

High ME1 Expression Is a Molecular Predictor of Post-Transplant Survival of Patients with Acute Myeloid Leukemia
 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Gene Expression Profiling
2.2. Searching Strategy and Derivation of Prognosis Associated Genes
2.3. Biological Pathways, Cells Signatures, and Drug Sensitivity Prediction
2.4. Cell Lines and Drugs
2.5. Lentiviral Vectors and Lentivirus Production
2.6. In Vitro Assays
2.6.1. Apoptosis Assay
2.6.2. Oxygen Consumption (OCR) and Extracellular Acidification Rate (ECAR) Measurements
2.6.3. T-Cell Mediated Cytotoxicity Assay
2.7. Statistical Analysis
3. Results
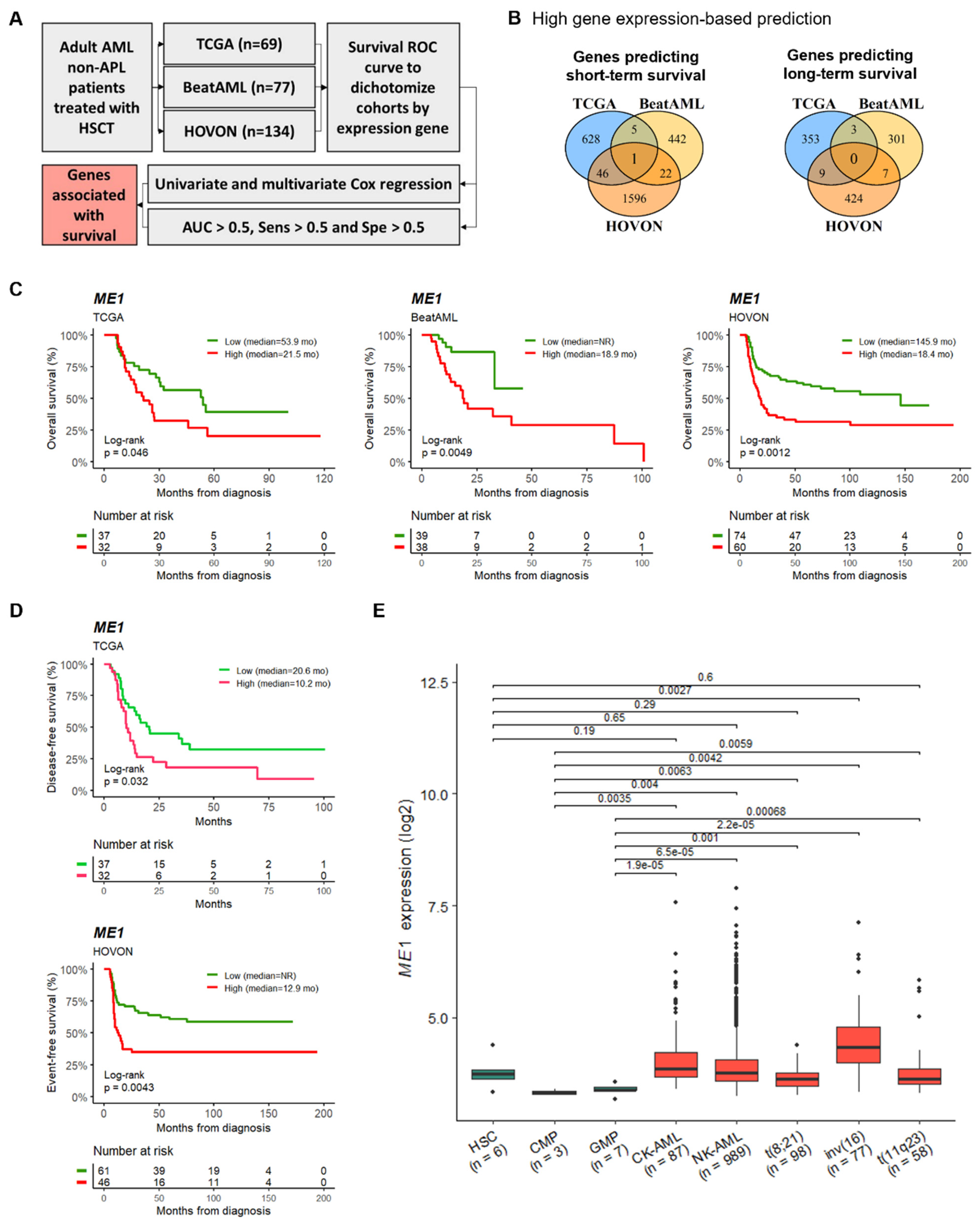
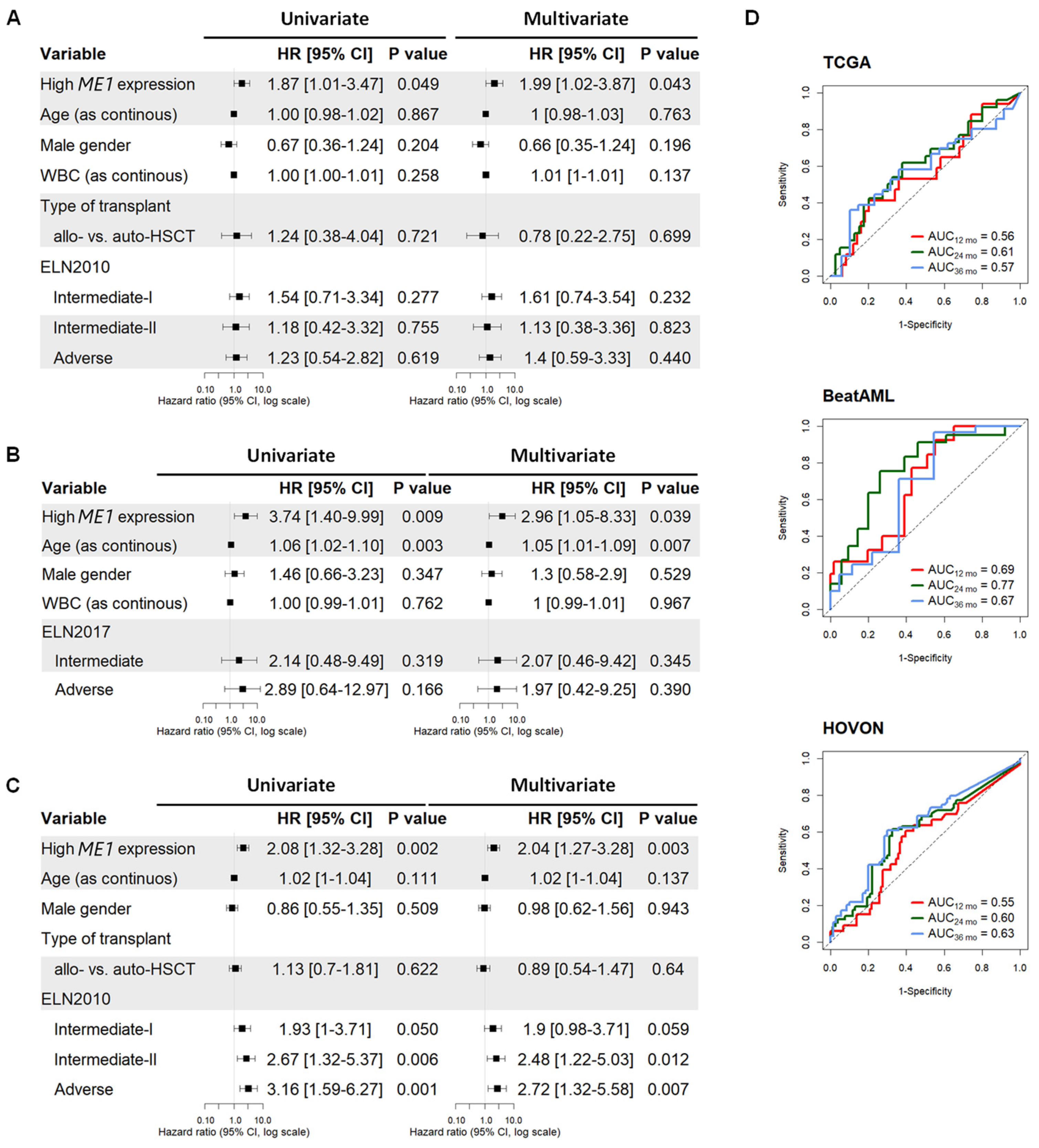
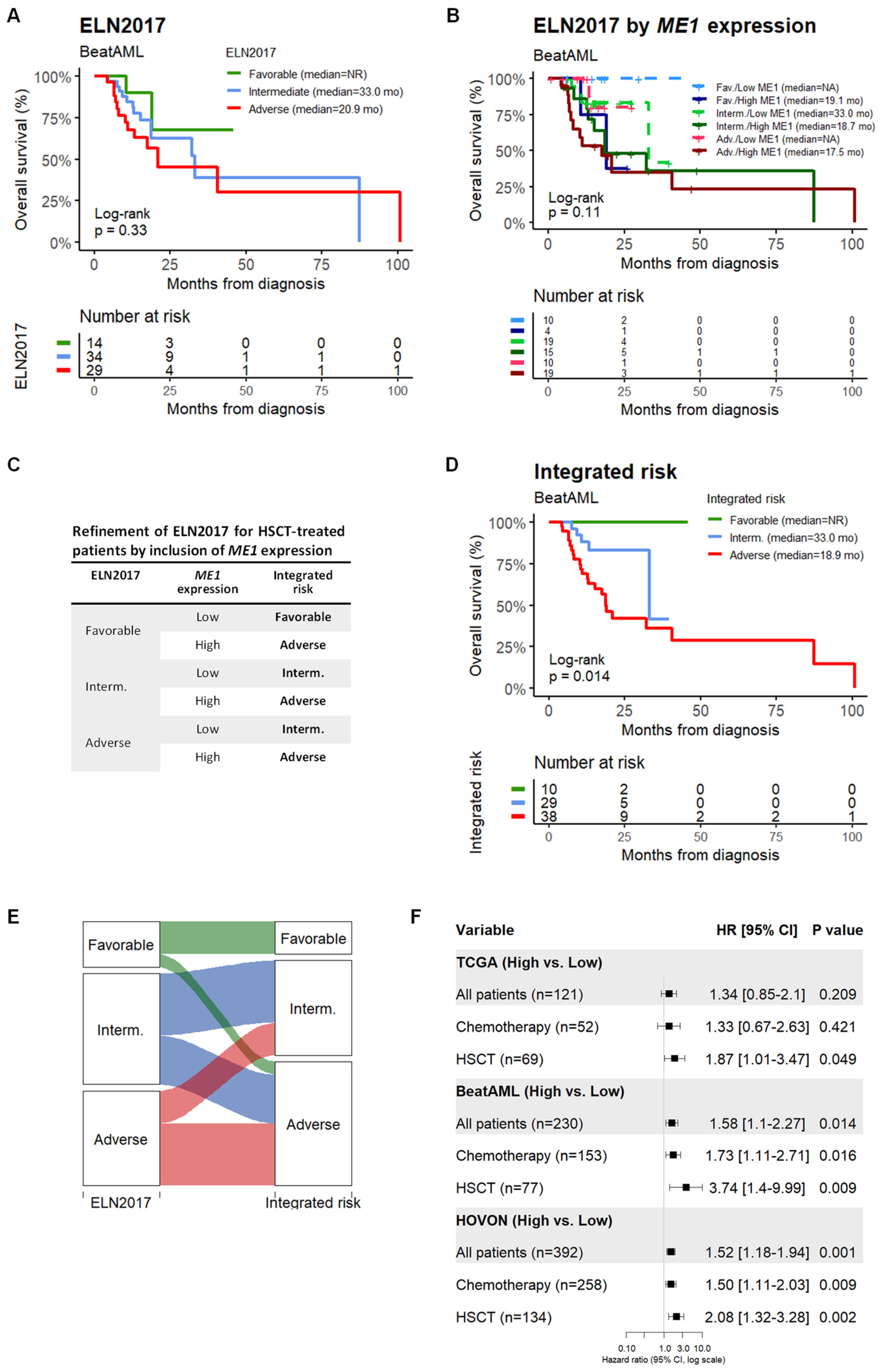
3.1. ME1 Gene Was Identified as an Independent Factor to Predict the Survival of AML Patients Subjected to HSCT
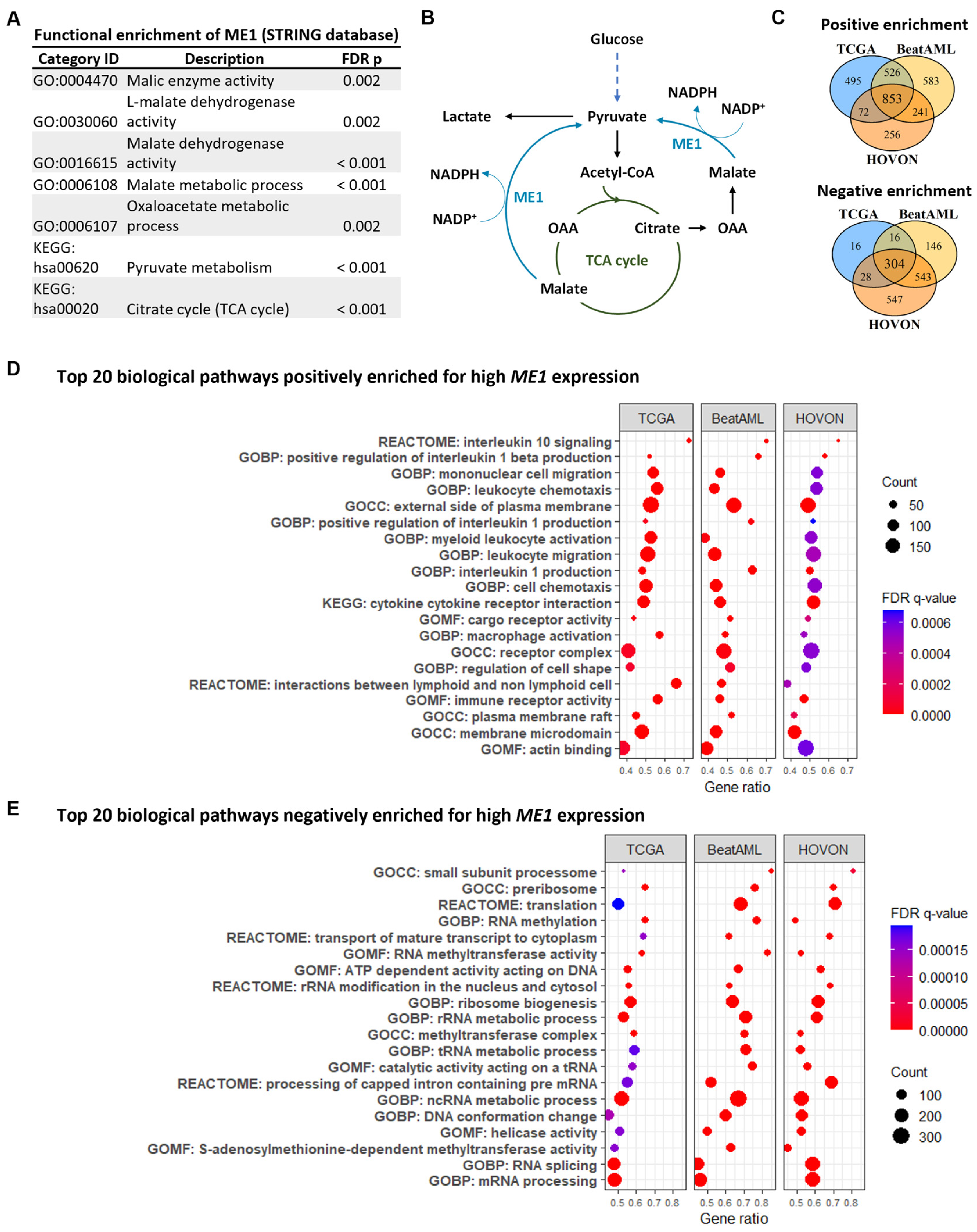
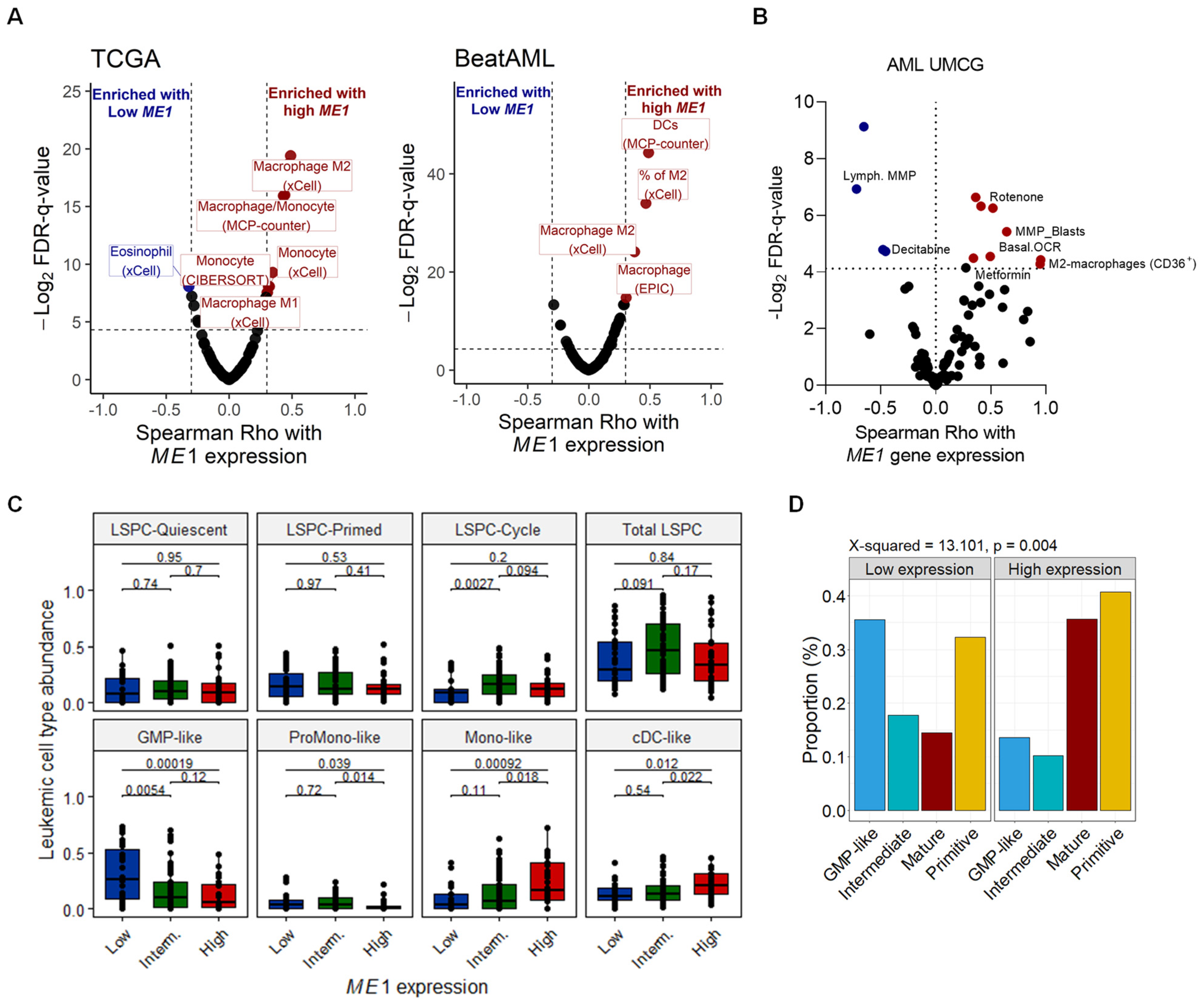
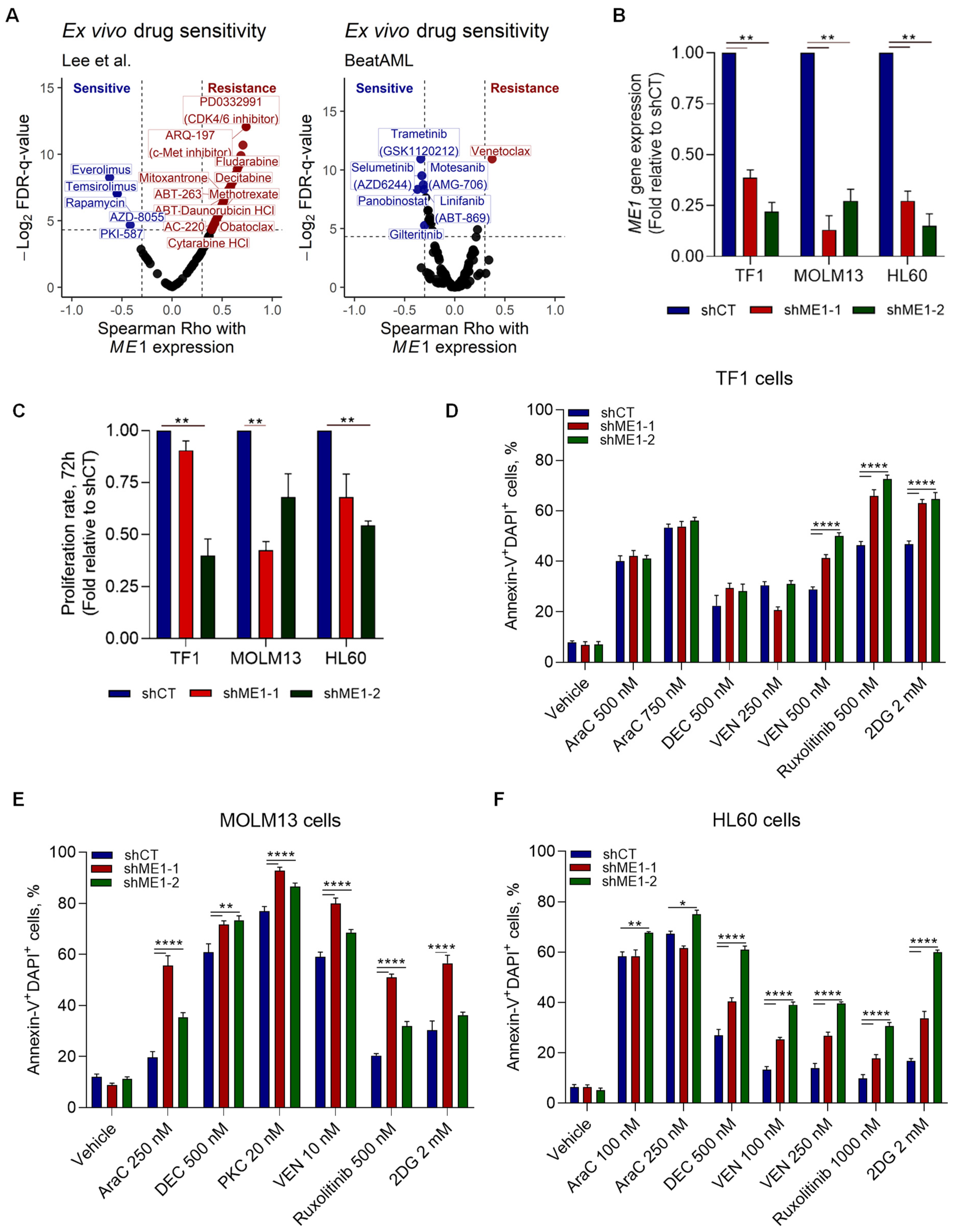
3.2. ME1 Is Associated with an Immunosuppressive TME, Mono/cDC-like Leukemia, and Increased Sensitivity to a Specific Set of Drugs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Appelbaum, F.R. Hematopoietic-cell transplantation at 50. N. Engl. J. Med. 2007, 357, 1472–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loke, J.; Buka, R.; Craddock, C. Allogeneic stem cell transplantation for acute myeloid leukemia: Who, when, and how? Front. Immunol. 2021, 12, 659595. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, J.J.; Breems, D.; van Putten, W.L.J.; Gratwohl, A.A.; Passweg, J.R.; Pabst, T.; Maertens, J.; Beverloo, H.B.; van Marwijk Kooy, M.; Wijermans, P.W.; et al. Comparative analysis of the value of allogeneic hematopoietic stem-cell transplantation in acute myeloid leukemia with monosomal karyotype versus other cytogenetic risk categories. J. Clin. Oncol. 2012, 30, 2140–2146. [Google Scholar] [CrossRef]
- Styczyński, J.; Tridello, G.; Koster, L.; Iacobelli, S.; van Biezen, A.; van der Werf, S.; Mikulska, M.; Gil, L.; Cordonnier, C.; Ljungman, P.; et al. Infectious Diseases Working Party EBMT Death after hematopoietic stem cell transplantation: Changes over calendar year time, infections and associated factors. Bone Marrow Transplant. 2020, 55, 126–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, J.; Jentzsch, M.; Bill, M.; Goldmann, K.; Schulz, J.; Niederwieser, D.; Platzbecker, U.; Schwind, S. Prognostic impact of the ELN2017 risk classification in patients with AML receiving allogeneic transplantation. Blood Adv. 2020, 4, 3864–3874. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Salvatore, D.; Labopin, M.; Ruggeri, A.; Battipaglia, G.; Ghavamzadeh, A.; Ciceri, F.; Blaise, D.; Arcese, W.; Sociè, G.; Bourhis, J.H.; et al. Outcomes of hematopoietic stem cell transplantation from unmanipulated haploidentical versus matched sibling donor in patients with acute myeloid leukemia in first complete remission with intermediate or high-risk cytogenetics: A study from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Haematologica 2018, 103, 1317–1328. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.-R.; Xu, L.-P.; Zhang, X.-H.; Liu, K.-Y.; Chang, Y.-J.; Lv, M.; Yan, C.-H.; Chen, Y.-H.; Han, W.; Wang, F.-R.; et al. Allogeneic hematopoietic stem cell transplantation for intermediate-risk acute myeloid leukemia in the first remission: Outcomes using haploidentical donors are similar to those using matched siblings. Ann. Hematol. 2021, 100, 555–562. [Google Scholar] [CrossRef]
- Ferrara, F.; Picardi, A. Is there still a role for autologous stem cell transplantation for the treatment of acute myeloid leukemia? Cancers 2019, 12, 59. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Chen, X.; Feng, S. Autologous hematopoietic stem cell transplantation in acute myelogenous leukemia. Biol. Blood Marrow Transplant. 2019, 25, e285–e292. [Google Scholar] [CrossRef]
- Chen, S.; Zeiser, R. Novel biomarkers for outcome after allogeneic hematopoietic stem cell transplantation. Front. Immunol. 2020, 11, 1854. [Google Scholar] [CrossRef] [PubMed]
- Ino, K.; Fuji, S.; Tajima, K.; Tanaka, T.; Okinaka, K.; Inamoto, Y.; Kurosawa, S.; Kim, S.-W.; Katayama, N.; Fukuda, T. Clinical Utility of Wilms’ Tumor 1 Monitoring in Patients with Myeloid Malignancy and Prior Allogeneic Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2017, 23, 1780–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rautenberg, C.; Pechtel, S.; Hildebrandt, B.; Betz, B.; Dienst, A.; Nachtkamp, K.; Kondakci, M.; Geyh, S.; Wieczorek, D.; Haas, R.; et al. Wilms’ Tumor 1 Gene Expression Using a Standardized European LeukemiaNet-Certified Assay Compared to Other Methods for Detection of Minimal Residual Disease in Myelodysplastic Syndrome and Acute Myelogenous Leukemia after Allogeneic Blood Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2018, 24, 2337–2343. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Xie, Y.; Wang, C.; Han, Y.; Bao, X.; Ma, S.; Yilmaz, A.; Yang, B.; Ji, Y.; Xu, J.; et al. Prediction of acute GVHD and relapse by metabolic biomarkers after allogeneic hematopoietic stem cell transplantation. JCI Insight 2018, 3, e99672. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.V.; Jorgensen, J.L.; Saliba, R.M.; Wang, S.A.; Alousi, A.M.; Andersson, B.S.; Bashir, Q.; Ciurea, S.O.; Kebriaei, P.; Marin, D.; et al. Early Post-Transplant Minimal Residual Disease Assessment Improves Risk Stratification in Acute Myeloid Leukemia. Biol. Blood Marrow Transplant. 2018, 24, 1514–1520. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.G.; Hoadley, K.; Triche, T.J.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Wouters, B.J.; Erpelinck, C.A.J.; Abbas, S.; Beverloo, H.B.; Lugthart, S.; Löwenberg, B.; Delwel, R.; Valk, P.J.M. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica 2009, 94, 131–134. [Google Scholar] [CrossRef] [Green Version]
- de Jonge, H.J.M.; Valk, P.J.M.; Veeger, N.J.G.M.; ter Elst, A.; den Boer, M.L.; Cloos, J.; de Haas, V.; van den Heuvel-Eibrink, M.M.; Kaspers, G.J.L.; Zwaan, C.M.; et al. High VEGFC expression is associated with unique gene expression profiles and predicts adverse prognosis in pediatric and adult acute myeloid leukemia. Blood 2010, 116, 1747–1754. [Google Scholar] [CrossRef] [Green Version]
- Weinhäuser, I.; Pereira-Martins, D.A.; Almeida, L.Y.; Hilberink, J.R.; Ortiz, C.; Silveira, D.R.A.; Quek, L.; Araujo, C.L.; Bianco, T.M.; Lucena-Araujo, A.; et al. M2-polarized macrophages control LSC fate by enhancing stemness, homing, immune evasion and metabolic reprogramming. bioRxiv 2022. [Google Scholar] [CrossRef]
- Pereira-Martins, D.A.; Griessinger, E.; Weinhäuser, I.; Coelho-Silva, J.; Silveira, D.R.A.; Quek, L.; Erdem, A.; Hilberink, J.R.; de Paula, E.V.; Olalla Saad, S.T.; et al. P439: Associating ex vivo drug sensitivity with metabolic status identifies effective combination strategies in acute myeloid leukemia. HemaSphere 2022, 6, 339–340. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Racle, J.; Gfeller, D. EPIC: A Tool to Estimate the Proportions of Different Cell Types from Bulk Gene Expression Data. Methods Mol. Biol. 2020, 2120, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Zeng, A.G.X.; Bansal, S.; Jin, L.; Mitchell, A.; Chen, W.C.; Abbas, H.A.; Chan-Seng-Yue, M.; Voisin, V.; van Galen, P.; Tierens, A.; et al. A cellular hierarchy framework for understanding heterogeneity and predicting drug response in acute myeloid leukemia. Nat. Med. 2022, 28, 1212–1223. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Lee, S.-I.; Celik, S.; Logsdon, B.A.; Lundberg, S.M.; Martins, T.J.; Oehler, V.G.; Estey, E.H.; Miller, C.P.; Chien, S.; Dai, J.; et al. A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia. Nat. Commun. 2018, 9, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira-Martins, D.A.; Weinhäuser, I.; Coelho-Silva, J.L.; França-Neto, P.L.; Almeida, L.Y.; Bianco, T.M.; Silva, C.L.; França, R.F.; Traina, F.; Rego, E.M.; et al. MLL5 improves ATRA driven differentiation and promotes xenotransplant engraftment in acute promyelocytic leukemia model. Cell Death Dis. 2021, 12, 371. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, E.; Zolov, S.N.; Huang, K.; Bonifant, C.L. T-cell Activity against AML Improved by Dual-Targeted T Cells Stimulated through T-cell and IL7 Receptors. Cancer Immunol. Res. 2019, 7, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Simmen, F.A.; Alhallak, I.; Simmen, R.C.M. Malic enzyme 1 (ME1) in the biology of cancer: It is not just intermediary metabolism. J. Mol. Endocrinol. 2020, 65, R77–R90. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Pongratz, R.L.; Kibbey, R.G.; Shulman, G.I.; Cline, G.W. Cytosolic and mitochondrial malic enzyme isoforms differentially control insulin secretion. J. Biol. Chem. 2007, 282, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Kreitz, J.; Schönfeld, C.; Seibert, M.; Stolp, V.; Alshamleh, I.; Oellerich, T.; Steffen, B.; Schwalbe, H.; Schnütgen, F.; Kurrle, N.; et al. Metabolic plasticity of acute myeloid leukemia. Cells 2019, 8, 805. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Du, W.; Mancuso, A.; Wellen, K.E.; Yang, X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013, 493, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.; Lu, W.; Du, Y.; Yan, W.; Bao, Q.; Tian, Y.; Wang, G.; Ye, H.; Hao, H. Cytosolic ME1 integrated with mitochondrial IDH2 supports tumor growth and metastasis. Redox Biol. 2020, 36, 101685. [Google Scholar] [CrossRef]
- Liao, R.; Ren, G.; Liu, H.; Chen, X.; Cao, Q.; Wu, X.; Li, J.; Dong, C. ME1 promotes basal-like breast cancer progression and associates with poor prognosis. Sci. Rep. 2018, 8, 16743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Zhou, S.; Wang, P.; Guo, Y.; Xie, B.; Ding, S. Malic enzyme 1 (ME1) is a potential oncogene in gastric cancer cells and is associated with poor survival of gastric cancer patients. Oncol. Targets Ther. 2019, 12, 5589–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, C.; Yamamoto, K.; Fujiwara-Tani, R.; Luo, Y.; Matsushima, S.; Fujii, K.; Ohmori, H.; Sasahira, T.; Sasaki, T.; Kitadai, Y.; et al. Expression of cytosolic malic enzyme (ME1) is associated with disease progression in human oral squamous cell carcinoma. Cancer Sci. 2018, 109, 2036–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, G. Mutant KRAS associated malic enzyme 1 expression is a predictive marker for radiation therapy response in non-small cell lung cancer. Radiat. Oncol. 2015, 10, 145. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Xing, C.; Cui, K.; Li, Y.; Zhang, J.; Du, R.; Zhang, X.; Li, Y. MicroRNA-30a attenuates mutant KRAS-driven colorectal tumorigenesis via direct suppression of ME1. Cell Death Differ. 2017, 24, 1253–1262. [Google Scholar] [CrossRef]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef]
- Kim, Y.K.; Kim, S.E.; Chang Park, H.; Hwang, J.H.; Lee, H.T. Human recombinant IL-10 reduces xenogenic cytotoxicity via macrophage M2 polarization. Biochem. Biophys. Rep. 2020, 24, 100857. [Google Scholar] [CrossRef]
- Chen, N.; Xu, Y.; Mou, J.; Rao, Q.; Xing, H.; Tian, Z.; Tang, K.; Wang, M.; Wang, J. Targeting of IL-10R on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood Cancer J. 2021, 11, 144. [Google Scholar] [CrossRef]
- Grimwade, D.; Walker, H.; Oliver, F.; Wheatley, K.; Harrison, C.; Harrison, G.; Rees, J.; Hann, I.; Stevens, R.; Burnett, A.; et al. The importance of diagnostic cytogenetics on outcome in AML: Analysis of 1612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood 1998, 92, 2322–2333. [Google Scholar] [CrossRef] [Green Version]
- Kottaridis, P.D.; Gale, R.E.; Frew, M.E.; Harrison, G.; Langabeer, S.E.; Belton, A.A.; Walker, H.; Wheatley, K.; Bowen, D.T.; Burnett, A.K.; et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: Analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood 2001, 98, 1752–1759. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Ravandi, F.; Agresta, S.; Konopleva, M.; Takahashi, K.; Kadia, T.; Routbort, M.; Patel, K.P.; Brandt, M.; Pierce, S.; et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am. J. Hematol. 2015, 90, 732–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, T.; Wu, S.; Zhao, H.; Xu, K.; Fu, H.; Cheng, Z.; Pang, Y.; Han, Y.; Chen, L.; Wang, C.; et al. Molecular predictors of post-transplant survival in acute myeloid leukemia. Blood Cancer J. 2017, 7, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Märklin, M.; Hagelstein, I.; Hinterleitner, C.; Salih, H.R.; Kauer, J.; Heitmann, J.S. CD105 (endoglin) as risk marker in AML patients undergoing stem cell transplantation. Int. J. Hematol. 2020, 112, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.; Labopin, M.; Socié, G.; Daguindau, E.; Volin, L.; Huynh, A.; Bourhis, J.H.; Milpied, N.; Cornelissen, J.; Chevallier, P.; et al. Acute Leukemia Working Party of the European Group of Blood and Bone Marrow Transplantation Outcome of pa-tients with distinct molecular genotypes and cytogenetically normal AML after allogeneic transplantation. Blood 2015, 126, 2062–2069. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz Rojas, C.A.; Costa-Neto, A.; Pereira-Martins, D.A.; Le, D.M.; Sternadt, D.; Weinhäuser, I.; Huls, G.; Schuringa, J.J.; Magalhães Rego, E. High ME1 Expression Is a Molecular Predictor of Post-Transplant Survival of Patients with Acute Myeloid Leukemia. Cancers 2023, 15, 296. https://doi.org/10.3390/cancers15010296
Ortiz Rojas CA, Costa-Neto A, Pereira-Martins DA, Le DM, Sternadt D, Weinhäuser I, Huls G, Schuringa JJ, Magalhães Rego E. High ME1 Expression Is a Molecular Predictor of Post-Transplant Survival of Patients with Acute Myeloid Leukemia. Cancers. 2023; 15(1):296. https://doi.org/10.3390/cancers15010296
Chicago/Turabian StyleOrtiz Rojas, César Alexander, Abel Costa-Neto, Diego A. Pereira-Martins, Duy Minh Le, Dominique Sternadt, Isabel Weinhäuser, Gerwin Huls, Jan Jacob Schuringa, and Eduardo Magalhães Rego. 2023. "High ME1 Expression Is a Molecular Predictor of Post-Transplant Survival of Patients with Acute Myeloid Leukemia" Cancers 15, no. 1: 296. https://doi.org/10.3390/cancers15010296
APA StyleOrtiz Rojas, C. A., Costa-Neto, A., Pereira-Martins, D. A., Le, D. M., Sternadt, D., Weinhäuser, I., Huls, G., Schuringa, J. J., & Magalhães Rego, E. (2023). High ME1 Expression Is a Molecular Predictor of Post-Transplant Survival of Patients with Acute Myeloid Leukemia. Cancers, 15(1), 296. https://doi.org/10.3390/cancers15010296