
BTK Isoforms p80 and p65 Are Expressed in Head and Neck Squamous Cell Carcinoma (HNSCC) and Involved in Tumor Progression

 , , , , , , , ,
, , , , , , , ,  ,
,  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. RNA Isolation
2.3. Quantitative Expression Analysis of BTK-p77 and BTK-p80/p65
2.4. Isolation of CD19+ B Cells from PBMC
2.5. Immunoblot Analysis
Research Subjects
2.6. Proliferation Assay
2.7. Cell Cycle Analysis
2.8. Chorio-Allantoic Membrane (CAM) Assay and IHC Analysis
2.9. Short Hairpin RNA Vector Cloning and Lentiviral Infection
2.10. Scratch Assay/Wound Closure Assay
2.11. Transwell Migration Assay
2.12. VEGFA ELISA
2.13. TCGA Dataset & Data Analysis
2.14. Statistical Analysis
3. Results
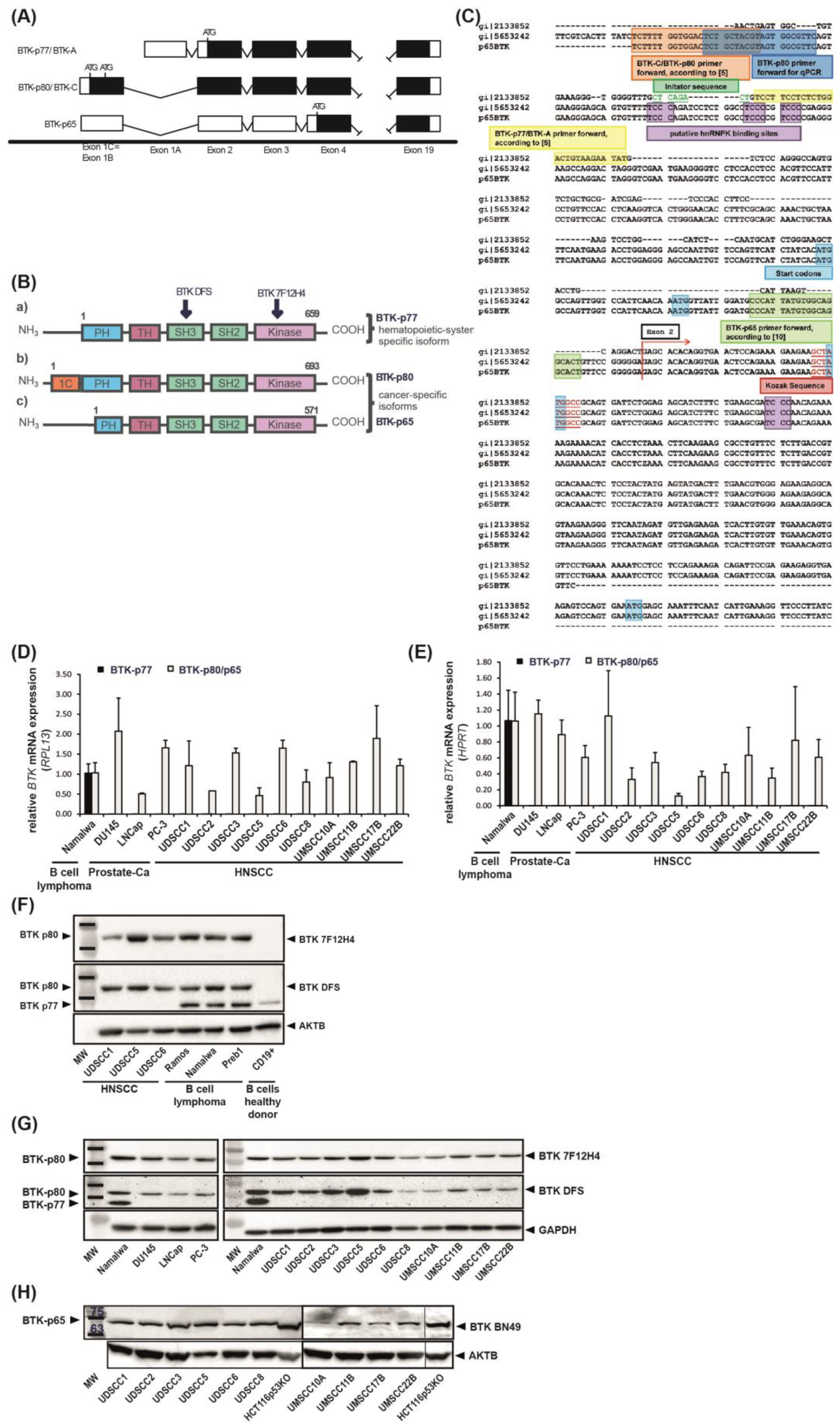
3.1. BTK-p80 and BTK-p65 Isoforms Are Expressed in HNSCC-Derived Cell Lines
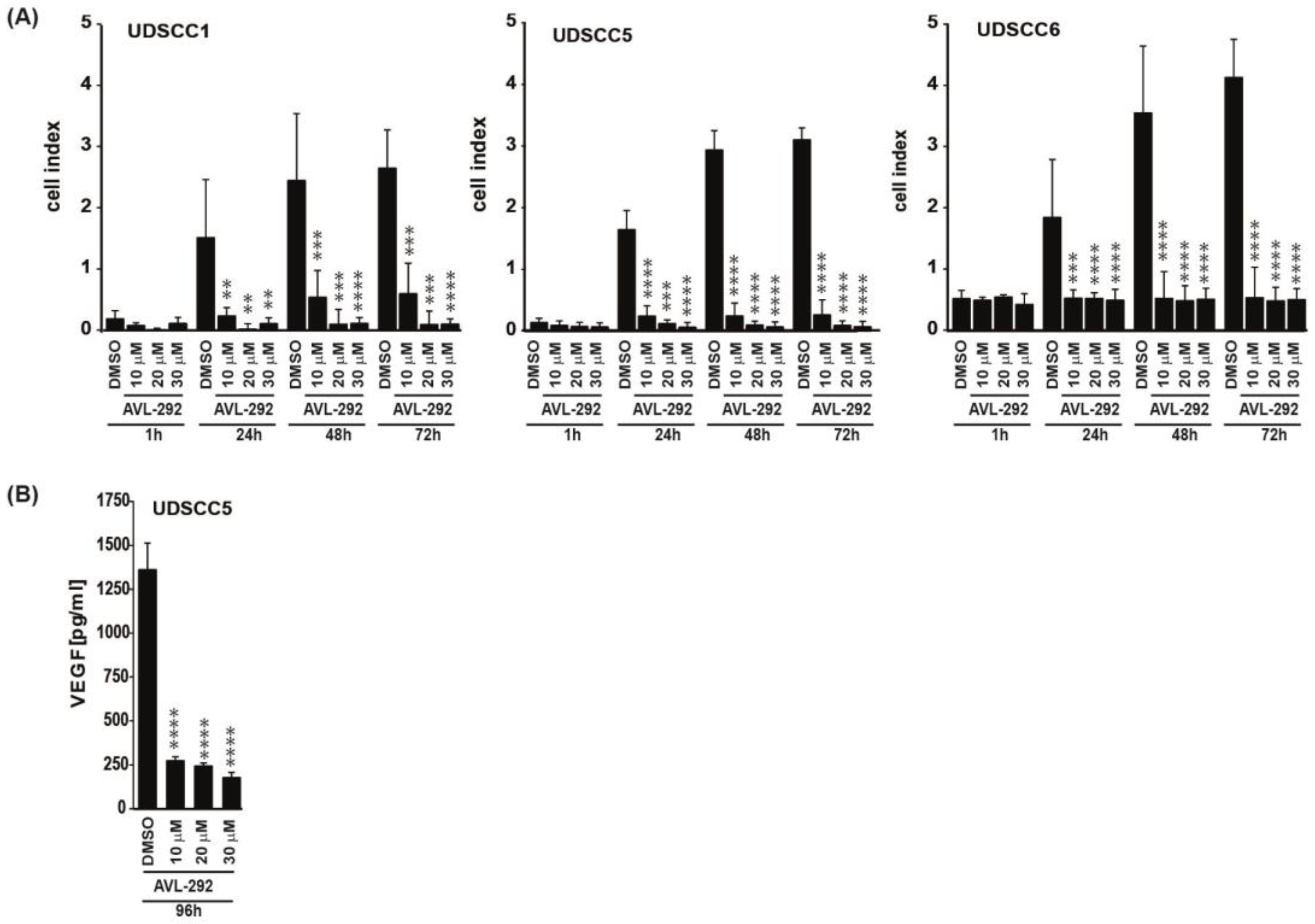
3.2. BTK Inhibition Affects Proliferation, Transmigration and VEGFA Secretion of HNSCC Derived Cell Lines
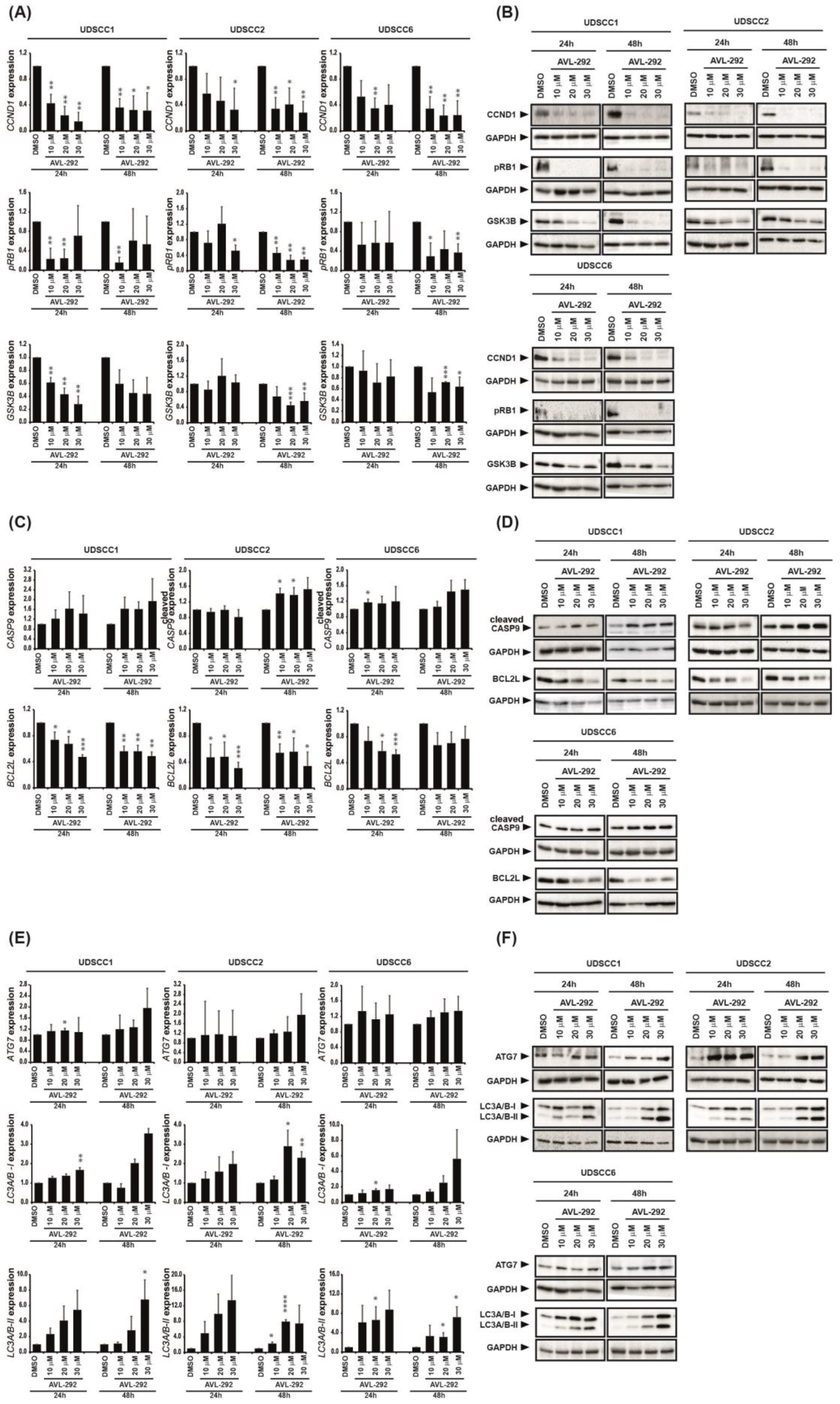
3.3. BTK Inhibition Induces Cell Cycle Arrest, Apoptosis and Autophagy in HNSCC-Derived Cell Lines
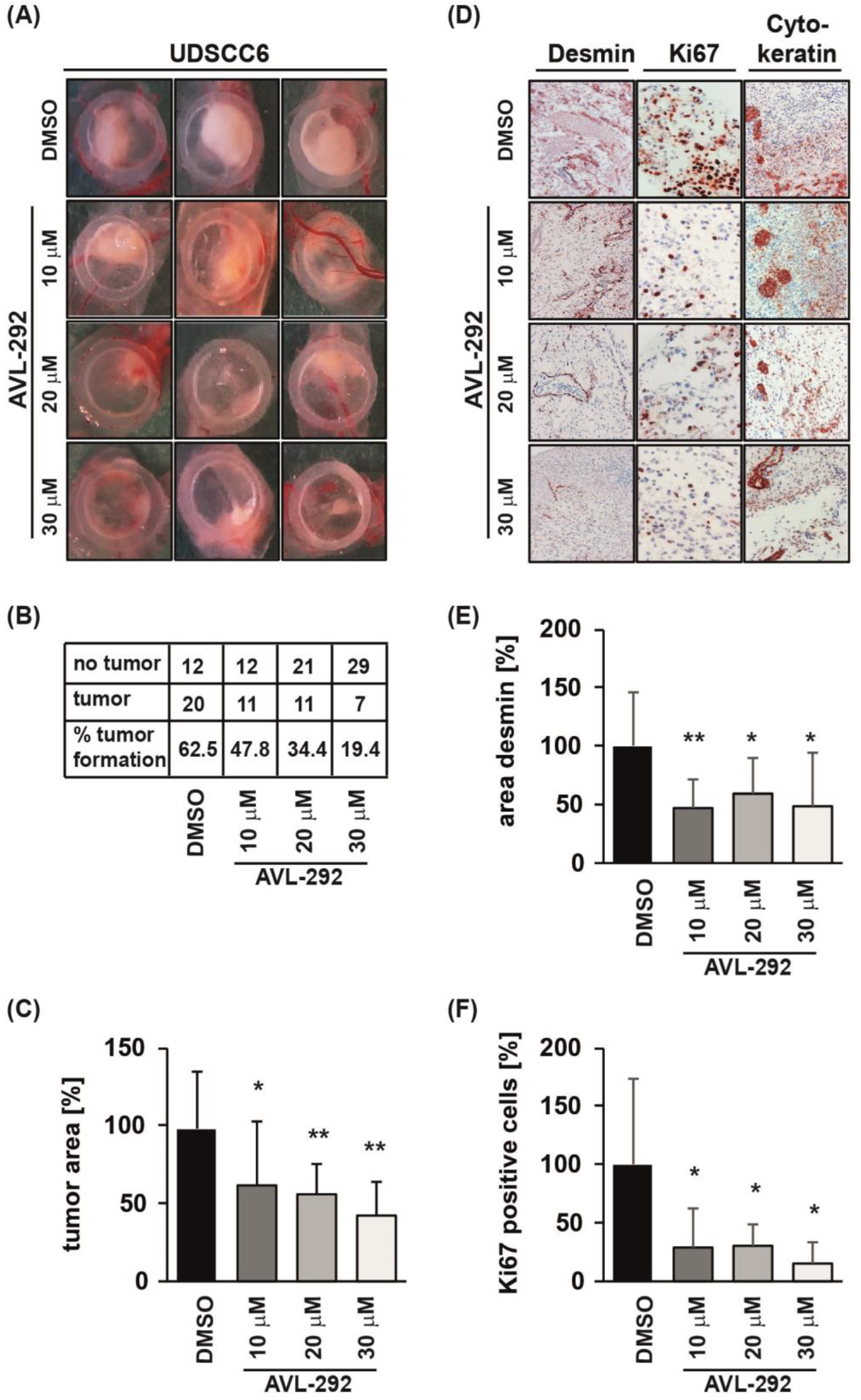
3.4. BTK Inhibition Impairs Tumor Growth and Angiogenesis In Vivo
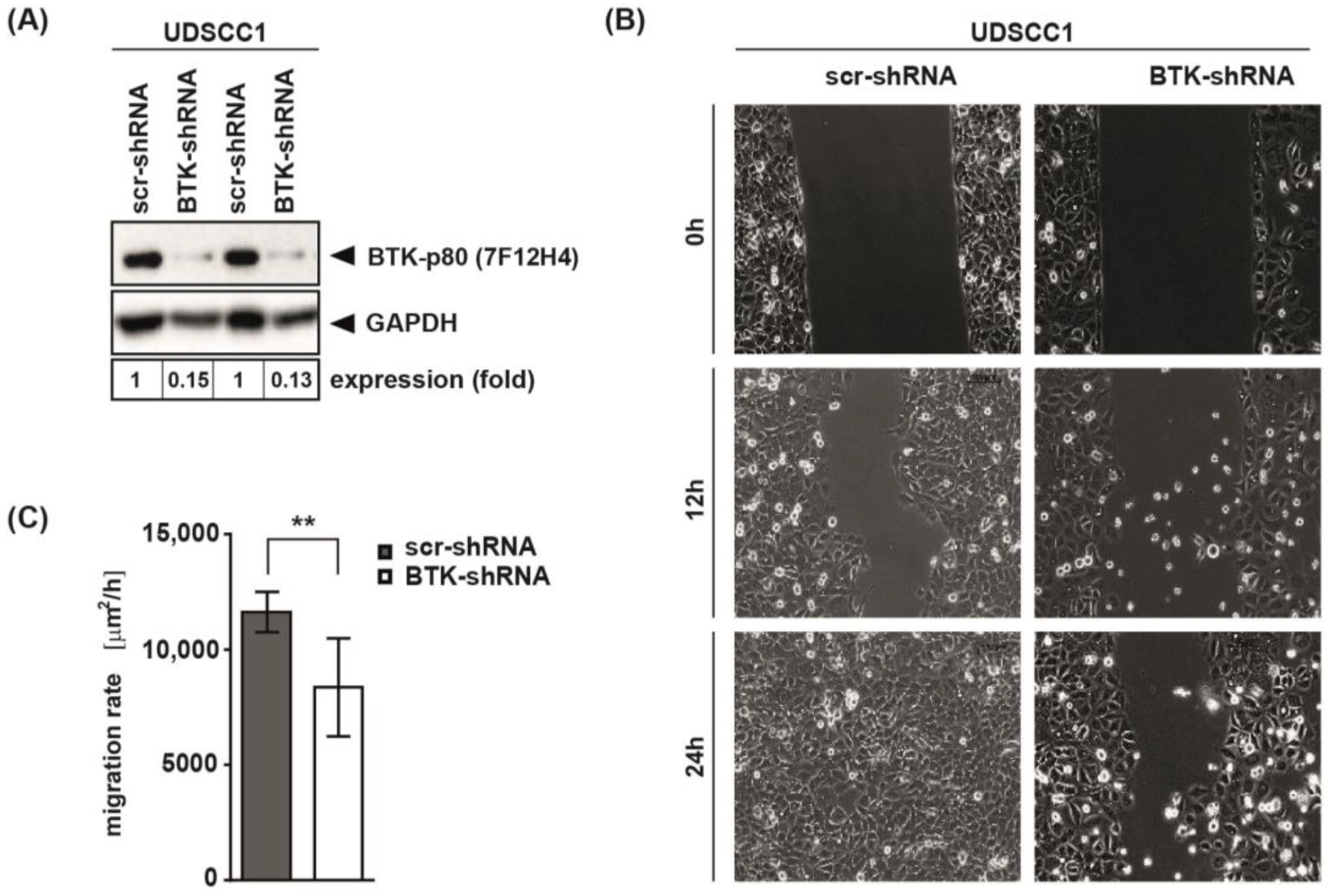
3.5. BTK Inhibition and Its Genetic Abrogation Delayed Wound Closure of HNSCC-Derived Cell Lines
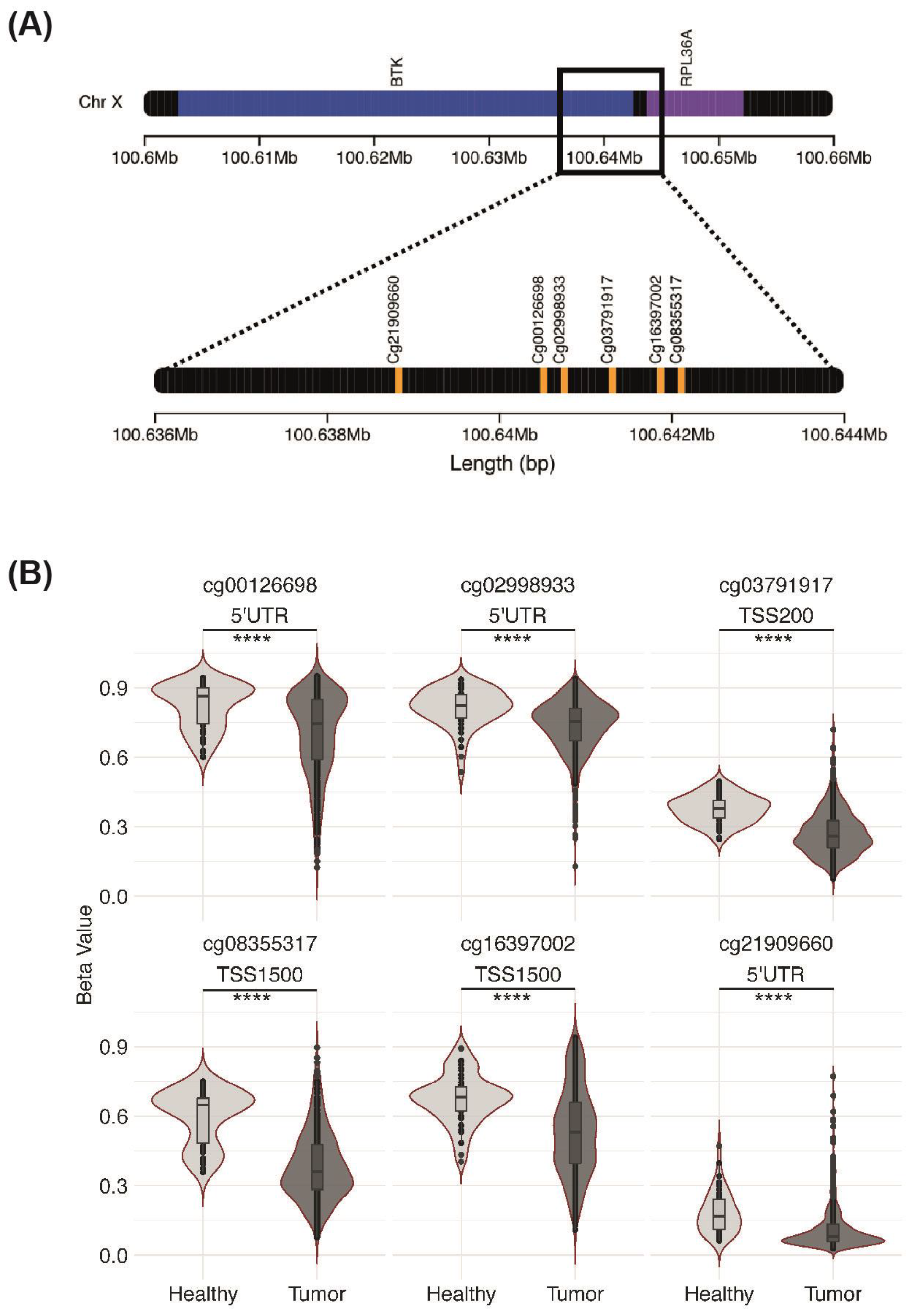
3.6. Association between BTK Expression and Methylation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giamas, G.; Man, Y.L.; Hirner, H.; Bischof, J.; Kramer, K.; Khan, K.; Ahmed, S.S.; Stebbing, J.; Knippschild, U. Kinases as targets in the treatment of solid tumors. Cell. Signal. 2010, 22, 984–1002. [Google Scholar] [CrossRef]
- Tsukada, S.; Saffran, D.C.; Rawlings, D.J.; Parolini, O.; Allen, R.C.; Klisak, I.; Sparkes, R.S.; Kubagawa, H.; Mohandas, T.; Quan, S.; et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993, 72, 279–290. [Google Scholar] [CrossRef]
- Vetrie, D.; Vorechovský, I.; Sideras, P.; Holland, J.; Davies, A.; Flinter, F.; Hammarström, L.; Kinnon, C.; Levinsky, R.; Bobrow, M.; et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 1993, 361, 226–233. [Google Scholar] [CrossRef]
- McDonald, C.; Xanthopoulos, C.; Kostareli, E. The role of Bruton’s tyrosine kinase in the immune system and disease. Immunology 2021, 164, 722–736. [Google Scholar] [CrossRef]
- de Weers, M.; Brouns, G.S.; Hinshelwood, S.; Kinnon, C.; Schuurman, R.K.; Hendriks, R.W.; Borst, J. B-cell antigen receptor stimulation activates the human Bruton’s tyrosine kinase, which is deficient in X-linked agammaglobulinemia. J. Biol. Chem. 1994, 269, 23857–23860. [Google Scholar] [CrossRef]
- Weber, A.N.R.; Bittner, Z.; Liu, X.; Dang, T.-M.; Radsak, M.P.; Brunner, C. Bruton’s Tyrosine Kinase: An Emerging Key Player in Innate Immunity. Front. Immunol. 2017, 8, 1454. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Ibrutinib inhibition of Bruton protein-tyrosine kinase (BTK) in the treatment of B cell neoplasms. Pharmacol. Res. 2016, 113, 395–408. [Google Scholar] [CrossRef]
- Eifert, C.; Wang, X.; Kokabee, L.; Kourtidis, A.; Jain, R.; Gerdes, M.J.; Conklin, D.S. A novel isoform of the B cell tyrosine kinase BTK protects breast cancer cells from apoptosis. Genes Chromosom. Cancer 2013, 52, 961–975. [Google Scholar] [CrossRef] [Green Version]
- Zucha, M.A.; Wu, A.T.; Lee, W.H.; Wang, L.S.; Lin, W.W.; Yuan, C.C.; Yeh, C.T. Bruton’s tyrosine kinase (Btk) inhibitor ibrutinib suppresses stem-like traits in ovarian cancer. Oncotarget 2015, 6, 13255–13268. [Google Scholar] [CrossRef] [Green Version]
- Grassilli, E.; Cerrito, M.G.; Bonomo, S.; Giovannoni, R.; Conconi, D.; Lavitrano, M. p65BTK Is a Novel Biomarker and Therapeutic Target in Solid Tumors. Front. Cell Dev. Biol. 2021, 9, 690365. [Google Scholar] [CrossRef]
- Kokabee, L.; Wang, X.; Sevinsky, C.J.; Wang, W.L.; Cheu, L.; Chittur, S.V.; Karimipoor, M.; Tenniswood, M.; Conklin, D.S. Bruton’s tyrosine kinase is a potential therapeutic target in prostate cancer. Cancer Biol. Ther. 2015, 16, 1604–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Liu, R.; Bhardwaj, G.; Yang, J.C.; Changou, C.; Ma, A.H.; Mazloom, A.; Chintapalli, S.; Xiao, K.; Xiao, W.; et al. Targeting Btk/Etk of prostate cancer cells by a novel dual inhibitor. Cell Death Dis. 2014, 5, e1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassilli, E.; Pisano, F.; Cialdella, A.; Bonomo, S.; Missaglia, C.; Cerrito, M.G.; Masiero, L.; Ianzano, L.; Giordano, F.; Cicirelli, V.; et al. A novel oncogenic BTK isoform is overexpressed in colon cancers and required for RAS-mediated transformation. Oncogene 2016, 35, 4368–4378. [Google Scholar] [CrossRef] [Green Version]
- Basile, D.; Gerratana, L.; Buonadonna, A.; Garattini, S.K.; Perin, T.; Grassilli, E.; Miolo, G.; Cerrito, M.G.; Belluco, C.; Bertola, G.; et al. Role of Bruton’s Tyrosine Kinase in Stage III Colorectal Cancer. Cancers 2019, 11, 880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.D.; Chen, X.Y.; Ji, K.W.; Tao, F. Targeting Btk with ibrutinib inhibit gastric carcinoma cells growth. Am. J. Transl. Res. 2016, 8, 3003–3012. [Google Scholar]
- Wei, L.; Su, Y.K.; Lin, C.M.; Chao, T.Y.; Huang, S.P.; Huynh, T.T.; Jan, H.J.; Whang-Peng, J.; Chiou, J.F.; Wu, A.T.; et al. Preclinical investigation of ibrutinib, a Bruton’s kinase tyrosine (Btk) inhibitor, in suppressing glioma tumorigenesis and stem cell phenotypes. Oncotarget 2016, 7, 69961–69975. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, X.; Hong, Y.; Wang, S.; Chen, P.; Gu, A.; Guo, X.; Zhao, P. Ibrutinib, a Bruton’s tyrosine kinase inhibitor, exhibits antitumoral activity and induces autophagy in glioblastoma. J. Exp. Clin. Cancer Res. 2017, 36, 96. [Google Scholar] [CrossRef] [Green Version]
- Sala, L.; Cirillo, G.; Riva, G.; Romano, G.; Giussani, C.; Cialdella, A.; Todisco, A.; Virtuoso, A.; Cerrito, M.G.; Bentivegna, A.; et al. Specific Expression of a New Bruton Tyrosine Kinase Isoform (p65BTK) in the Glioblastoma Gemistocytic Histotype. Front. Mol. Neurosci. 2019, 12, 2. [Google Scholar] [CrossRef] [Green Version]
- Giordano, F.; Vaira, V.; Cortinovis, D.; Bonomo, S.; Goedmakers, J.; Brena, F.; Cialdella, A.; Ianzano, L.; Forno, I.; Cerrito, M.G.; et al. p65BTK is a novel potential actionable target in KRAS-mutated/EGFR-wild type lung adenocarcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 260. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, A.J.; Yu, L.; Bäckesjö, C.M.; Vargas, L.; Faryal, R.; Aints, A.; Christensson, B.; Berglöf, A.; Vihinen, M.; Nore, B.F.; et al. Bruton’s tyrosine kinase (Btk): Function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 2009, 228, 58–73. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Alsahafi, E.; Begg, K.; Amelio, I.; Raulf, N.; Lucarelli, P.; Sauter, T.; Tavassoli, M. Clinical update on head and neck cancer: Molecular biology and ongoing challenges. Cell Death Dis. 2019, 10, 540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lansford, C.D.; Grenman, R.; Bier, H.; Somers, K.D.; Kim, S.Y.; Whiteside, T.L.; Clayman, G.L.; Welkoborsky, H.-J.; Carey, T.E. Head and Neck Cancers. In Human Cell Culture: Cancer Cell Lines Part 2; Masters, J.R.W., Palsson, B., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 2002; pp. 185–255. [Google Scholar]
- Balló, H.; Koldovsky, P.; Hoffmann, T.; Balz, V.; Hildebrandt, B.; Gerharz, C.D.; Bier, H. Establishment and characterization of four cell lines derived from human head and neck squamous cell carcinomas for an autologous tumor-fibroblast in vitro model. Anticancer Res. 1999, 19, 3827–3836. [Google Scholar]
- Wilkat, M.; Bast, H.; Drees, R.; Dünser, J.; Mahr, A.; Azoitei, N.; Marienfeld, R.; Frank, F.; Brhel, M.; Ushmorov, A.; et al. Adenosine receptor 2B activity promotes autonomous growth, migration as well as vascularization of head and neck squamous cell carcinoma cells. Int. J. Cancer 2020, 147, 202–217. [Google Scholar] [CrossRef] [Green Version]
- Kibe, T.; Kishida, M.; Kamino, M.; Iijima, M.; Chen, L.; Habu, M.; Miyawaki, A.; Hijioka, H.; Nakamura, N.; Kiyono, T.; et al. Immortalization and characterization of normal oral epithelial cells without using HPV and SV40 genes. Oral Sci. Int. 2011, 8, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Moffatt-Jauregui, C.E.; Robinson, B.; de Moya, A.V.; Brockman, R.D.; Roman, A.V.; Cash, M.N.; Culp, D.J.; Lamont, R.J. Establishment and characterization of a telomerase immortalized human gingival epithelial cell line. J. Periodontal Res. 2013, 48, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Azoitei, N.; Diepold, K.; Brunner, C.; Rouhi, A.; Genze, F.; Becher, A.; Kestler, H.; van Lint, J.; Chiosis, G.; Koren, J., 3rd; et al. HSP90 supports tumor growth and angiogenesis through PRKD2 protein stabilization. Cancer Res. 2014, 74, 7125–7136. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wong, J.; Sevinsky, C.J.; Kokabee, L.; Khan, F.; Sun, Y.; Conklin, D.S. Bruton’s Tyrosine Kinase Inhibitors Prevent Therapeutic Escape in Breast Cancer Cells. Mol. Cancer Ther. 2016, 15, 2198–2208. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wahl, M.I.; Eguinoa, A.; Stephens, L.R.; Hawkins, P.T.; Witte, O.N. Phosphatidylinositol 3-kinase-γ activates Bruton’s tyrosine kinase in concert with Src family kinases. Proc. Natl. Acad. Sci. USA 1997, 94, 13820–13825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.-C.; Wu, Y.-C.; Huang, C.-M.; Hsieh, M.-S.; Huang, T.-Y.; Huang, C.-S.; Hsu, T.-N.; Huang, M.-S.; Lee, W.-H.; Yeh, C.-T.; et al. Inhibition of Bruton’s tyrosine kinase as a therapeutic strategy for chemoresistant oral squamous cell carcinoma and potential suppression of cancer stemness. Oncogenesis 2021, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A. Bruton’s Tyrosine Kinase (BTK) Inhibitors in Clinical Trials. Curr. Hematol. Malig. Rep. 2014, 9, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.K.; Tester, R.; Aslanian, S.; Karp, R.; Sheets, M.; Labenski, M.T.; Witowski, S.R.; Lounsbury, H.; Chaturvedi, P.; Mazdiyasni, H.; et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J. Pharmacol. Exp. Ther. 2013, 346, 219–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Kawakami, Y.; Kawakami, T. The pleckstrin homology domain of Bruton tyrosine kinase interacts with protein kinase C. Proc. Natl. Acad. Sci. USA 1994, 91, 9175–9179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Quinto, I.; Chen, X.; Palmieri, C.; Rabin, R.L.; Schwartz, O.M.; Nelson, D.L.; Scala, G. Direct inhibition of Bruton’s tyrosine kinase by IBtk, a Btk-binding protein. Nat. Immunol. 2001, 2, 939–946. [Google Scholar] [CrossRef]
- Yu, L.; Mohamed, A.J.; Vargas, L.; Berglöf, A.; Finn, G.; Lu, K.P.; Smith, C.I. Regulation of Bruton tyrosine kinase by the peptidylprolyl isomerase Pin1. J. Biol. Chem. 2006, 281, 18201–18207. [Google Scholar] [CrossRef] [Green Version]
- Joseph, R.E.; Wales, T.E.; Fulton, D.B.; Engen, J.R.; Andreotti, A.H. Achieving a Graded Immune Response: BTK Adopts a Range of Active/Inactive Conformations Dictated by Multiple Interdomain Contacts. Structure 2017, 25, 1481–1494.e1484. [Google Scholar] [CrossRef] [Green Version]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef]
- Brunner, C.; Müller, B.; Wirth, T. Bruton’s Tyrosine Kinase is involved in innate and adaptive immunity. Histol. Histopathol. 2005, 20, 945–955. [Google Scholar]
- Kim, J.-M.; Park, J.; Noh, E.-M.; Song, H.-K.; Kang, S.Y.; Jung, S.H.; Kim, J.-S.; Youn, H.J.; Lee, Y.-R. Downregulation of matriptase suppresses the PAR-2/PLCγ2/PKC-mediated invasion and migration abilities of MCF-7 breast cancer cells. Oncol. Rep. 2021, 46, 247. [Google Scholar] [CrossRef] [PubMed]
- Kriegs, M.; Clauditz, T.S.; Hoffer, K.; Bartels, J.; Buhs, S.; Gerull, H.; Zech, H.B.; Bußmann, L.; Struve, N.; Rieckmann, T.; et al. Analyzing expression and phosphorylation of the EGF receptor in HNSCC. Sci. Rep. 2019, 9, 13564. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Mateos, J.; Seijas-Tamayo, R.; Mesía, R.; Taberna, M.; Pastor Borgoñón, M.; Pérez-Ruiz, E.; Adansa Klain, J.C.; Vázquez Fernández, S.; del Barco Morillo, E.; Lozano, A.; et al. Epidermal growth factor receptor (EGFR) pathway polymorphisms as predictive markers of cetuximab toxicity in locally advanced head and neck squamous cell carcinoma (HNSCC) in a Spanish population. Oral Oncol. 2016, 63, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Ngan, H.-L.; Liu, Y.; Fong, A.Y.; Poon, P.H.Y.; Yeung, C.K.; Chan, S.S.M.; Lau, A.; Piao, W.; Li, H.; Tse, J.S.W.; et al. MAPK pathway mutations in head and neck cancer affect immune microenvironments and ErbB3 signaling. Life Sci. Alliance 2020, 3, e201900545. [Google Scholar] [CrossRef]
- Wang, L.; Liu, T.; Nishioka, M.; Aguirre, R.L.; Win, S.S.; Okada, N. Activation of ERK1/2 and cyclin D1 expression in oral tongue squamous cell carcinomas: Relationship between clinicopathological appearances and cell proliferation. Oral Oncol. 2006, 42, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Molinolo, A.A.; Amornphimoltham, P.; Squarize, C.H.; Castilho, R.M.; Patel, V.; Gutkind, J.S. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009, 45, 324–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, N.; Utsumi, T.; Takagi, Y. Tissue-specific expression of the human aromatase cytochrome P-450 gene by alternative use of multiple exons 1 and promoters, and switching of tissue-specific exons 1 in carcinogenesis. Proc. Natl. Acad. Sci. USA 1993, 90, 11312–11316. [Google Scholar] [CrossRef] [Green Version]
- Archey, W.B.; Sweet, M.P.; Alig, G.C.; Arrick, B.A. Methylation of CpGs as a Determinant of Transcriptional Activation at Alternative Promoters for Transforming Growth Factor-β3. Cancer Res. 1999, 59, 2292–2296. [Google Scholar]
- Li, T.W.H.; Ting, J.-H.T.; Yokoyama, N.N.; Bernstein, A.; van de Wetering, M.; Waterman, M.L. Wnt activation and alternative promoter repression of LEF1 in colon cancer. Mol. Cell. Biol. 2006, 26, 5284–5299. [Google Scholar] [CrossRef] [Green Version]
- Thorsen, K.; Schepeler, T.; Øster, B.; Rasmussen, M.H.; Vang, S.; Wang, K.; Hansen, K.Q.; Lamy, P.; Pedersen, J.S.; Eller, A.; et al. Tumor-specific usage of alternative transcription start sites in colorectal cancer identified by genome-wide exon array analysis. BMC Genom. 2011, 12, 505. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Betzler, A.C.; Strobel, H.; Abou Kors, T.; Ezić, J.; Lesakova, K.; Pscheid, R.; Azoitei, N.; Sporleder, J.; Staufenberg, A.-R.; Drees, R.; et al. BTK Isoforms p80 and p65 Are Expressed in Head and Neck Squamous Cell Carcinoma (HNSCC) and Involved in Tumor Progression. Cancers 2023, 15, 310. https://doi.org/10.3390/cancers15010310
Betzler AC, Strobel H, Abou Kors T, Ezić J, Lesakova K, Pscheid R, Azoitei N, Sporleder J, Staufenberg A-R, Drees R, et al. BTK Isoforms p80 and p65 Are Expressed in Head and Neck Squamous Cell Carcinoma (HNSCC) and Involved in Tumor Progression. Cancers. 2023; 15(1):310. https://doi.org/10.3390/cancers15010310
Chicago/Turabian StyleBetzler, Annika C., Hannah Strobel, Tsima Abou Kors, Jasmin Ezić, Kristina Lesakova, Ronja Pscheid, Ninel Azoitei, Johanna Sporleder, Anna-Rebekka Staufenberg, Robert Drees, and et al. 2023. "BTK Isoforms p80 and p65 Are Expressed in Head and Neck Squamous Cell Carcinoma (HNSCC) and Involved in Tumor Progression" Cancers 15, no. 1: 310. https://doi.org/10.3390/cancers15010310
APA StyleBetzler, A. C., Strobel, H., Abou Kors, T., Ezić, J., Lesakova, K., Pscheid, R., Azoitei, N., Sporleder, J., Staufenberg, A. -R., Drees, R., Weissinger, S. E., Greve, J., Doescher, J., Theodoraki, M. -N., Schuler, P. J., Laban, S., Kibe, T., Kishida, M., Kishida, S., ... Brunner, C. (2023). BTK Isoforms p80 and p65 Are Expressed in Head and Neck Squamous Cell Carcinoma (HNSCC) and Involved in Tumor Progression. Cancers, 15(1), 310. https://doi.org/10.3390/cancers15010310