
Therapeutic Drug Monitoring of Tyrosine Kinase Inhibitors in the Treatment of Advanced Renal Cancer

 ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. General Considerations for TDM of Anticancer Drugs
3. Pharmacokinetics of TKIs and Their Variabilities
3.1. Pharmacokinetics
3.2. Interindividual Pharmacokinetic Variability
3.3. Intrapatient Pharmacokinetic Variability
4. Pharmacokinetic–Pharmacodynamic Relationships for TKI in Treating Renal Cancer
4.1. Limits of Standard Doses
4.2. Pharmacokinetic–Pharmacodynamic Relationships

{kind=link}
| Number of Patients | PK Marker | PD Efficacy | PD Tolerability | Ref. | |
|---|---|---|---|---|---|
| Sorafenib | 52 | Cmin,ss | Threshold Cmin,ss for onset of grade ≥ 2 hand–foot skin reaction and hypertension: 5.78 and 4.78 μg/mL, respectively | [43] | |
| Sunitinib | 149 | Composite AUCss (sunitinib+SU12662) | Positive relationship between composite AUCss probability of response (partial of complete) or stable disease Association between higher composite AUCss and longer TTP and OS | Negative relationship between cumulated composite AUCss at day 28 and absolute neutrophil count | [12] |
| 55 | AUCss Composite AUCss (sunitinib+SU12662) | Longer median OS in patients with composite AUCss > 1973 ng/mL∙h: 35.2 (CI 95%, 26.5–ND) vs. 16.7 (CI 95%, 4.3–ND) months; p = 0.0051 Trend for longer median PFS in patients with composite AUCss > 1973 ng/mL∙h: 35.2 (CI 95%, 8.0–ND) vs. 8.4 (CI 95%, 3.7–ND) months; p = 0.15 | High sunitinib AUCss: independent risk factor of grade ≥ 3 acute toxicity (OR = 1.16 (CI 95%, 1.05–1.28); p = 0.005) High SU12662 AUCss: independent risk factor of grade ≥ 2 thrombocytopenia (OR = 1.27 (CI 95%, 1.03–1.57); p = 0.028) | [44] | |
| 21 | Composite Cmin,ss (sunitinib+SU12662) | Patients with composite Cmin,ss < 100 ng/mL vs. those with composite Cmin,SS >100 ng/mL: Longer median TTF: 590 vs. 71 days, respectively; p = 0.04 Longer median PFS: 748 vs. 238 days, respectively; p = 0.02 Trend for longer median: 939 vs. 570 days; p = 0.07 | Positive relationship between increased composite Cmin,ss and severity of anorexia (p < 0.05) and fatigue (p < 0.05) Inverse correlation between composite Cmin,ss and platelet counts (p < 0.05) Higher incidence of grade ≥ 3 toxicities in patients with composite Cmin,ss > 100 ng/mL (75.0% vs. 23.1%, respectively) | [30] | |
| 20 | Composite Cmin,ss (sunitinib+SU12662) | Patients with composite Cmin,ss < 50 ng/mL vs. those with Cmin,ss > 50 ng/mL: Longer median TTF: 743 (CI 95%, 217–1583) vs. 56 (CI 95%, 21–179) days; p < 0.001 Longer median PFS: 731 (CI 95%, 197–1576) vs. 95 (CI 95%, 197–1576) days; p < 0.001 | Higher median composite Cmin,ss within 6 weeks in patients with DLT than in those without: 92.7 (range 52.7–196.9) vs. 43.4 (38.3–54.1) ng/mL; p = 0.001 | [45] | |
| 63 | Composite AUCss (sunitinib+SU12662) | At disease progression, trend to a lower composite AUCss than the one during the first cycle: 1678 vs. 2004 ng/mL.h, respectively; p = 0.072 Median PFS not statistically longer in patients with composite AUCss > 2150 ng/mL.h at cycle 1: 14.8 (CI 95%, 2.7–26.9) vs. 11.4 (CI 95%, 5.8–17.0) months; p = 0.45 | High composite AUCss: independent risk factor of grade ≥ 3 acute toxicity (OR = 2.72 (CI 95%, 1.84–4.02); p < 0.0001) → Target composite AUCss < 2150 ng/mL.h to prevent onset of grade ≥ 3 acute toxicity | [46] | |
| Cabozantinib | 319 | Cmin,SS | Average Cmin,ss: 375, 750 and 1125 ng/mL for 20-, 40-, and 60-mg day, respectively. HR for risk of progressive disease or death:
| Association between an increase in average Cmin,ss and increased risk:
| [20] |
| 76 | Cmin,ss AUCss Cmax,ss | Lower median exposure in patients with progressive disease vs. patients with best disease control
| Higher median exposure in patients with DLT than in those without
| [31] | |
| 25 | Cmin,SS | No difference in PFS in patients with Cmin,SS < 573 ng/mL with others: 19.0 (CI 95%, 0–45.7) vs. 34 (CI 95%, 32.6–35.5) weeks, respectively | Trend for higher median Cmin,ss in patients with DLT than in those without: 769 ng/mL (CI 95%, 663–893) vs. 568 ng/mL (CI 95%, 384–842), respectively; p = 0.079 | [39] | |
| 59 | Cmin,ss | No statistical difference in PFS in patients with average Cmin,SS ≥ 750 ng/mL (over the whole treatment period) compared to others: 19 (CI 95%, 0–40) vs. 52 (CI 95%, 34–70) weeks, respectively; p = 0.2 Trend for longer PFS in patients with average Cmin,SS ≤ 572 ng/mL (over the whole treatment period) compared to others: 65 (CI 95%, not reached) vs. 42 (CI 95%, 20–64) weeks, respectively; p = 0.055 No statistical difference in OS in patients with average Cmin,SS ≤ 572 ng/mL (over the whole treatment period) compared to others | Higher median Cmin,ss in patients with DLT than in those without: 831 (CI 95%, 711–1040) vs. 569 ng/mL (CI 95%, 494–754); p = 0.001 Higher DLT incidence in patients with a Cmin,ss ≥ 750 ng/mL at start dose compared to patients with an exposure < 750 ng/mL (78.6% vs. 38.7%; p = 0.003) | [38] | |
| Pazopanib | 177 | Cmin,ss | Longer median PFS in patients with Cmin,ss > 20.5 μg/mL at week 4: 52.0 vs. 19.6 weeks, respectively; p = 0.00378 Five-fold greater median observed tumour shrinkage in patients with Cmin,ss > 20.5 μg/mL at week 4: 37.9% vs. 6.9%. | Increased incidence of grade ≥ 3 toxicity in patients with Cmin,ss in the fourth quartile (36 to 85 μg/mL) | [35] |
| 35 | Cmin,ss | Longer median PFS in patients with Cmin,ss > 20 μg/mL: 34.1 vs. 12.5 weeks, respectively; p = 0.0271; Cmin,SS > 20 μg/mL: independent protective factor for death, HR 0.25 (CI 95%, 0.076–0.81); p = 0.021 | [36] | ||
| 311 | Cmin,ss | Patients achieving early or late Cmin,ss > 20.5 μg/mL had significantly longer disease-free survival:
| Increased incidence of grade ≥ 3 hypertension according to quartile in group early Cmin,ss | [37] | |
| 27 | Cmin,SS | Objective response rate (complete response or partial response) similar in patients with Cmin,SS between 20.5 to 50.3 μg/mL and those with Cmin,SS ≥ 50.3 μg/mL (45.5 vs. 46.2%) No objective response observed in patients with Cmin,ss < 20.5 μg/mL | Positive relationship between increased Cmin,ss and severity (grade 0–1 vs. grade ≥ 2) of anorexia (p < 0.05), fatigue (p < 0.05) and hypertension (p < 0.05) Higher incidence of grade ≥ 3 toxicity in patients with a Cmin,ss ≥ 50.3 μg/mL (61.5% vs. 7.1%) → Target Cmin,ss< 50.3 μg/mL to prevent onset of grade ≥ 3 toxicities | [32] | |
| Axitinib | 168 | AUCss | Longer median PFS in patients with AUCss ≥ 300 ng/mL∙h: 13.8 vs. 7.4 months; HR 0.558 (95% CI, 0.379–0.823); p = 0.003 Longer median OS in patients with AUCss ≥ 300 ng/mL∙h: 37.4 vs. 15.8 months; HR 0.489 (95% CI, 0.324–0.738); p < 0.001 | Weak correlation between AUCss and blood diastolic pressure (r2 < 0.10) | [41] |
| 33 | Cmin,ss AUCss | Longer OS in patients with Cmin,ss ≥ 5 ng/mL: median not reached vs. 299 days, respectively; p = 0.022) Longer OS in patients with AUCss ≥ 300 ng/mL∙h: median not reached vs. 409 days, respectively; p = 0.045) No relationship between PFS and AUCss or Cmin,ss | Threshold value Cmin,ss: 6.6 and 7.1 ng/mL to predict grade ≥ 2 hypothyroidism (p = 0.005) and grade ≥ 2 anorexia (p = 0.035), respectively | [42] | |
| 20 | Cmax,ss | Higher Cmax,ss higher in responders (complete or partial response) than in non-responders (stable or progression disease); p = 0.013 Longer median PFS in patients with Cmax,ss > 12.4 ng/mL: 799 (95% CI, 140–not estimable) vs. 336 days (95% CI, 70–not estimable) days; p = 0.047 | Higher cumulative incidence of DLT in patients with Cmax,ss ≥ 40.2 ng/mL: sub-hazard ratio, 4.13 (95% CI, 1.27–13.5); p = 0.019 | [33] | |
| 35 | Cmin,ss,first (2 weeks after treatment start) Cmin,ss,1-3m (mean Cmin,ss between 1 and 3 months after treatment start) | Statistical association between best response and plasma exposure (Cmin,ss,first and Cmin,ss,1-3m) Higher plasma exposure in patients with PFS ≥ 5 months:
| Higher plasma exposure in patients with grade ≥ 3 toxicity:
| [34] | |
| Tivozanib | 432 | Caverage over a 4 weeks treatment period | Logistic regression between Caverage and probability for hand–foot syndrome: OR = 1.2 [95%CI: 1.00–1.03] | [47] | |
| Lenvatinib | 260 | Cmin,ss | Plasma exposure based on starting dose was a significant predictor for the occurrence of any grade proteinuria, nausea, and vomiting, and for Grade 3 or higher hypertension | [19] |
5. Benefit of TDM in Clinical Practice
5.1. TDM as a Predictive Factor for Clinical Outcome
5.2. Patients with Poor Adherence
5.3. Management of Drug–Drug Interactions
6. Clinical Practices and Practical Issues
6.1. AUCτ,ss vs. Cmin,ss as the Best Marker of Systemic Exposure
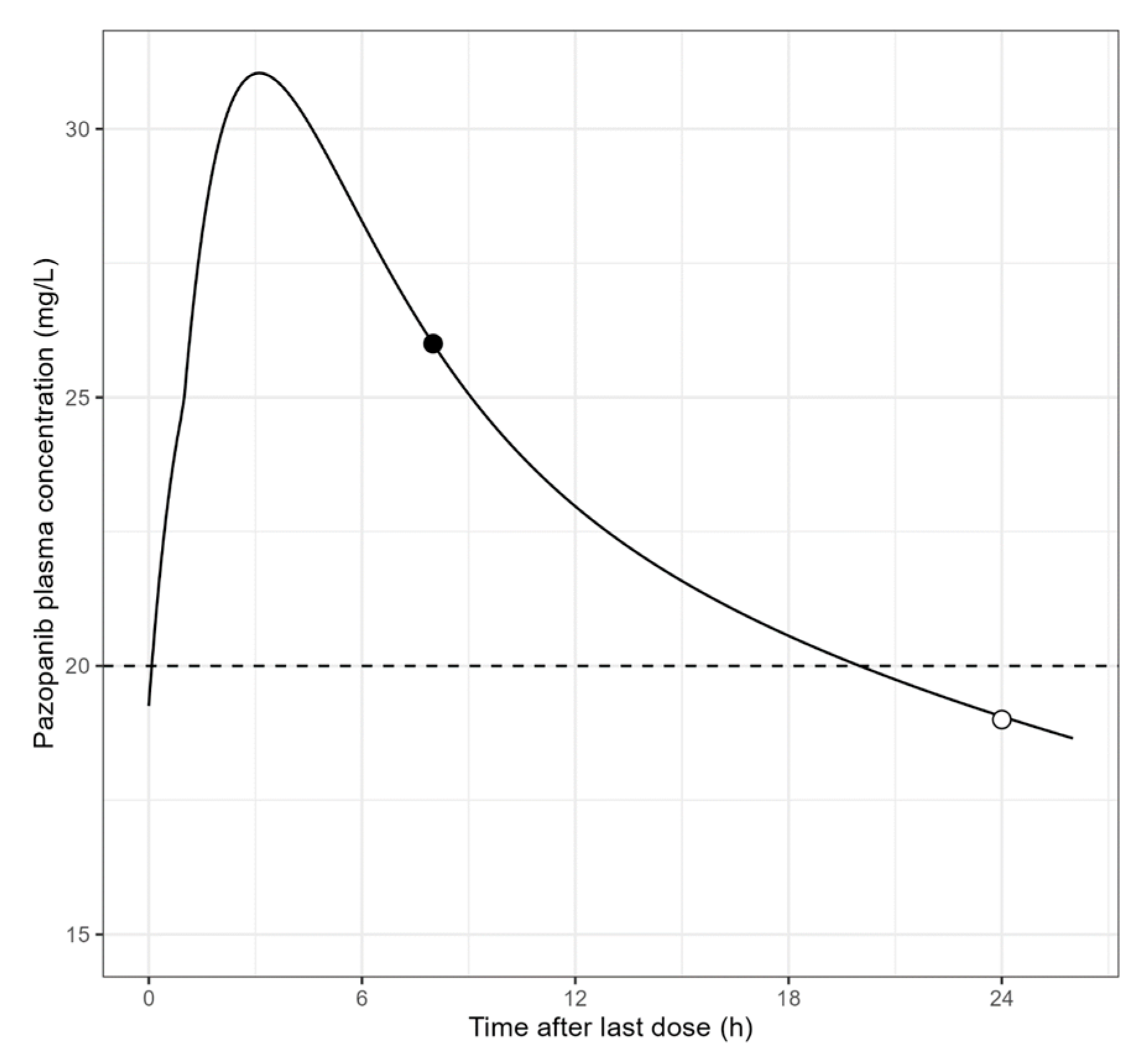
6.2. Estimation of Cmin,ss from a Blood Sample Collected during the Dosing Interval
6.3. Pre-Analytical Aspects and Assay
6.4. Interpretation of TDM Results
6.5. Combinations of Tyrosine Kinase Inhibitors with Immunotherapy
6.6. Plasma Protein Binding
6.7. Intrapatient Pharmacokinetic Variability
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Michaelis, J.; Grabbert, M.; Sigle, A.; Yilmaz, M.; Schlager, D.; Gratzke, C.; Miernik, A.; Schoeb, D.S. Tyrosine Kinase Inhibitors in the Treatment of Metastasised Renal Cell Carcinoma-Future or the Past? Cancers 2022, 14, 3777. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juárez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 384, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.; Alekseev, B.; Rha, S.-Y.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Kopyltsov, E.; Méndez-Vidal, M.J.; et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Apolo, A.B.; Powles, T.; Escudier, B.; Burotto, M.; Zhang, J.; Simsek, B.; Scheffold, C.; Motzer, R.J.; Choueiri, T.K. Nivolumab plus ipilimumab plus cabozantinib triplet combination for patients with previously untreated advanced renal cell carcinoma: Results from a discontinued arm of the phase III CheckMate 9ER trial. Eur. J. Cancer 2022, 177, 63–71. [Google Scholar] [CrossRef]
- Clarke, W.A.; Chatelut, E.; Fotoohi, A.K.; Larson, R.A.; Martin, J.H.; Mathijssen, R.H.J.; Salamone, S.J. Therapeutic drug monitoring in oncology: International Association of Therapeutic Drug Monitoring and Clinical Toxicology consensus guidelines for imatinib therapy. Eur. J. Cancer 2021, 157, 428–440. [Google Scholar] [CrossRef]
- Widmer, N.; Bardin, C.; Chatelut, E.; Paci, A.; Beijnen, J.; Levêque, D.; Veal, G.; Astier, A. Review of therapeutic drug monitoring of anticancer drugs part two--targeted therapies. Eur. J. Cancer 2014, 50, 2020–2036. [Google Scholar] [CrossRef]
- Chatelut, E.; Puisset, F. The scientific basis of body surface area-based dosing. Clin. Pharm. Ther. 2014, 95, 359–361. [Google Scholar] [CrossRef]
- Chatelut, E.; Hendrikx, J.J.M.A.; Martin, J.; Ciccolini, J.; Moes, D.J.A.R. Unraveling the complexity of therapeutic drug monitoring for monoclonal antibody therapies to individualize dose in oncology. Pharm. Res. Perspect. 2021, 9, e00757. [Google Scholar] [CrossRef]
- Fogli, S.; Porta, C.; Del Re, M.; Crucitta, S.; Gianfilippo, G.; Danesi, R.; Rini, B.I.; Schmidinger, M. Optimizing treatment of renal cell carcinoma with VEGFR-TKIs: A comparison of clinical pharmacology and drug-drug interactions of anti-angiogenic drugs. Cancer Treat Rev. 2020, 84, 101966. [Google Scholar] [CrossRef]
- Houk, B.E.; Bello, C.L.; Poland, B.; Rosen, L.S.; Demetri, G.D.; Motzer, R.J. Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: Results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother. Pharm. 2010, 66, 357–371. [Google Scholar] [CrossRef]
- Jain, L.; Woo, S.; Gardner, E.R.; Dahut, W.L.; Kohn, E.C.; Kummar, S.; Mould, D.R.; Giaccone, G.; Yarchoan, R.; Venitz, J.; et al. Population pharmacokinetic analysis of sorafenib in patients with solid tumours. Br. J. Clin. Pharm. 2011, 72, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Faivre, S.; Delbaldo, C.; Vera, K.; Robert, C.; Lozahic, S.; Lassau, N.; Bello, C.; Deprimo, S.; Brega, N.; Massimini, G.; et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J. Clin. Oncol. 2006, 24, 25–35. [Google Scholar] [CrossRef]
- Goodman, V.L.; Rock, E.P.; Dagher, R.; Ramchandani, R.P.; Abraham, S.; Gobburu, J.V.S.; Booth, B.P.; Verbois, S.L.; Morse, D.E.; Liang, C.Y.; et al. Approval summary: Sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin. Cancer Res. 2007, 13, 1367–1373. [Google Scholar]
- Ozbey, A.C.; Combarel, D.; Poinsignon, V.; Lovera, C.; Saada, E.; Mir, O.; Paci, A. Population Pharmacokinetic Analysis of Pazopanib in Patients and Determination of Target AUC. Pharmaceuticals 2021, 14, 927. [Google Scholar] [CrossRef]
- Hurwitz, H.I.; Dowlati, A.; Saini, S.; Savage, S.; Suttle, A.B.; Gibson, D.M.; Hodge, J.P.; Merkle, E.M.; Pandite, L. Phase I trial of pazopanib in patients with advanced cancer. Clin. Cancer Res. 2009, 15, 4220–4227. [Google Scholar] [CrossRef] [Green Version]
- FDA. C for DE and R: Axitinib Clinical Pharmacology and Biopharmaceutic Review [Internet]. 2012. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202324orig1s000clinpharmr.pdf (accessed on 2 November 2022).
- EMA. C for MP for HU (CHMP): Lenvima-EPAR Public Assessment Report [Internet]. 2015. Available online: https://www.ema.europa.eu/en/documents/assessment-report/lenvima-epar-public-assessment-report_en.pdf (accessed on 2 November 2022).
- Lacy, S.; Nielsen, J.; Yang, B.; Miles, D.; Nguyen, L.; Hutmacher, M. Population exposure-response analysis of cabozantinib efficacy and safety endpoints in patients with renal cell carcinoma. Cancer Chemother. Pharm. 2018, 81, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Brave, M.; Beaver, J.A.; Cheng, J.; Tang, S.; Zahalka, E.; Palmby, T.R.; Venugopal, R.; Song, P.; Liu, Q.; et al. U.S. Food and Drug Administration Approval: Cabozantinib for the Treatment of Advanced Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 330–335. [Google Scholar] [CrossRef] [Green Version]
- EMA. C for MP for HU (CHMP): Fotvida-EPAR Public Assessment Report [Internet]. 2017. Available online: https://www.ema.europa.eu/en/documents/assessment-report/fotivda-epar-public-assessment-report_en.pdf (accessed on 2 November 2022).
- Ornstein, M.C.; Rini, B.I. Pharmacokinetically Guided Dosing of Oral Drugs: True Precision Oncology? Clin. Cancer Res. 2016, 22, 5626–5628. [Google Scholar] [CrossRef] [Green Version]
- Imbs, D.-C.; Négrier, S.; Cassier, P.; Hollebecque, A.; Varga, A.; Blanc, E.; Lafont, T.; Escudier, B.; Soria, J.-C.; Pérol, D.; et al. Pharmacokinetics of pazopanib administered in combination with bevacizumab. Cancer Chemother. Pharm. 2014, 73, 1189–1196. [Google Scholar]
- FDA. Cabometyx Summary of Product Characteristics [Internet]. 2015. Available online: https://www.ema.europa.eu/en/documents/product-information/cabometyx-epar-product-information_en.pdf (accessed on 2 November 2022).
- Cohen, R.B.; Oudard, S. Antiangiogenic therapy for advanced renal cell carcinoma: Management of treatment-related toxicities. Investig. New Drugs 2012, 30, 2066–2079. [Google Scholar] [CrossRef] [PubMed]
- Postel-Vinay, S.; Collette, L.; Paoletti, X.; Rizzo, E.; Massard, C.; Olmos, D.; Fowst, C.; Levy, B.; Mancini, P.; Lacombe, D.; et al. Towards new methods for the determination of dose limiting toxicities and the assessment of the recommended dose for further studies of molecularly targeted agents--dose-Limiting Toxicity and Toxicity Assessment Recommendation Group for Early Trials of Targeted therapies, an European Organisation for Research and Treatment of Cancer-led study. Eur. J. Cancer 2014, 50, 2040–2049. [Google Scholar] [PubMed]
- Roda, D.; Jimenez, B.; Banerji, U. Are Doses and Schedules of Small-Molecule Targeted Anticancer Drugs Recommended by Phase I Studies Realistic? Clin. Cancer Res. 2016, 22, 2127–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groenland, S.L.; van Eerden, R.A.G.; Westerdijk, K.; Meertens, M.; Koolen, S.L.W.; Moes, D.J.A.R.; de Vries, N.; Rosing, H.; Otten, H.; Vulink, A.J.E.; et al. Therapeutic drug monitoring-based precision dosing of oral targeted therapies in oncology: A prospective multicenter study. Ann. Oncol. 2022, 33, 1071–1082. [Google Scholar] [CrossRef]
- Noda, S.; Otsuji, T.; Baba, M.; Yoshida, T.; Kageyama, S.; Okamoto, K.; Okada, Y.; Kawauchi, A.; Onishi, H.; Hira, D.; et al. Assessment of Sunitinib-Induced Toxicities and Clinical Outcomes Based on Therapeutic Drug Monitoring of Sunitinib for Patients with Renal Cell Carcinoma. Clin. Genitourin. Cancer 2015, 13, 350–358. [Google Scholar] [CrossRef]
- Cerbone, L.; Combarel, D.; Geraud, A.; Auclin, E.; Foulon, S.; Alves Costa Silva, C.; Colomba, E.; Carril, L.; Derosa, L.; Flippot, R.; et al. Association of cabozantinib pharmacokinetics, progression and toxicity in metastatic renal cell carcinoma patients: Results from a pharmacokinetics/pharmacodynamics study. ESMO Open 2021, 6, 100312. [Google Scholar] [CrossRef]
- Noda, S.; Yoshida, T.; Hira, D.; Murai, R.; Tomita, K.; Tsuru, T.; Kageyama, S.; Kawauchi, A.; Ikeda, Y.; Morita, S.-Y.; et al. Exploratory Investigation of Target Pazopanib Concentration Range for Patients with Renal Cell Carcinoma. Clin. Genitourin. Cancer 2019, 17, e306–e313. [Google Scholar] [CrossRef]
- Fukudo, M.; Tamaki, G.; Azumi, M.; Kakizaki, H.; Matsumoto, S.; Tasaki, Y. Absorption of the orally active multikinase inhibitor axitinib as a therapeutic index to guide dose titration in metastatic renal cell carcinoma. Investig. New Drugs. 2021, 39, 595–604. [Google Scholar] [CrossRef]
- Synowiec, Z.; Sobańska, K.; Synowiec, T.; Teżyk, A.; Tomczak, P.; Jabłecka, A. Axitinib Trough Concentration and its Influence on the Efficacy and Toxicity of Second-line Renal Cell Carcinoma Treatment. Clin. Genitourin. Cancer 2022, 20, 390.e1–390.e8. [Google Scholar] [CrossRef]
- Suttle, A.B.; Ball, H.A.; Molimard, M.; Hutson, T.E.; Carpenter, C.; Rajagopalan, D.; Lin, Y.; Swann, S.; Amado, R.; Pandite, L. Relationships between pazopanib exposure and clinical safety and efficacy in patients with advanced renal cell carcinoma. Br. J. Cancer 2014, 111, 1909–1916. [Google Scholar] [CrossRef] [Green Version]
- Verheijen, R.B.; Swart, L.E.; Beijnen, J.H.; Schellens, J.H.M.; Huitema, A.D.R.; Steeghs, N. Exposure-survival analyses of pazopanib in renal cell carcinoma and soft tissue sarcoma patients: Opportunities for dose optimization. Cancer Chemother. Pharm. 2017, 80, 1171–1178. [Google Scholar]
- Sternberg, C.N.; Donskov, F.; Haas, N.B.; Doehn, C.; Russo, P.; Elmeliegy, M.; Baneyx, G.; Banerjee, H.; Aimone, P.; Motzer, R.J. Pazopanib Exposure Relationship with Clinical Efficacy and Safety in the Adjuvant Treatment of Advanced Renal Cell Carcinoma. Clin. Cancer Res. 2018, 24, 3005–3013. [Google Scholar] [CrossRef]
- Krens, S.D.; van Erp, N.P.; Groenland, S.L.; Moes, D.J.A.R.; Mulder, S.F.; Desar, I.M.E.; van der Hulle, T.; Steeghs, N.; van Herpen, C.M.L. Exposure-response analyses of cabozantinib in patients with metastatic renal cell cancer. BMC Cancer 2022, 22, 228. [Google Scholar]
- Krens, S.D.; van Boxtel, W.; Uijen, M.J.M.; Jansman, F.G.A.; Desar, I.M.E.; Mulder, S.F.; van Herpen, C.M.L.; van Erp, N.P. Exposure-toxicity relationship of cabozantinib in patients with renal cell cancer and salivary gland cancer. Int. J. Cancer 2022, 150, 308–316. [Google Scholar] [CrossRef]
- Mendel, D.B.; Laird, A.D.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar]
- Rini, B.I.; Garrett, M.; Poland, B.; Dutcher, J.P.; Rixe, O.; Wilding, G.; Stadler, W.M.; Pithavala, Y.K.; Kim, S.; Tarazi, J.; et al. Axitinib in metastatic renal cell carcinoma: Results of a pharmacokinetic and pharmacodynamic analysis. J. Clin. Pharm. 2013, 53, 491–504. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, R.; Inoue, T.; Fujiyama, N.; Tsuchiya, N.; Numakura, K.; Kagaya, H.; Saito, M.; Narita, S.; Satoh, S.; Niioka, T.; et al. Contribution of UGT1A1 genetic polymorphisms related to axitinib pharmacokinetics to safety and efficacy in patients with renal cell carcinoma. Med. Oncol. 2018, 35, 51. [Google Scholar] [CrossRef]
- Fukudo, M.; Ito, T.; Mizuno, T.; Shinsako, K.; Hatano, E.; Uemoto, S.; Kamba, T.; Yamasaki, T.; Ogawa, O.; Seno, H.; et al. Exposure-toxicity relationship of sorafenib in Japanese patients with renal cell carcinoma and hepatocellular carcinoma. Clin Pharm. 2014, 53, 185–196. [Google Scholar] [CrossRef]
- Narjoz, C.; Cessot, A.; Thomas-Schoemann, A.; Golmard, J.L.; Huillard, O.; Boudou-Rouquette, P.; Behouche, A.; Taieb, F.; Durand, J.P.; Dauphin, A.; et al. Role of the lean body mass and of pharmacogenetic variants on the pharmacokinetics and pharmacodynamics of sunitinib in cancer patients. Investig. New Drugs 2015, 33, 257–268. [Google Scholar] [CrossRef]
- Takasaki, S.; Kawasaki, Y.; Kikuchi, M.; Tanaka, M.; Suzuka, M.; Noda, A.; Sato, Y.; Yamashita, S.; Mitsuzuka, K.; Saito, H.; et al. Relationships between sunitinib plasma concentration and clinical outcomes in Japanese patients with metastatic renal cell carcinoma. Int. J. Clin. Oncol. 2018, 23, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Cabel, L.; Blanchet, B.; Thomas-Schoemann, A.; Huillard, O.; Bellesoeur, A.; Cessot, A.; Giroux, J.; Boudou-Rouquette, P.; Coriat, R.; Vidal, M.; et al. Drug monitoring of sunitinib in patients with advanced solid tumors: A monocentric observational French study. Fundam. Clin. Pharm. 2018, 32, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA. C for DE and R: Tivozanib Clinical and Pharmacology Review [Internet]. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/212904Orig1s000MultidisciplineR.pdf (accessed on 2 November 2022).
- Verheijen, R.B.; Bins, S.; Mathijssen, R.H.J.; Lolkema, M.P.; van Doorn, L.; Schellens, J.H.M.; Beijnen, J.H.; Langenberg, M.H.G.; Huitema, A.D.R.; Steeghs, N.; et al. Individualized Pazopanib Dosing: A Prospective Feasibility Study in Cancer Patients. Clin. Cancer Res. 2016, 22, 5738–5746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, F.; Chauvin, J.; DeVictor, B.; Lacarelle, B.; Deville, J.-L.; Ciccolini, J. Clinical-Based vs. Model-Based Adaptive Dosing Strategy: Retrospective Comparison in Real-World mRCC Patients Treated with Sunitinib. Pharmaceuticals 2021, 14, 494. [Google Scholar] [CrossRef] [PubMed]
- Lankheet, N.A.G.; Kloth, J.S.L.; Gadellaa-van Hooijdonk, C.G.M.; Cirkel, G.A.; Mathijssen, R.H.J.; Lolkema, M.P.J.K.; Schellens, J.H.M.; Voest, E.E.; Sleijfer, S.; De Jonge, M.J.A.; et al. Pharmacokinetically guided sunitinib dosing: A feasibility study in patients with advanced solid tumours. Br. J. Cancer 2014, 110, 2441–2449. [Google Scholar] [CrossRef]
- Sabanathan, D.; Zhang, A.; Fox, P.; Coulter, S.; Gebski, V.; Balakrishnar, B.; Chan, M.; Liddle, C.; Gurney, H. Dose individualization of sunitinib in metastatic renal cell cancer: Toxicity-adjusted dose or therapeutic drug monitoring. Cancer Chemother. Pharm. 2017, 80, 385–393. [Google Scholar] [CrossRef]
- Yu, H.; van Erp, N.; Bins, S.; Mathijssen, R.H.J.; Schellens, J.H.M.; Beijnen, J.H.; Steeghs, N.; Huitema, A.D.R. Development of a Pharmacokinetic Model to Describe the Complex Pharmacokinetics of Pazopanib in Cancer Patients. Clin. Pharm. 2017, 56, 293–303. [Google Scholar] [CrossRef]
- Groenland, S.L.; van Eerden, R.A.G.; Verheijen, R.B.; de Vries, N.; Thijssen, B.; Rosing, H.; Beijnen, J.H.; Koolen, S.L.W.; Mathijssen, R.H.J.; Huitema, A.D.R.; et al. Cost-Neutral Optimization of Pazopanib Exposure by Splitting Intake Moments: A Prospective Pharmacokinetic Study in Cancer Patients. Clin. Pharm. 2020, 59, 941–948. [Google Scholar] [CrossRef]
- Hornecker, M.; Blanchet, B.; Billemont, B.; Sassi, H.; Ropert, S.; Taieb, F.; Mir, O.; Abbas, H.; Harcouet, L.; Coriat, R.; et al. Saturable absorption of sorafenib in patients with solid tumors: A population model. Investig. New Drugs 2012, 30, 1991–2000. [Google Scholar] [CrossRef]
- Gotta, V.; Widmer, N.; Decosterd, L.A.; Chalandon, Y.; Heim, D.; Gregor, M.; Benz, R.; Leoncini-Franscini, L.; Baerlocher, G.M.; Duchosal, M.A.; et al. Clinical usefulness of therapeutic concentration monitoring for imatinib dosage individualization: Results from a randomized controlled trial. Cancer Chemother. Pharm. 2014, 74, 1307–1319. [Google Scholar] [CrossRef]
- Marin, D.; Bazeos, A.; Mahon, F.-X.; Eliasson, L.; Milojkovic, D.; Bua, M.; Apperley, J.F.; Szydlo, R.; Desai, R.; Kozlowski, K.; et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J. Clin. Oncol. 2010, 28, 2381–2388. [Google Scholar] [CrossRef]
- Karvaly, G.B.; Karádi, I.; Vincze, I.; Neely, M.N.; Trojnár, E.; Prohászka, Z.; Imreh, É.; Vásárhelyi, B.; Zsáry, A. A pharmacokinetics-based approach to the monitoring of patient adherence to atorvastatin therapy. Pharm. Res. Perspect. 2021, 9, e00856. [Google Scholar] [CrossRef]
- Azam, C.; Claraz, P.; Chevreau, C.; Vinson, C.; Cottura, E.; Mourey, L.; Pouessel, D.; Guibaud, S.; Pollet, O.; Le Goff, M.; et al. Association between clinically relevant toxicities of pazopanib and sunitinib and the use of weak CYP3A4 and P-gp inhibitors. Eur. J. Clin. Pharm. 2020, 76, 579–587. [Google Scholar] [CrossRef]
- Agarwal, M.; Thareja, N.; Benjamin, M.; Akhondi, A.; Mitchell, G.D. Tyrosine Kinase Inhibitor-Induced Hypertension. Curr. Oncol. Rep. 2018, 20, 65. [Google Scholar] [CrossRef]
- Le Louedec, F.; Puisset, F.; Thomas, F.; Chatelut, É.; White-Koning, M. Easy and reliable maximum a posteriori Bayesian estimation of pharmacokinetic parameters with the open-source R package mapbayr. CPT Pharmacomet. Syst. Pharm. 2021, 10, 1208–1220. [Google Scholar] [CrossRef]
- Yu, H.; Steeghs, N.; Nijenhuis, C.M.; Schellens, J.H.M.; Beijnen, J.H.; Huitema, A.D.R. Practical guidelines for therapeutic drug monitoring of anticancer tyrosine kinase inhibitors: Focus on the pharmacokinetic targets. Clin. Pharm. 2014, 53, 305–325. [Google Scholar] [CrossRef]
- Simon, F.; Gautier-Veyret, E.; Truffot, A.; Chenel, M.; Payen, L.; Stanke-Labesque, F.; Tod, M. Modeling Approach to Predict the Impact of Inflammation on the Pharmacokinetics of CYP2C19 and CYP3A4 Substrates. Pharm. Res. 2021, 38, 415–428. [Google Scholar] [CrossRef]
- Mir, O.; Coriat, R.; Blanchet, B.; Durand, J.-P.; Boudou-Rouquette, P.; Michels, J.; Ropert, S.; Vidal, M.; Pol, S.; Chaussade, S.; et al. Sarcopenia predicts early dose-limiting toxicities and pharmacokinetics of sorafenib in patients with hepatocellular carcinoma. PLoS ONE 2012, 7, e37563. [Google Scholar] [CrossRef]
- Veerman, G.D.M.; Hussaarts, K.G.A.M.; Jansman, F.G.A.; Koolen, S.W.L.; van Leeuwen, R.W.F.; Mathijssen, R.H.J. Clinical implications of food-drug interactions with small-molecule kinase inhibitors. Lancet Oncol. 2020, 21, e265–e279. [Google Scholar] [CrossRef]
- Bins, S.; Huitema, A.D.R.; Laven, P.; Bouazzaoui, S.E.; Yu, H.; van Erp, N.; van Herpen, C.; Hamberg, P.; Gelderblom, H.; Steeghs, N.; et al. Impact of CYP3A4*22 on Pazopanib Pharmacokinetics in Cancer Patients. Clin. Pharm. 2019, 58, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Mir, O.; Touati, N.; Lia, M.; Litière, S.; Le Cesne, A.; Sleijfer, S.; Blay, J.-Y.; Leahy, M.; Young, R.; Mathijssen, R.H.J.; et al. Impact of Concomitant Administration of Gastric Acid-Suppressive Agents and Pazopanib on Outcomes in Soft-Tissue Sarcoma Patients Treated within the EORTC 62043/62072 Trials. Clin. Cancer Res. 2019, 25, 1479–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peer, C.J.; Sissung, T.M.; Kim, A.; Jain, L.; Woo, S.; Gardner, E.R.; Kirkland, C.T.; Troutman, S.M.; English, B.C.; Richardson, E.D.; et al. Sorafenib is an inhibitor of UGT1A1 but is metabolized by UGT1A9, implications of genetic variants on pharmacokinetics and hyperbilirubinemia. Clin. Cancer Res. 2012, 18, 2099–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudou-Rouquette, P.; Narjoz, C.; Golmard, J.L.; Thomas-Schoemann, A.; Mir, O.; Taieb, F.; Durand, J.-P.; Coriat, R.; Dauphin, A.; Vidal, M.; et al. Early sorafenib-induced toxicity is associated with drug exposure and UGTIA9 genetic polymorphism in patients with solid tumors: A preliminary study. PLoS ONE 2012, 7, e42875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, T.; Fukudo, M.; Terada, T.; Kamba, T.; Nakamura, E.; Ogawa, O.; Inui, K.-I.; Katsura, T. Impact of genetic variation in breast cancer resistance protein (BCRP/ABCG2) on sunitinib pharmacokinetics. Drug Metab. Pharm. 2012, 27, 631–639. [Google Scholar] [CrossRef]
- Powles, T.; Plimack, E.R.; Soulières, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef]
- Apolo, A.B.; Nadal, R.; Girardi, D.M.; Niglio, S.A.; Ley, L.; Cordes, L.M.; Steinberg, S.M.; Sierra Ortiz, O.; Cadena, J.; Diaz, C.; et al. Phase I Study of Cabozantinib and Nivolumab Alone or with Ipilimumab for Advanced or Metastatic Urothelial Carcinoma and Other Genitourinary Tumors. J. Clin. Oncol. 2020, 38, 3672–3684. [Google Scholar] [CrossRef]
- Arrondeau, J.; Mir, O.; Boudou-Rouquette, P.; Coriat, R.; Ropert, S.; Dumas, G.; Rodrigues, M.J.; Rousseau, B.; Blanchet, B.; Goldwasser, F. Sorafenib exposure decreases over time in patients with hepatocellular carcinoma. Investig. New Drugs 2012, 30, 2046–2049. [Google Scholar] [CrossRef]

| CL/F (L/h) IIV (%) | F (%) | Tmax (h) | Unbound Plasma Fraction (%) | Metabolic Pathways | Drug Transporters | Elimination Half-Life (h) | Ref | |
|---|---|---|---|---|---|---|---|---|
| Sorafenib | 8.13 (18) | ND | 3 | <1 | CYP3A4, UGT1A9 | MRP2 | 25–48 | [13] |
| Sunitinib | 46.4 (46) | ND | 8.5 | 5 | CYP3A4 | BCRP | 40–60 | [14,15] |
| Pazopanib | 0.458 (71) | 21.5 | 3 | <0.1 | CYP3A4 | P-gp; BCRP | 27–36 | [16,17] |
| Axitinib | 32.3 (57) | 58 | 3.9–6.0 | 1 | CYP3A4, CYP1A2, CYP2C19, UGT1A1 | P-gp; BCRP | 2.5–6.1 (43) | [18] |
| Lenvatinib | 6.5 (25.5) | ND | 1–4 | 1–2 | CYP3A4 | P-gp; BCRP | 20.6–34.3 | [19] |
| Cabozantinib | 2.2 (46) | ND | 2–3 | <1 | CYP3A4, CYP2C9 | MRP2, OAT3 | 120 | [20,21] |
| Tivozanib | 0.862 (men); 0.651 (women) (40.5) | ND | 3 | <1 | CYP3A4, UGT? | - | 108–121 | [22] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puisset, F.; Mseddi, M.; Mourey, L.; Pouessel, D.; Blanchet, B.; Chatelut, E.; Chevreau, C. Therapeutic Drug Monitoring of Tyrosine Kinase Inhibitors in the Treatment of Advanced Renal Cancer. Cancers 2023, 15, 313. https://doi.org/10.3390/cancers15010313
Puisset F, Mseddi M, Mourey L, Pouessel D, Blanchet B, Chatelut E, Chevreau C. Therapeutic Drug Monitoring of Tyrosine Kinase Inhibitors in the Treatment of Advanced Renal Cancer. Cancers. 2023; 15(1):313. https://doi.org/10.3390/cancers15010313
Chicago/Turabian StylePuisset, Florent, Mourad Mseddi, Loïc Mourey, Damien Pouessel, Benoit Blanchet, Etienne Chatelut, and Christine Chevreau. 2023. "Therapeutic Drug Monitoring of Tyrosine Kinase Inhibitors in the Treatment of Advanced Renal Cancer" Cancers 15, no. 1: 313. https://doi.org/10.3390/cancers15010313
APA StylePuisset, F., Mseddi, M., Mourey, L., Pouessel, D., Blanchet, B., Chatelut, E., & Chevreau, C. (2023). Therapeutic Drug Monitoring of Tyrosine Kinase Inhibitors in the Treatment of Advanced Renal Cancer. Cancers, 15(1), 313. https://doi.org/10.3390/cancers15010313