388–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Cao, Q.; Wen, W.; Wang, H. Targeted therapy for hepatocellular carcinoma: Challenges and opportunities. Cancer Lett. 2019, 460, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Tebbutt, N. Systemic treatment of advanced hepatocellular cancer: New hope on the horizon. Expert Rev. Anticancer. Ther. 2019, 19, 343–353. [Google Scholar] [CrossRef]
- Chung, J.H.; Dewal, N.; Sokol, E.; Mathew, P.; Whitehead, R.; Millis, S.Z.; Frampton, G.M.; Bratslavsky, G.; Pal, S.K.; Lee, R.J.; et al. Prospective Comprehensive Genomic Profiling of Primary and Metastatic Prostate Tumors. JCO Precis. Oncol. 2019, 3, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Tucker, M.; Marin, D.; Gupta, R.T.; Healy, P.; Humeniuk, M.; Jarvis, C.; Zhang, T.; McNamara, M.; George, D.J.; et al. Clinical utility of FoundationOne tissue molecular profiling in men with metastatic prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2019, 37, 813.e1–813.e9. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Lyons, E.; DeArbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sahai, V.; Sohal, D.P.S.; et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: A retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020, 21, 508–518. [Google Scholar] [CrossRef]
- Foundation Medicine FoundationOne®CDx Technical Information. Available online: https://info.foundationmedicine.com/hubfs/FMI%20Labels/FoundationOne_CDx_Label_Technical_Info.pdf (accessed on 20 September 2022).
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Compeau, P.E.C.; A Pevzner, P.; Tesler, G. How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 2011, 29, 987–991. [Google Scholar] [CrossRef] [Green Version]
- Van Loo, P.; Nordgard, S.H.; Lingjærde, O.C.; Russnes, H.G.; Rye, I.H.; Sun, W.; Weigman, V.J.; Marynen, P.; Zetterberg, A.; Naume, B.; et al. Allele-specific copy number analysis of tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 16910–16915. [Google Scholar] [CrossRef]
- Sun, J.X.; He, Y.; Sanford, E.; Montesion, M.; Frampton, G.M.; Vignot, S.; Soria, J.-C.; Ross, J.S.; Miller, V.A.; Stephens, P.J.; et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS Comput. Biol. 2018, 14, e1005965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hoff, D.D.; Stephenson, J.J.S., Jr.; Rosen, P.; Loesch, D.M.; Borad, M.J.; Anthony, S.; Jameson, G.S.; Brown, S.; Cantafio, N.; Richards, D.A.; et al. Pilot Study Using Molecular Profiling of Patients’ Tumors to Find Potential Targets and Select Treatments for Their Refractory Cancers. J. Clin. Oncol. 2010, 28, 4877–4883. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.H.; Thomas, J.W.; Chalmers, Z.R.; Schrock, A.B.; Tapias, C.A.; Frampton, G.M.; Kramer, K.; Miller, V.A.; Ali, S.M.; Tan, B.A.; et al. Comparison of comprehensive genomic profiling (CGP) and hotspot next generation sequencing (NGS) assays in identifying treatment options for care of patients with metastatic cancer in in the community setting. J. Clin. Oncol. 2016, 34, e23120. [Google Scholar] [CrossRef]
- Wheler, J.J.; Janku, F.; Naing, A.; Li, Y.; Stephen, B.; Zinner, R.; Subbiah, V.; Fu, S.; Karp, D.; Falchook, G.S.; et al. Cancer Therapy Directed by Comprehensive Genomic Profiling: A Single Center Study. Cancer Res. 2016, 76, 3690–3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Alsop, K.; Fereday, S.; Meldrum, C.; DeFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation–Positive Women with Ovarian Cancer: A Report From the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.S.; Rothermundt, C.; Thomas, K.; Bancroft, E.; Eeles, R.; Shanley, S.; Ardern-Jones, A.; Norman, A.; Kaye, S.B.; Gore, M.E. “BRCAness” syndrome in ovarian cancer: A case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J. Clin. Oncol. 2008, 26, 5530–5536. [Google Scholar] [CrossRef]
- Norquist, B.M.; Brady, M.F.; Harrell, M.I.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Burger, R.A.; Tewari, K.S.; et al. Mutations in Homologous Recombination Genes and Outcomes in Ovarian Carcinoma Patients in GOG 218: An NRG Oncology/Gynecologic Oncology Group Study. Clin. Cancer Res. 2018, 24, 777–783. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Khan, S.; Sun, Y.; Hess, K.; Shmulevich, I.; Sood, A.K.; Zhang, W. Association of BRCA1 and BRCA2 Mutations with Survival, Chemotherapy Sensitivity, and Gene Mutator Phenotype in Patients with Ovarian Cancer. JAMA 2011, 306, 1557–1565. [Google Scholar] [CrossRef]
- Gallagher, D.; Konner, J.; Bell-McGuinn, K.; Bhatia, J.; Sabbatini, P.; Aghajanian, C.; Offit, K.; Barakat, R.; Spriggs, D.; Kauff, N. Survival in epithelial ovarian cancer: A multivariate analysis incorporating BRCA mutation status and platinum sensitivity. Ann. Oncol. 2011, 22, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Eoh, K.J.; Park, H.S.; Park, J.S.; Lee, S.-T.; Han, J.; Lee, J.-Y.; Kim, S.W.; Kim, S.; Kim, Y.T.; Nam, E.J. Comparison of Clinical Outcomes of BRCA1/2 Pathologic Mutation, Variants of Unknown Significance, or Wild Type Epithelial Ovarian Cancer Patients. Cancer Res. Treat. 2017, 49, 408–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safra, T.; Lai, W.C.; Borgato, L.; Nicoletto, M.O.; Berman, T.; Reich, E.; Alvear, M.; Haviv, I.; Muggia, F.M. BRCA mutations and outcome in epithelial ovarian cancer (EOC): Experience in ethnically diverse groups. Ann. Oncol. 2013, 24, viii63–viii68. [Google Scholar] [CrossRef] [PubMed]
- Chetrit, A.; Hirsh-Yechezkel, G.; Ben-David, Y.; Lubin, F.; Friedman, E.; Sadetzki, S. Effect of BRCA1/2 Mutations on Long-Term Survival of Patients with Invasive Ovarian Cancer: The National Israeli Study of Ovarian Cancer. J. Clin. Oncol. 2008, 26, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e296. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, N.; Nakayama, K.; Shamima, Y.; Ishikawa, M.; Katagiri, A.; Iida, K.; Miyazaki, K. Gene amplification CCNE1 is related to poor survival and potential therapeutic target in ovarian cancer. Cancer 2010, 116, 2621–2634. [Google Scholar] [CrossRef]
- Yuan, J.; Hu, Z.; Mahal, B.A.; Zhao, S.D.; Kensler, K.H.; Pi, J.; Hu, X.; Zhang, Y.; Wang, Y.; Jiang, J.; et al. Integrated Analysis of Genetic Ancestry and Genomic Alterations across Cancers. Cancer Cell 2018, 34, 549–560.e9. [Google Scholar] [CrossRef] [Green Version]
- Etemadmoghadam, D.; Defazio, A.; Beroukhim, R.; Mermel, C.; George, J.; Getz, G.; Tothill, R.; Okamoto, A.; Raeder, M.B.; Harnett, P.; et al. Integrated Genome-Wide DNA Copy Number and Expression Analysis Identifies Distinct Mechanisms of Primary Chemoresistance in Ovarian Carcinomas. Clin. Cancer Res. 2009, 15, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Ratner, E.S.; Keane, F.K.; Lindner, R.; A Tassi, R.; Paranjape, T.; Glasgow, M.; Nallur, S.; Deng, Y.; Lu, L.; Steele, L.; et al. A KRAS variant is a biomarker of poor outcome, platinum chemotherapy resistance and a potential target for therapy in ovarian cancer. Oncogene 2011, 31, 4559–4566. [Google Scholar] [CrossRef] [Green Version]
- Colombo, I.; Lheureux, S.; Oza, A.M. Rucaparib: A novel PARP inhibitor for BRCA advanced ovarian cancer. Drug Des. Dev. Ther. 2018, 12, 605–617. [Google Scholar] [CrossRef]
- Bi, F.; Chen, Y.; Yang, Q. Significance of tumor mutation burden combined with immune infiltrates in the progression and prognosis of ovarian cancer. Cancer Cell Int. 2020, 20, 373. [Google Scholar] [CrossRef] [PubMed]
- Choucair, K.; Morand, S.; Stanbery, L.; Edelman, G.; Dworkin, L.; Nemunaitis, J. TMB: A promising immune-response biomarker, and potential spearhead in advancing targeted therapy trials. Cancer Gene Ther. 2020, 27, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Bhaumik, S.; Dhodapkar, K.; Grivel, J.-C.J.B.; Gupta, S.; A Hanks, B.; Janetzki, S.; O Kleen, T.; Koguchi, Y.; Lund, A.W.; et al. SITC cancer immunotherapy resource document: A compass in the land of biomarker discovery. J. Immunother. Cancer 2020, 8, e000705. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2018, 30, 44–56. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, J.-Y.; Kim, S. How to use immune checkpoint inhibitor in ovarian cancer? J. Gynecol. Oncol. 2019, 30, e105. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Gao, X.; Qin, Q.; Li, H.; Yuan, Z.; Zhao, S. Association between tumor mutation burden and immune infiltration in ovarian cancer. Int. Immunopharmacol. 2020, 89, 107126. [Google Scholar] [CrossRef]
- Feinberg, J.; Elvin, J.; Bellone, S.; Santin, A. Identification of ovarian cancer patients for immunotherapy by concurrent assessment of tumor mutation burden (TMB), microsatellite instability (MSI) status, and targetable genomic alterations (GA). Gynecol. Oncol. 2018, 149, 36. [Google Scholar] [CrossRef]
- VanderWalde, A.; Spetzler, D.; Xiao, N.; Gatalica, Z.; Marshall, J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018, 7, 746–756. [Google Scholar] [CrossRef]
- Schwaederle, M.; Parker, B.A.; Schwab, R.B.; Daniels, G.A.; Piccioni, D.E.; Kesari, S.; Helsten, T.L.; Bazhenova, L.A.; Romero, J.; Fanta, P.T.; et al. Precision Oncology: The UC San Diego Moores Cancer Center PREDICT Experience. Mol. Cancer Ther. 2016, 15, 743–752. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | CGP N = 108 | Control N = 838 | All N = 946 | p Value |
|---|---|---|---|---|
| Age at diagnosis (years) | 62.7 (34.1–80.2) | 61.3 (21.5–93.1) | 61.4 (21.5–93.1) | 0.373 |
| Stage | ||||
| Stages I + II | 11/107 (10.3%) | 127/824 (15.4%) | 138/931 (14.8%) | 0.358 |
| Stage III | 76/107 (71.0%) | 560/824 (68.0%) | 636/931 (68.3%) | |
| Stage IV | 20/107 (18.7%) | 137/824 (16.6%) | 157/931 (16.9%) | |
| Histology | ||||
| Serous papillary | 84/107 (78.5%) | 529/820 (64.5%) | 613/927 (66.1%) | 0.002 |
| Endometrioid/poorly differentiated | 17/107 (15.9%) | 265/820 (32.3%) | 282/927 (30.4%) | |
| Mucinous/clear cell/carcinosarcoma | 6/107 (5.6%) | 26/820 (3.2%) | 32/927 (3.5%) | |
| BRCA mutation status | ||||
| BRCA wildtype | 76 (70.4%) | 343/546 (62.8%) | 419/654 (64.1%) | 0.309 |
| BRCA1 mutation | 22 (20.4%) | 146/546 (26.7%) | 168/654 (26.7%) | |
| BRCA2 mutation | 10 (9.3%) | 57/546 (10.4%) | 67/654 (10.2%) | |
| Unknown | 0 (0%) | 297/837 (34.7%) | 290/945 (30.7%) | |
| Mutation | ||||
| Germline | 24 (22.2%) | 193/548 (35.2%) | 217/656 (33.1%) | 0.001 |
| Somatic | 8 (7.4%) | 12/548 (2.2%) | 20/656 (3.1%) | |
| Ethnicity | ||||
| Ashkenazi Jewish | 75 (69.4%) | 422/832 (50.7%) | 497/940 (52.9%) | 0.0007 |
| PARP inhibitors | 33 (30.6%) | 75 (9.0%) | 108 (11.4%) | <0.0001 |
| Current Study N = 108 | PanCancer Atlas TCGA Ovarian * N = 489 | |||||
|---|---|---|---|---|---|---|
| Mutation | Number of Samples | (%) | Mutation | Number of Samples | (%) | |
| 1 | TP53 | 88 | (81.5) | TP53 | 306 | (95.9) |
| 2 | BRCA1 | 23 | (21.3) | TTN | 62 | (17.0) |
| 3 | CCNE1 | 21 | (19.4) | BRCA 1 | 37 | (11.7) |
| 4 | KRAS | 13 | (12.0) | BRCA2 | 35 | (10.8) |
| 5 | BRCA2 | 12 | (11.1) | USH2A | 20 | (6.3) |
| 6 | MYC | 12 | (11.1) | CSMD3 | 19 | (6.0) |
| 7 | NF1 | 9 | (8.3) | FAT3 | 19 | (5.7) |
| 8 | PIK3CA | 9 | (8.3) | MUC16 | 20 | (5.7)) |
| 9 | RB1 | 8 | (7.4) | LRP2 | 16 | (4.7) |
| 10 | ERBB2 | 6 | (5.6) | RYR2 | 15 | (4.4) |
| 11 | ARID1A | 6 | (5.6) | HMCN1 | 15 | (4.4) |
| 12 | APC | 6 | (5.6) | LRP1B | 14 | (4.0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peleg Hasson, S.; Hershkovitz, D.; Adar, L.; Brezis, M.; Shachar, E.; Aks, R.; Galmor, L.; Raviv, Y.; Ben Neriah, S.; Merimsky, O.; et al. Implementation of Comprehensive Genomic Profiling in Ovarian Cancer Patients: A Retrospective Analysis. Cancers 2023, 15, 218. https://doi.org/10.3390/cancers15010218
Peleg Hasson S, Hershkovitz D, Adar L, Brezis M, Shachar E, Aks R, Galmor L, Raviv Y, Ben Neriah S, Merimsky O, et al. Implementation of Comprehensive Genomic Profiling in Ovarian Cancer Patients: A Retrospective Analysis. Cancers. 2023; 15(1):218. https://doi.org/10.3390/cancers15010218
Chicago/Turabian StylePeleg Hasson, Shira, Dov Hershkovitz, Lyri Adar, Miriam Brezis, Eliya Shachar, Rona Aks, Lee Galmor, Yuval Raviv, Shira Ben Neriah, Ofer Merimsky, and et al. 2023. "Implementation of Comprehensive Genomic Profiling in Ovarian Cancer Patients: A Retrospective Analysis" Cancers 15, no. 1: 218. https://doi.org/10.3390/cancers15010218
APA StylePeleg Hasson, S., Hershkovitz, D., Adar, L., Brezis, M., Shachar, E., Aks, R., Galmor, L., Raviv, Y., Ben Neriah, S., Merimsky, O., Sabo, E., Wolf, I., & Safra, T. (2023). Implementation of Comprehensive Genomic Profiling in Ovarian Cancer Patients: A Retrospective Analysis. Cancers, 15(1), 218. https://doi.org/10.3390/cancers15010218