
Tackling of Immunorefractory Tumors by Targeting Alternative Immune Checkpoints

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
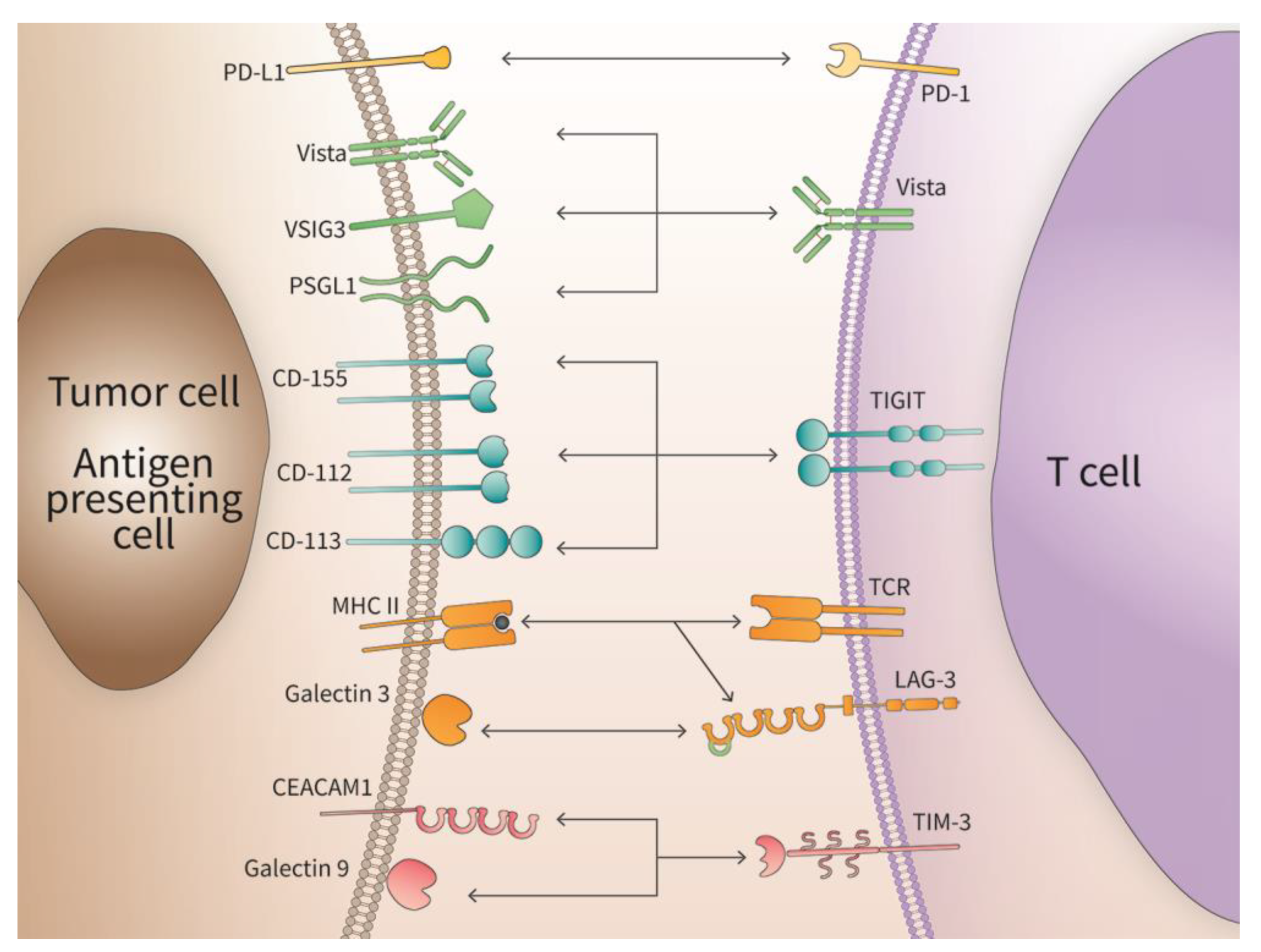
2. VISTA
3. TIGIT
4. LAG 3
5. TIM-3
6. PD-1
7. Overcoming the Resistance to Immunotherapy
7.1. Expression of Alternate ICs
7.2. Impaired Antigen-Stimulated T Lymphocyte Response
7.3. Disrupted IFN-γ Signaling in Tumor Cells
7.4. T Lymphocyte Exhaustion
7.5. Dysregulation of Oncogenic Signaling Pathways
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM | A disintegrin and metalloprotease |
| APC | Antigen presenting cells |
| BAT3 | B-associated transcript |
| BC | Breast cancer |
| BLIMP1 | B-lymphocyte induced maturation protein |
| BTLA | B and T lymphocyte attenuator |
| CDK | Cell division kinase |
| CEACAM1 | carcinoembryonic antigen cell adhesion molecule 1 |
| CTLA-4 | Cytotoxic T lymphocyte antigen 4 |
| DC | Dendritic cells |
| DNAM1 | DNAX accessory molecule 1 |
| DNMT | DNA methyltransferase |
| ECM | Extracellular matrix |
| ERK | Extracellular signal regulated kinase |
| FDA | Food and drug administration |
| FGL1 | Fibrinogen-like protein 1 |
| FOX | Forkhead box protein |
| Gal-3 | Galectin 3 |
| HLA-B | Human leukocyte antigen B |
| HMGB1 | High mobility group B1 |
| IC | Immune checkpoint |
| ICI | Immune checkpoint inhibition |
| IDO1 | Indoleamine 2,3 -dioxygenase 1 |
| IFN-g | Interferon-g |
| IRF | Interferon regulatory factor |
| JAK | Janus Kinase |
| LAG-3 | lymphocyte activation gene 3 |
| MAPK | Mitogen activated protein kinase |
| MDSC | Myeloid-derived suppressor cells |
| MHC II | Major histocompatibility complex class II |
| NACT | Neoadjuvant chemotherapy |
| NECL | Nectin-like |
| NF-kB | Nuclear factor kappa of B lymphocytes |
| NFAT | Nuclear factor of activated T cell transcription factor |
| NK Cells | Natural killer cells |
| NSCLC | Non-small cell lung carcinoma |
| PAK4 | p21 activated kinase 4 |
| PD-L1 | Programmed death ligand 1 |
| PD-1 | Programmed death receptor 1 |
| PI3K | Phosphatidylinositol-3-kinase |
| PtdSer | Phosphatidyl serine |
| PSGL1 | P-selectin glycoprotein ligand 1 |
| PVR | Polio virus receptor |
| SHIP1 | SH3 domain containing inhibitory phosphatase 1 |
| STAT1 | Signal transducers and activators of transcription 1 |
| STAT3 | Signal transducers and activators of transcription 3 |
| TAM | Tumor associated macrophages |
| TAN | Tumor associated neutrophils |
| TCR | T-cell receptor |
| TIGIT | T lymphocyte immunoreceptor with Ig and ITIM domains |
| TILs | Tumor infiltrating lymphocytes |
| TIM-3 | T lymphocyte immunoglobulin mucin 3 |
| TIME | Tumor immune microenvironment |
| TLR | Toll-like receptor |
| TNBC | Triple-negative breast cancer |
| TNF-a | Tumor necrosis factor a |
| TME | Tumor microenvironment |
| Tregs | Regulatory T cells |
| VEGF | Vascular endothelial growth factor |
| VISTA | V-domain Ig suppressor of T cell activation |
| VSIG3 | V-set and immunoglobulin domain containing 3 |
| YY1 | Yin Yang 1 |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Pantel, K.; Brakenhoff, R.H. Dissecting the metastatic cascade. Nat. Rev. Cancer 2004, 4, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- ESahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. CB 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Koliaraki, V.; Prados, A.; Armaka, M.; Kollias, G. The mesenchymal context in inflammation, immunity and cancer. Nat. Immunol. 2020, 21, 974–982. [Google Scholar] [CrossRef]
- Schreiber, K.; Karrison, T.G.; Wolf, S.P.; Kiyotani, K.; Steiner, M.; Littmann, E.R.; Pamer, E.G.; Kammertoens, T.; Schreiber, H.; Leisegang, M. Impact of TCR Diversity on the Development of Transplanted or Chemically Induced Tumors. Cancer Immunol. Res. 2020, 8, 192–202. [Google Scholar] [CrossRef]
- Ramadoss, N.S.; Robinson, W.H. Characterizing the BCR repertoire in immune-mediated diseases. Nat. Rev. Rheumatol. 2020, 16, 7–8. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- Kammertoens, T.; Friese, C.; Arina, A.; Idel, C.; Briesemeister, D.; Rothe, M.; Ivanov, A.; Szymborska, A.; Patone, G.; Kunz, S.; et al. Tumour ischaemia by interferon-γ resembles physiological blood vessel regression. Nature 2017, 545, 98–102. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.; Kastenmüller, W. CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Miggelbrink, A.M.; Jackson, J.D.; Lorrey, S.J.; Srinivasan, E.S.; Waibl-Polania, J.; Wilkinson, D.S.; Fecci, P.E. CD4 T-Cell Exhaustion: Does It Exist and What Are Its Roles in Cancer? Clin. Cancer Res. An. Off. J. Am. Assoc. Cancer Res. 2021, 27, 5742. [Google Scholar] [CrossRef]
- Saravia, J.; Chapman, N.M.; Chi, H. Helper T cell differentiation. Cell. Mol. Immunol. 2019, 16, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years on. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef]
- Ribas, A. Adaptive Immune Resistance: How Cancer Protects from Immune Attack. Cancer Discov. 2015, 5, 915–919. [Google Scholar] [CrossRef]
- Sukumar, M.; Roychoudhuri, R.; Restifo, N.P. Nutrient Competition: A New Axis of Tumor Immunosuppression. Cell 2015, 162, 1206–1208. [Google Scholar] [CrossRef]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The cancer metabolic reprogramming and immune response. Mol. Cancer 2021, 20, 28. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Dufva, O.; Pölönen, P.; Brück, O.; Keränen, M.A.; Klievink, J.; Mehtonen, J.; Huuhtanen, J.; Kumar, A.; Malani, D.; Siitonen, S.; et al. Immunogenomic Landscape of Hematological Malignancies. Cancer Cell 2020, 38, 380–399. [Google Scholar] [CrossRef] [PubMed]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Su, L.J.; Pinckney, J.; Critton, D.; Boyer, E.; Krishnakumar, A.; Corbett, M.; Rankin, A.L.; Dibella, R.; Campbell, L.; et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature 2019, 574, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Boles, K.S.; Vermi, W.; Facchetti, F.; Fuchs, A.; Wilson, T.J.; Diacovo, T.G.; Cella, M.; Colonna, M. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur. J. Immunol. 2009, 39, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef]
- Huard, B.; Mastrangeli, R.; Prigent, P.; Bruniquel, D.; Donini, S.; El-Tayar, N.; Maigret, B.; Dréano, M.; Triebel, F. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc. Natl. Acad. Sci. USA 1997, 94, 5744–5749. [Google Scholar] [CrossRef]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. Embo J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Wang, J.; Wu, G.; Manick, B.; Hernandez, V.; Renelt, M.; Erickson, C.; Guan, J.; Singh, R.; Rollins, S.; Solorz, A.; et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology 2019, 156, 74–85. [Google Scholar] [CrossRef]
- ElTanbouly, M.A.; Zhao, Y.; Nowak, E.; Li, J.; Schaafsma, E.; Le Mercier, I.; Ceeraz, S.; Lines, J.L.; Peng, C.; Carriere, C.; et al. VISTA is a checkpoint regulator for naïve T cell quiescence and peripheral tolerance. Science 2020, 367, eaay0524. [Google Scholar] [CrossRef] [PubMed]
- Baranzini, S.E. The role of antiproliferative gene Tob1 in the immune system. Clin. Exp. Neuroimmunol. 2014, 5, 132–136. [Google Scholar] [CrossRef]
- Carlson, C.M.; Endrizzi, B.T.; Wu, J.; Ding, X.; Weinreich, M.A.; Walsh, E.R.; Wani, M.A.; Lingrel, J.B.; Hogquist, K.A.; Jameson, S.C. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature 2006, 442, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, I.; Fruman, D.A. Regulation of quiescence in lymphocytes. Trends Immunol. 2003, 24, 380–386. [Google Scholar] [CrossRef]
- Xu, W.; Hiếu, T.; Malarkannan, S.; Wang, L. The structure, expression, and multifaceted role of immune-checkpoint protein VISTA as a critical regulator of anti-tumor immunity, autoimmunity, and inflammation. Cell. Mol. Immunol. 2018, 15, 438–446. [Google Scholar] [CrossRef]
- Prodeus, A.; Abdul-Wahid, A.; Sparkes, A.; Fischer, N.W.; Cydzik, M.; Chiang, N.; Alwash, M.; Ferzoco, A.; Vacaresse, N.; Julius, M.; et al. VISTA.COMP—An engineered checkpoint receptor agonist that potently suppresses T cell-mediated immune responses. JCI Insight 2017, 2, e94308. [Google Scholar] [CrossRef]
- Harada, H.; Suzu, S.; Hayashi, Y.; Okada, S. BT-IgSF, a novel immunoglobulin superfamily protein, functions as a cell adhesion molecule. J. Cell. Physiol. 2005, 204, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, R.; Neubert, E.N.; Stairiker, C.J.; Henriquez, M.L.; Bradley, L.M. PSGL-1 Is a T Cell Intrinsic Inhibitor That Regulates Effector and Memory Differentiation and Responses During Viral Infection. Front. Immunol. 2021, 12, 677824. [Google Scholar] [CrossRef]
- DeRogatis, J.M.; Viramontes, K.M.; Neubert, E.N.; Tinoco, R. PSGL-1 Immune Checkpoint Inhibition for CD4(+) T Cell Cancer Immunotherapy. Front. Immunol. 2021, 12, 636238. [Google Scholar] [CrossRef] [PubMed]
- Le Mercier, I.; Chen, W.; Lines, J.L.; Day, M.; Li, J.; Sergent, P.; Noelle, R.J.; Wang, L. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. 2014, 74, 1933–1944. [Google Scholar] [CrossRef]
- Zong, L.; Mo, S.; Yu, S.; Zhou, Y.; Zhang, M.; Chen, J.; Xiang, Y. Expression of the immune checkpoint VISTA in breast cancer. Cancer Immunol. Immunother. 2020, 69, 1437–1446. [Google Scholar] [CrossRef]
- Xie, X.; Zhang, J.; Shi, Z.; Liu, W.; Hu, X.; Qie, C.; Chen, W.; Wang, Y.; Wang, L.; Jiang, J.; et al. The Expression Pattern and Clinical Significance of the Immune Checkpoint Regulator VISTA in Human Breast Cancer. Front. Immunol. 2020, 11, 563044. [Google Scholar] [CrossRef]
- Phase 1 Study of CI-8993 Anti-VISTA Antibody in Patients with Advanced Solid Tumor Malignancies. Available online: https://ClinicalTrials.gov/show/NCT04475523 (accessed on 5 July 2022).
- A Study of CA-170 (Oral PD-L1, PD-L2 and VISTA Checkpoint Antagonist) in Patients with Advanced Tumors and Lymphomas. Available online: https://ClinicalTrials.gov/show/NCT02812875 (accessed on 5 July 2022).
- A Study of HMBD-002, a Monoclonal Antibody Targeting VISTA, as Monotherapy and Combined with Pembrolizumab. Available online: https://ClinicalTrials.gov/show/NCT05082610 (accessed on 5 July 2022).
- Niebel, D.; Fröhlich, A.; Zarbl, R.; Fietz, S.; de Vos, L.; Vogt, T.J.; Dietrich, J.; Sirokay, J.; Kuster, P.; Saavedra, G.; et al. DNA methylation regulates TIGIT expression within the melanoma microenvironment, is prognostic for overall survival, and predicts progression-free survival in patients treated with anti-PD-1 immunotherapy. Clin. Epigenetics 2022, 14, 50. [Google Scholar] [CrossRef]
- Levin, S.D.; Taft, D.W.; Brandt, C.S.; Bucher, C.; Howard, E.D.; Chadwick, E.M.; Johnston, J.; Hammond, A.; Bontadelli, K.; Ardourel, D.; et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur. J. Immunol. 2011, 41, 902–915. [Google Scholar] [CrossRef] [PubMed]
- Pende, D.; Bottino, C.; Castriconi, R.; Cantoni, C.; Marcenaro, S.; Rivera, P.; Spaggiari, G.M.; Dondero, A.; Carnemolla, B.; Reymond, N.; et al. PVR (CD155) and Nectin-2 (CD112) as ligands of the human DNAM-1 (CD226) activating receptor: Involvement in tumor cell lysis. Mol. Immunol. 2005, 42, 463–469. [Google Scholar] [CrossRef]
- Seth, S.; Maier, M.K.; Qiu, Q.; Ravens, I.; Kremmer, E.; Förster, R.; Bernhardt, G. The murine pan T cell marker CD96 is an adhesion receptor for CD155 and nectin-1. Biochem. Biophys. Res. Commun. 2007, 364, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD96: New checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Ramagopal, U.A.; Rubinstein, R.; Vigdorovich, V.; Nathenson, S.G.; Almo, S.C. Structure of Nectin-2 reveals determinants of homophilic and heterophilic interactions that control cell-cell adhesion. Proc. Natl. Acad. Sci. USA 2012, 109, 14836–14840. [Google Scholar] [CrossRef]
- Fujito, T.; Ikeda, W.; Kakunaga, S.; Minami, Y.; Kajita, M.; Sakamoto, Y.; Monden, M.; Takai, Y. Inhibition of cell movement and proliferation by cell-cell contact-induced interaction of Necl-5 with nectin-3. J. Cell. Biol. 2005, 171, 165–173. [Google Scholar] [CrossRef]
- Stengel, K.F.; Harden-Bowles, K.; Yu, X.; Rouge, L.; Yin, J.; Comps-Agrar, L.; Wiesmann, C.; Bazan, J.F.; Eaton, D.L.; Grogan, J.L. Structure of TIGIT immunoreceptor bound to poliovirus receptor reveals a cell-cell adhesion and signaling mechanism that requires cis-trans receptor clustering. Proc. Natl. Acad. Sci. USA 2012, 109, 5399–5404. [Google Scholar] [CrossRef] [PubMed]
- M-Rabe, M.; Cabaud, O.; Josselin, E.; Finetti, P.; Castellano, R.; Farina, A.; Agavnian-Couquiaud, E.; Saviane, G.; Collette, Y.; Viens, P.; et al. Nectin-4: A new prognostic biomarker for efficient therapeutic targeting of primary and metastatic triple-negative breast cancer. Ann. Oncol. 2017, 28, 769–776. [Google Scholar] [CrossRef]
- Nishiwada, S.; Sho, M.; Yasuda, S.; Shimada, K.; Yamato, I.; Akahori, T.; Kinoshita, S.; Nagai, M.; Konishi, N.; Nakajima, Y. Nectin-4 expression contributes to tumor proliferation, angiogenesis and patient prognosis in human pancreatic cancer. J. Exp. Clin. Cancer Res. 2015, 34, 30. [Google Scholar] [CrossRef]
- Takano, A.; Ishikawa, N.; Nishino, R.; Masuda, K.; Yasui, W.; Inai, K.; Nishimura, H.; Ito, H.; Nakayama, H.; Miyagi, Y.; et al. Identification of nectin-4 oncoprotein as a diagnostic and therapeutic target for lung cancer. Cancer Res. 2009, 69, 6694–6703. [Google Scholar] [CrossRef]
- Li, M.; Xia, P.; Du, Y.; Liu, S.; Huang, G.; Chen, J.; Zhang, H.; Hou, N.; Cheng, X.; Zhou, L.; et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-γ production of natural killer cells via β-arrestin 2-mediated negative signaling. J. Biol. Chem. 2014, 289, 17647–17657. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013, 20, 456–464. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef]
- Martinet, L.; Smyth, M.J. Balancing natural killer cell activation through paired receptors. Nat. Rev. Immunol. 2015, 15, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V.; et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Luo, Z.; Ismtula, D.; Bi, X.; Kong, H.; Wang, Y.; Yang, Z.; Mao, X. TIGIT is a Novel Prognostic Marker and Correlate for Immune Infiltration in Invasive Breast Cancer. Comb. Chem. High Throughput Screen. 2023, 26, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Stamm, H.; Oliveira-Ferrer, L.; Grossjohann, E.M.; Muschhammer, J.; Thaden, V.; Brauneck, F.; Kischel, R.; Müller, V.; Bokemeyer, C.; Fiedler, W.; et al. Targeting the TIGIT-PVR immune checkpoint axis as novel therapeutic option in breast cancer. Oncoimmunology 2019, 8, e1674605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gao, C.; Shao, J.; Wang, Z. TIGIT-related transcriptome profile and its association with tumor immune microenvironment in breast cancer. Biosci. Rep. 2021, 41, BSR20204340. [Google Scholar] [CrossRef]
- The Recombinant Humanized Anti-TIGIT Monoclonal Antibody (JS006) Monotherapy and in Combination with Toripalimab in Patients with Advanced Tumor. Available online: https://ClinicalTrials.gov/show/NCT05061628 (accessed on 7 July 2022).
- A Study to Evaluate the Efficacy of IBI939 in Combination with Sintilimab in Patients with Advanced NSCLC. Available online: https://ClinicalTrials.gov/show/NCT04672369 (accessed on 7 July 2022).
- COM902 (A TIGIT Inhibitor) in Subjects with Advanced Malignancies. Available online: https://ClinicalTrials.gov/show/NCT04354246 (accessed on 7 July 2022).
- First in Human Study of M6223. Available online: https://ClinicalTrials.gov/show/NCT04457778 (accessed on 7 July 2022).
- Assessment of Safety and Preliminary Efficacy with BAT6021 in Solid Tumor Patients. Available online: https://ClinicalTrials.gov/show/NCT05073484 (accessed on 7 July 2022).
- Zimberelimab and Domvanalimab in Combination with Chemotherapy Versus Pembrolizumab with Chemotherapy in Patients with Untreated Metastatic Non-Small Cell Lung Cancer. Available online: https://ClinicalTrials.gov/show/NCT05502237 (accessed on 7 July 2022).
- COM701 in Combination with BMS-986207 and Nivolumab in Subjects with Advanced Solid Tumors. Available online: https://ClinicalTrials.gov/show/NCT04570839 (accessed on 7 July 2022).
- A Study of NTX-1088, a Monoclonal Antibody Targeting the Poliovirus Receptor (PVR, CD155), as Monotherapy and Combined with Pembrolizumab. Available online: https://ClinicalTrials.gov/show/NCT05378425 (accessed on 7 July 2022).
- A Study of OMP-313M32 in Subjects with Locally Advanced or Metastatic Solid Tumors. Available online: https://ClinicalTrials.gov/show/NCT03119428 (accessed on 7 July 2022).
- A Study of BMS-986207 Given Alone and in Combination with Nivolumab or with Nivolumab and Ipilimumab in Advanced Solid Tumors. Available online: https://ClinicalTrials.gov/show/NCT02913313 (accessed on 7 July 2022).
- A Study of Tiragolumab in Combination with Atezolizumab Compared with Placebo in Combination with Atezolizumab in Patients with Previously Untreated Locally Advanced Unresectable or Metastatic PD-L1-Selected Non-Small Cell Lung Cancer. Available online: https://ClinicalTrials.gov/show/NCT04294810 (accessed on 7 July 2022).
- Li, N.; Workman, C.J.; Martin, S.M.; Vignali, D.A. Biochemical analysis of the regulatory T cell protein lymphocyte activation gene-3 (LAG-3; CD223). J. Immunol. 2004, 173, 6806–6812. [Google Scholar] [CrossRef]
- Mastrangeli, R.; Micangeli, E.; Donini, S. Cloning of murine LAG-3 by magnetic bead bound homologous probes and PCR (gene-capture PCR). Anal. Biochem. 1996, 241, 93–102. [Google Scholar] [CrossRef]
- Workman, C.J.; Dugger, K.J.; Vignali, D.A. Cutting edge: Molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J. Immunol. 2002, 169, 5392–5395. [Google Scholar] [CrossRef]
- Workman, C.J.; Vignali, D.A. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur. J. Immunol. 2003, 33, 970–979. [Google Scholar] [CrossRef]
- Iouzalen, N.; Andreae, S.; Hannier, S.; Triebel, F. LAP, a lymphocyte activation gene-3 (LAG-3)-associated protein that binds to a repeated EP motif in the intracellular region of LAG-3, may participate in the down-regulation of the CD3/TCR activation pathway. Eur. J. Immunol. 2001, 31, 2885–2891. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Gaulard, P.; Faure, F.; Hercend, T.; Triebel, F. Cellular expression and tissue distribution of the human LAG-3-encoded protein, an MHC class II ligand. Immunogenetics 1994, 39, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Manetti, R.; Cosmi, L.; Galli, G.; Heusser, C.H.; Romagnani, S.; Maggi, E. Opposite role for interleukin-4 and interferon-gamma on CD30 and lymphocyte activation gene-3 (LAG-3) expression by activated naive T cells. Eur. J. Immunol. 1997, 27, 2239–2244. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Manetti, R.; Tomasévic, I.; Guidizi, M.G.; Biagiotti, R.; Giannò, V.; Germano, P.; Mavilia, C.; Maggi, E.; Romagnani, S. Expression and release of LAG-3-encoded protein by human CD4+ T cells are associated with IFN-gamma production. Faseb J. 1996, 10, 769–776. [Google Scholar] [CrossRef]
- Burton, B.R.; Britton, G.J.; Fang, H.; Verhagen, J.; Smithers, B.; Sabatos-Peyton, C.A.; Carney, L.J.; Gough, J.; Strobel, S.; Wraith, D.C. Sequential transcriptional changes dictate safe and effective antigen-specific immunotherapy. Nat. Commun. 2014, 5, 4741. [Google Scholar] [CrossRef]
- Huard, B.; Prigent, P.; Pagès, F.; Bruniquel, D.; Triebel, F. T cell major histocompatibility complex class II molecules down-regulate CD4+ T cell clone responses following LAG-3 binding. Eur. J. Immunol. 1996, 26, 1180–1186. [Google Scholar] [CrossRef]
- Dumic, J.; Dabelic, S.; Flögel, M. Galectin-3: An open-ended story. Biochim. Biophys. Acta 2006, 1760, 616–635. [Google Scholar] [CrossRef]
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol. Res. 2015, 3, 412–423. [Google Scholar] [CrossRef]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e312. [Google Scholar] [CrossRef]
- Li, N.; Wang, Y.; Forbes, K.; Vignali, K.M.; Heale, B.S.; Saftig, P.; Hartmann, D.; Black, R.A.; Rossi, J.J.; Blobel, C.P.; et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. Embo J. 2007, 26, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Qi, Y.; Zhai, J.; Kong, X.; Wang, X.; Wang, Z.; Fang, Y.; Wang, J. Molecular and Clinical Characterization of LAG3 in Breast Cancer Through 2994 Samples. Front. Immunol. 2021, 12, 599207. [Google Scholar] [CrossRef] [PubMed]
- This Study Tests the New Medicine BI 754111 Alone or in Combination with Another New Substance BI 754091 in Patients with Advanced Cancer. The Study Tests Different doses to Find the Best Dose for Continuous Treatment. Available online: https://ClinicalTrials.gov/show/NCT03156114 (accessed on 9 July 2022).
- PDR001 Plus LAG525 for Patients with Advanced Solid and Hematologic Malignancies. Available online: https://ClinicalTrials.gov/show/NCT03365791 (accessed on 9 July 2022).
- Anderson, A.C.; Anderson, D.E.; Bregoli, L.; Hastings, W.D.; Kassam, N.; Lei, C.; Chandwaskar, R.; Karman, J.; Su, E.W.; Hirashima, M.; et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 2007, 318, 1141–1143. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, L.C.; Lopez-Vergès, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef]
- Jiang, X.; Yu, J.; Shi, Q.; Xiao, Y.; Wang, W.; Chen, G.; Zhao, Z.; Wang, R.; Xiao, H.; Hou, C.; et al. Tim-3 promotes intestinal homeostasis in DSS colitis by inhibiting M1 polarization of macrophages. Clin. Immunol. 2015, 160, 328–335. [Google Scholar] [CrossRef]
- Yang, X.; Jiang, X.; Chen, G.; Xiao, Y.; Geng, S.; Kang, C.; Zhou, T.; Li, Y.; Guo, X.; Xiao, H.; et al. T cell Ig mucin-3 promotes homeostasis of sepsis by negatively regulating the TLR response. J. Immunol. 2013, 190, 2068–2079. [Google Scholar] [CrossRef]
- Phong, B.L.; Avery, L.; Sumpter, T.L.; Gorman, J.V.; Watkins, S.C.; Colgan, J.D.; Kane, L.P. Tim-3 enhances FcεRI-proximal signaling to modulate mast cell activation. J. Exp. Med. 2015, 212, 2289–2304. [Google Scholar] [CrossRef]
- Yang, R.; Sun, L.; Li, C.F.; Wang, Y.H.; Yao, J.; Li, H.; Yan, M.; Chang, W.C.; Hsu, J.M.; Cha, J.H.; et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 2021, 12, 832. [Google Scholar] [CrossRef]
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef]
- van de Weyer, P.S.; Muehlfeit, M.; Klose, C.; Bonventre, J.V.; Walz, G.; Kuehn, E.W. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem. Biophys. Res. Commun. 2006, 351, 571–576. [Google Scholar] [CrossRef]
- Rangachari, M.; Zhu, C.; Sakuishi, K.; Xiao, S.; Karman, J.; Chen, A.; Angin, M.; Wakeham, A.; Greenfield, E.A.; Sobel, R.A.; et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat. Med. 2012, 18, 1394–1400. [Google Scholar] [CrossRef]
- DeKruyff, R.H.; Bu, X.; Ballesteros, A.; Santiago, C.; Chim, Y.L.; Lee, H.H.; Karisola, P.; Pichavant, M.; Kaplan, G.G.; Umetsu, D.T.; et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J. Immunol. 2010, 184, 1918–1930. [Google Scholar] [CrossRef]
- Nakayama, M.; Akiba, H.; Takeda, K.; Kojima, Y.; Hashiguchi, M.; Azuma, M.; Yagita, H.; Okumura, K. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 2009, 113, 3821–3830. [Google Scholar] [CrossRef]
- Horst, A.K.; Bickert, T.; Brewig, N.; Ludewig, P.; van Rooijen, N.; Schumacher, U.; Beauchemin, N.; Ito, W.D.; Fleischer, B.; Wagener, C.; et al. CEACAM1+ myeloid cells control angiogenesis in inflammation. Blood 2009, 113, 6726–6736. [Google Scholar] [CrossRef]
- Kammerer, R.; Stober, D.; Singer, B.B.; Obrink, B.; Reimann, J. Carcinoembryonic antigen-related cell adhesion molecule 1 on murine dendritic cells is a potent regulator of T cell stimulation. J. Immunol. 2001, 166, 6537–6544. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Goverman, J.M. The influence of T cell Ig mucin-3 signaling on central nervous system autoimmune disease is determined by the effector function of the pathogenic T cells. J. Immunol. 2013, 190, 4991–4999. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, G.; Li, Y.; Wang, R.; Wang, L.; Lin, Z.; Gao, X.; Feng, J.; Ma, Y.; Shen, B.; et al. Involvement of T cell Ig Mucin-3 (Tim-3) in the negative regulation of inflammatory bowel disease. Clin. Immunol. 2010, 134, 169–177. [Google Scholar] [CrossRef]
- Liu, Y.; Shu, Q.; Gao, L.; Hou, N.; Zhao, D.; Liu, X.; Zhang, X.; Xu, L.; Yue, X.; Zhu, F.; et al. Increased Tim-3 expression on peripheral lymphocytes from patients with rheumatoid arthritis negatively correlates with disease activity. Clin. Immunol. 2010, 137, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Guo, X.; Jiang, X.; Zhou, P.; Xiao, Y.; Zhou, T.; Chen, G.; Zhao, Z.; Xiao, H.; Hou, C.; et al. Dysregulated Tim-3 expression and its correlation with imbalanced CD4 helper T cell function in ulcerative colitis. Clin. Immunol. 2012, 145, 230–240. [Google Scholar] [CrossRef]
- Yang, L.; Anderson, D.E.; Kuchroo, J.; Hafler, D.A. Lack of TIM-3 immunoregulation in multiple sclerosis. J. Immunol. 2008, 180, 4409–4414. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fueyo, A.; Tian, J.; Picarella, D.; Domenig, C.; Zheng, X.X.; Sabatos, C.A.; Manlongat, N.; Bender, O.; Kamradt, T.; Kuchroo, V.K.; et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 2003, 4, 1093–1101. [Google Scholar] [CrossRef]
- Cheng, S.; Han, F.; Xu, Y.; Qu, T.; Ju, Y. Expression of Tim-3 in breast cancer tissue promotes tumor progression. Int. J. Clin. Exp. Pathol. 2018, 11, 1157–1166. [Google Scholar] [PubMed]
- Zhang, H.; Xiang, R.; Wu, B.; Li, J.; Luo, G. T-cell immunoglobulin mucin-3 expression in invasive ductal breast carcinoma: Clinicopathological correlations and association with tumor infiltration by cytotoxic lymphocytes. Mol. Clin. Oncol. 2017, 7, 557–563. [Google Scholar] [CrossRef]
- Sym023 (Anti-TIM-3) in Patients with Advanced Solid Tumor Malignancies or Lymphomas. Available online: https://ClinicalTrials.gov/show/NCT03489343 (accessed on 11 July 2022).
- Study of Efficacy and Safety of MBG453 in Combination with Azacitidine in Subjects with Intermediate, High or Very High Risk Myelodysplastic Syndrome (MDS) as Per IPSS-R, or Chronic Myelomonocytic Leukemia-2 (CMML-2). Available online: https://ClinicalTrials.gov/show/NCT04266301 (accessed on 11 July 2022).
- A Study of LY3321367 Alone or with LY3300054 in Participants with Advanced Relapsed/Refractory Solid Tumors. Available online: https://ClinicalTrials.gov/show/NCT03099109 (accessed on 11 July 2022).
- A Study of Lomvastomig (RO7121661) and Tobemstomig (RO7247669) Compared with Nivolumab in Participants with Advanced or Metastatic Squamous Cell Carcinoma of the Esophagus. Available online: https://ClinicalTrials.gov/show/NCT04785820 (accessed on 7 July 2022).
- A Study to Assess the Safety and Efficacy of LB1410 in Participants with Advanced Solid Tumor or Lymphoma (Keyplus-001). Available online: https://www.ClinicalTrials.gov/show/NCT05357651 (accessed on 15 August 2022).
- TSR-022 (Anti-TIM-3 Antibody) and TSR-042 (Anti-PD-1 Antibody) in Patients with Liver Cancer. Available online: https://ClinicalTrials.gov/show/NCT03680508 (accessed on 15 August 2022).
- Study of BGB-A425 and LBL-007 in Combination with Tislelizumab in Advanced Solid Tumors. Available online: https://ClinicalTrials.gov/ct2/show/NCT03744468 (accessed on 15 August 2022).
- A Safety and Tolerability Study of INCAGN02390 in Select Advanced Malignancies. Available online: https://ClinicalTrials.gov/show/NCT03652077 (accessed on 15 August 2022).
- An Investigational Immunotherapy Study of BMS-986258 Alone and in Combination with Nivolumab in Participants with Solid Cancers That Are Advanced or Have Spread. Available online: https://ClinicalTrials.gov/show/NCT03446040 (accessed on 15 August 2022).
- Carreno, B.M.; Collins, M. The B7 family of ligands and its receptors: New pathways for costimulation and inhibition of immune responses. Annu. Rev. Immunol. 2002, 20, 29–53. [Google Scholar] [CrossRef]
- Zak, K.M.; Kitel, R.; Przetocka, S.; Golik, P.; Guzik, K.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure 2015, 23, 2341–2348. [Google Scholar] [CrossRef]
- Shinohara, T.; Taniwaki, M.; Ishida, Y.; Kawaichi, M.; Honjo, T. Structure and chromosomal localization of the human PD-1 gene (PDCD1). Genomics 1994, 23, 704–706. [Google Scholar] [CrossRef]
- Lipson, E.J.; Drake, C.G. Ipilimumab: An anti-CTLA-4 antibody for metastatic melanoma. Clin. Cancer Res. 2011, 17, 6958–6962. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Jiang, F.; Honjo, T.; Okazaki, T. PD-1 deficiency reveals various tissue-specific autoimmunity by H-2b and dose-dependent requirement of H-2g7 for diabetes in NOD mice. Proc. Natl. Acad. Sci. USA 2008, 105, 3533–3538. [Google Scholar] [CrossRef] [PubMed]
- Staron, M.M.; Gray, S.M.; Marshall, H.D.; Parish, I.A.; Chen, J.H.; Perry, C.J.; Cui, G.; Li, M.O.; Kaech, S.M. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity 2014, 41, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.; Oestreich, K.J.; Ha, S.J.; Duraiswamy, J.; Akondy, R.S.; West, E.E.; Wei, Z.; Lu, P.; Austin, J.W.; Riley, J.L.; et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity 2011, 35, 400–412. [Google Scholar] [CrossRef]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef]
- Lin, C.; He, H.; Liu, H.; Li, R.; Chen, Y.; Qi, Y.; Jiang, Q.; Chen, L.; Zhang, P.; Zhang, H.; et al. Tumour-associated macrophages-derived CXCL8 determines immune evasion through autonomous PD-L1 expression in gastric cancer. Gut 2019, 68, 1764–1773. [Google Scholar] [CrossRef]
- Kim, Y.J.; Won, C.H.; Lee, M.W.; Choi, J.H.; Chang, S.E.; Lee, W.J. Correlation Between Tumor-Associated Macrophage and Immune Checkpoint Molecule Expression and Its Prognostic Significance in Cutaneous Melanoma. J. Clin. Med. 2020, 9, 2500. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef]
- Shi, G.; Yang, Q.; Zhang, Y.; Jiang, Q.; Lin, Y.; Yang, S.; Wang, H.; Cheng, L.; Zhang, X.; Li, Y.; et al. Modulating the Tumor Microenvironment via Oncolytic Viruses and CSF-1R Inhibition Synergistically Enhances Anti-PD-1 Immunotherapy. Mol. Ther. 2019, 27, 244–260. [Google Scholar] [CrossRef]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. 2016, 9, 5023–5039. [Google Scholar] [CrossRef] [PubMed]
- Udall, M.; Rizzo, M.; Kenny, J.; Doherty, J.; Dahm, S.; Robbins, P.; Faulkner, E. PD-L1 diagnostic tests: A systematic literature review of scoring algorithms and test-validation metrics. Diagn. Pathol. 2018, 13, 12. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhao, X.; Fu, J.; Wang, H. Progress and Challenges in Precise Treatment of Tumors with PD-1/PD-L1 Blockade. Front. Immunol. 2020, 11, 339. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Stewart, R.L.; Updike, K.L.; Factor, R.E.; Henry, N.L.; Boucher, K.M.; Bernard, P.S.; Varley, K.E. A Multigene Assay Determines Risk of Recurrence in Patients with Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 3466–3478. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.; Gligorov, J.; André, F.; Cameron, D.; Schneeweiss, A.; Barrios, C.; Xu, B.; Wardley, A.; Kaen, D.; Andrade, L.; et al. Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann. Oncol. 2021, 32, 994–1004. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Gao, J.; Hou, B.; Zhu, Q.; Yang, L.; Jiang, X.; Zou, Z.; Li, X.; Xu, T.; Zheng, M.; Chen, Y.-H.; et al. Engineered bioorthogonal POLY-PROTAC nanoparticles for tumour-specific protein degradation and precise cancer therapy. Nat. Commun. 2022, 13, 4318. [Google Scholar] [CrossRef] [PubMed]
- Allaire, J.C.; Balk, M.; Azmi, S.; Handl, H.L.; Yang, K.; Barnes, G. Use of PD-1 and PD-L1 inhibitors after first-line therapy in esophageal cancer patients in the US. Curr. Med. Res. Opin. 2021, 37, 1403–1407. [Google Scholar] [CrossRef]
- Maher, C.M.; Thomas, J.D.; Haas, D.A.; Longen, C.G.; Oyer, H.M.; Tong, J.Y.; Kim, F.J. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol. Cancer Res. 2018, 16, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zheng, C.; Liu, Y.; Luo, W.; Deng, H.; Shen, J. Chitosan biguanide induced mitochondrial inhibition to amplify the efficacy of oxygen-sensitive tumor therapies. Carbohydr. Polym. 2022, 295, 119878. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Liu, Y.; Jiang, X.; Zheng, C.; Luo, W.; Xiang, X.; Qi, X.; Shen, J. Metformin modified chitosan as a multi-functional adjuvant to enhance cisplatin-based tumor chemotherapy efficacy. Int. J. Biol. Macromol. 2023, 224, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Long, G.V.; Robert, C.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; et al. Survival Outcomes in Patients With Previously Untreated BRAF Wild-Type Advanced Melanoma Treated With Nivolumab Therapy: Three-Year Follow-up of a Randomized Phase 3 Trial. JAMA Oncol. 2019, 5, 187–194. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Sosman, J.A.; Atkins, M.B.; Leming, P.D.; et al. Five-Year Survival and Correlates among Patients with Advanced Melanoma, Renal Cell Carcinoma, or Non-Small Cell Lung Cancer Treated with Nivolumab. JAMA Oncol. 2019, 5, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-Year Overall Survival for Patients with Advanced NonSmall-Cell Lung Cancer Treated with Pembrolizumab: Results from the Phase I KEYNOTE-001 Study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef]
- Karasarides, M.; Cogdill, A.P.; Robbins, P.B.; Bowden, M.; Burton, E.M.; Butterfield, L.H.; Cesano, A.; Hammer, C.; Haymaker, C.L.; Horak, C.E.; et al. Hallmarks of Resistance to Immune-Checkpoint Inhibitors. Cancer Immunol. Res. 2022, 10, 372–383. [Google Scholar] [CrossRef]
- Saleh, R.; Toor, S.M.; Khalaf, S.; Elkord, E. Breast Cancer Cells and PD-1/PD-L1 Blockade Upregulate the Expression of PD-1, CTLA-4, TIM-3 and LAG-3 Immune Checkpoints in CD4(+) T Cells. Vaccines 2019, 7, 149. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schreiner, J.; Muller, P.; Herzig, P.; Roller, A.; Belousov, A.; Umana, P.; Pisa, P.; Klein, C.; Bacac, M.; et al. Progression of Lung Cancer Is Associated with Increased Dysfunction of T Cells Defined by Coexpression of Multiple Inhibitory Receptors. Cancer Immunol. Res. 2015, 3, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Balkhi, M.Y.; Wittmann, G.; Xiong, F.; Junghans, R.P. YY1 Upregulates Checkpoint Receptors and Downregulates Type I Cytokines in Exhausted, Chronically Stimulated Human T Cells. iScience 2018, 2, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Sakuishi, K.; Xiao, S.; Sun, Z.; Zaghouani, S.; Gu, G.; Wang, C.; Tan, D.J.; Wu, C.; Rangachari, M.; et al. An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat. Commun. 2015, 6, 6072. [Google Scholar] [CrossRef]
- Curdy, N.; Lanvin, O.; Laurent, C.; Fournie, J.J.; Franchini, D.M. Regulatory Mechanisms of Inhibitory Immune Checkpoint Receptors Expression. Trends Cell Biol. 2019, 29, 777–790. [Google Scholar] [CrossRef] [PubMed]
- DeLong, J.H.; O’Hara Hall, A.; Rausch, M.; Moodley, D.; Perry, J.; Park, J.; Phan, A.T.; Beiting, D.P.; Kedl, R.M.; Hill, J.A.; et al. IL-27 and TCR Stimulation Promote T Cell Expression of Multiple Inhibitory Receptors. Immunohorizons 2019, 3, 13–25. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): A multicentre, randomised, open-label, phase 3 trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan Nair, V.; El Salhat, H.; Taha, R.Z.; John, A.; Ali, B.R.; Elkord, E. DNA methylation and repressive H3K9 and H3K27 trimethylation in the promoter regions of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, and PD-L1 genes in human primary breast cancer. Clin. Epigenetics 2018, 10, 78. [Google Scholar] [CrossRef]
- Allgauer, M.; Budczies, J.; Christopoulos, P.; Endris, V.; Lier, A.; Rempel, E.; Volckmar, A.L.; Kirchner, M.; von Winterfeld, M.; Leichsenring, J.; et al. Implementing tumor mutational burden (TMB) analysis in routine diagnostics-a primer for molecular pathologists and clinicians. Transl. Lung Cancer Res. 2018, 7, 703–715. [Google Scholar] [CrossRef]
- Vega, D.M.; Yee, L.M.; McShane, L.M.; Williams, P.M.; Chen, L.; Vilimas, T.; Fabrizio, D.; Funari, V.; Newberg, J.; Bruce, L.K.; et al. Aligning tumor mutational burden (TMB) quantification across diagnostic platforms: Phase II of the Friends of Cancer Research TMB Harmonization Project. Ann. Oncol. 2021, 32, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients with Non-Small-Cell Lung Cancer Profiled with Targeted Next-Generation Sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Malouf, G.G.; Jacobs, I.; Chou, J.; Liu, L.; Johnson, M.L. Addressing resistance to immune checkpoint inhibitor therapy: An urgent unmet need. Future Oncol. 2021, 17, 1401–1439. [Google Scholar] [CrossRef] [PubMed]
- Maleki Vareki, S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 157. [Google Scholar] [CrossRef]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Sucker, A.; Horn, S.; Heeke, C.; Bielefeld, N.; Schrors, B.; Bicker, A.; Lindemann, M.; Roesch, A.; Gaudernack, G.; et al. Melanoma Lesions Independently Acquire T-cell Resistance during Metastatic Latency. Cancer Res. 2016, 76, 4347–4358. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Pereira, C.; Gimenez-Xavier, P.; Pros, E.; Pajares, M.J.; Moro, M.; Gomez, A.; Navarro, A.; Condom, E.; Moran, S.; Gomez-Lopez, G.; et al. Genomic Profiling of Patient-Derived Xenografts for Lung Cancer Identifies B2M Inactivation Impairing Immunorecognition. Clin. Cancer Res. 2017, 23, 3203–3213. [Google Scholar] [CrossRef] [PubMed]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef] [PubMed]
- Sucker, A.; Zhao, F.; Pieper, N.; Heeke, C.; Maltaner, R.; Stadtler, N.; Real, B.; Bielefeld, N.; Howe, S.; Weide, B.; et al. Acquired IFNgamma resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat. Commun. 2017, 8, 15440. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef]
- Wei, F.; Zhong, S.; Ma, Z.; Kong, H.; Medvec, A.; Ahmed, R.; Freeman, G.J.; Krogsgaard, M.; Riley, J.L. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc. Natl. Acad. Sci. USA 2013, 110, E2480–E2489. [Google Scholar] [CrossRef]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef] [PubMed]
- Radziewicz, H.; Ibegbu, C.C.; Fernandez, M.L.; Workowski, K.A.; Obideen, K.; Wehbi, M.; Hanson, H.L.; Steinberg, J.P.; Masopust, D.; Wherry, E.J.; et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J. Virol. 2007, 81, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Chen, L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C. PD-1: A Driver or Passenger of T Cell Exhaustion? Mol. Cell 2020, 77, 930–931. [Google Scholar] [CrossRef]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-cell exhaustion: Characteristics, causes and conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef]
- Ma, J.; Zheng, B.; Goswami, S.; Meng, L.; Zhang, D.; Cao, C.; Li, T.; Zhu, F.; Ma, L.; Zhang, Z.; et al. PD1(Hi) CD8(+) T cells correlate with exhausted signature and poor clinical outcome in hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 331. [Google Scholar] [CrossRef]
- Sznol, M.; Chen, L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin. Cancer Res. 2013, 19, 1021–1034. [Google Scholar] [CrossRef]
- Lei, Q.; Wang, D.; Sun, K.; Wang, L.; Zhang, Y. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front. Cell Dev. Biol. 2020, 8, 672. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Mittra, A.; Naqash, A.R.; Takebe, N. A review of mechanisms of resistance to immune checkpoint inhibitors and potential strategies for therapy. Cancer Drug Resist. 2020, 3, 252–275. [Google Scholar] [CrossRef]
- Dai, X.; Bu, X.; Gao, Y.; Guo, J.; Hu, J.; Jiang, C.; Zhang, Z.; Xu, K.; Duan, J.; He, S.; et al. Energy status dictates PD-L1 protein abundance and anti-tumor immunity to enable checkpoint blockade. Mol. Cell 2021, 81, 2317–2331.e2316. [Google Scholar] [CrossRef]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, A.; Lei, Q.; Zhang, Y. Tumor-intrinsic signaling pathways: Key roles in the regulation of the immunosuppressive tumor microenvironment. J. Hematol. Oncol. 2019, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef]
- Stutvoet, T.S.; Kol, A.; de Vries, E.G.; de Bruyn, M.; Fehrmann, R.S.; Terwisscha van Scheltinga, A.G.; de Jong, S. MAPK pathway activity plays a key role in PD-L1 expression of lung adenocarcinoma cells. J. Pathol. 2019, 249, 52–64. [Google Scholar] [CrossRef]
- Sumimoto, H.; Takano, A.; Teramoto, K.; Daigo, Y. RAS-Mitogen-Activated Protein Kinase Signal Is Required for Enhanced PD-L1 Expression in Human Lung Cancers. PLoS ONE 2016, 11, e0166626. [Google Scholar] [CrossRef]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015, 7, 279ra241. [Google Scholar] [CrossRef]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef]
- Zheng, C.; Luo, W.; Liu, Y.; Chen, J.; Deng, H.; Zhou, Z.; Shen, J. Killing three birds with one stone: Multi-stage metabolic regulation mediated by clinically usable berberine liposome to overcome photodynamic immunotherapy resistance. Chem. Eng. J. 2023, 454, 140164. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, J.; Liu, Y.; Zheng, C.; Luo, W.; Chen, L.; Zhou, S.; Li, Z.; Shen, J. Cascade two-stage tumor re-oxygenation and immune re-sensitization mediated by self-assembled albumin-sorafenib nanoparticles for enhanced photodynamic immunotherapy. Acta Pharm. Sin. B 2022, 12, 4204–4223. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e924. [Google Scholar] [CrossRef]
- Fujita, S.; Sato, Y.; Sato, K.; Eizumi, K.; Fukaya, T.; Kubo, M.; Yamashita, N.; Sato, K. Regulatory dendritic cells protect against cutaneous chronic graft-versus-host disease mediated through CD4+CD25+Foxp3+ regulatory T cells. Blood 2007, 110, 3793–3803. [Google Scholar] [CrossRef]
- Wakkach, A.; Fournier, N.; Brun, V.; Breittmayer, J.-P.; Cottrez, F.; Groux, H. Characterization of Dendritic Cells that Induce Tolerance and T Regulatory 1 Cell Differentiation In Vivo. Immunity 2003, 18, 605–617. [Google Scholar] [CrossRef]
- Sato, K.; Yamashita, N.; Baba, M.; Matsuyama, T. Modified myeloid dendritic cells act as regulatory dendritic cells to induce anergic and regulatory T cells. Blood 2003, 101, 3581–3589. [Google Scholar] [CrossRef]
- Yaguchi, T.; Goto, Y.; Kido, K.; Mochimaru, H.; Sakurai, T.; Tsukamoto, N.; Kudo-Saito, C.; Fujita, T.; Sumimoto, H.; Kawakami, Y. Immune suppression and resistance mediated by constitutive activation of Wnt/beta-catenin signaling in human melanoma cells. J. Immunol. 2012, 189, 2110–2117. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e4. [Google Scholar] [CrossRef]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Holtzhausen, A.; Zhao, F.; Evans, K.S.; Tsutsui, M.; Orabona, C.; Tyler, D.S.; Hanks, B.A. Melanoma-Derived Wnt5a Promotes Local Dendritic-Cell Expression of IDO and Immunotolerance: Opportunities for Pharmacologic Enhancement of Immunotherapy. Cancer Immunol. Res. 2015, 3, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Chon, S.Y.; Hassanain, H.H.; Gupta, S.L. Cooperative role of interferon regulatory factor 1 and p91 (STAT1) response elements in interferon-gamma-inducible expression of human indoleamine 2,3-dioxygenase gene. J. Biol. Chem. 1996, 271, 17247–17252. [Google Scholar] [CrossRef]
- Yentz, S.; Smith, D. Indoleamine 2,3-Dioxygenase (IDO) Inhibition as a Strategy to Augment Cancer Immunotherapy. BioDrugs 2018, 32, 311–317. [Google Scholar] [CrossRef]
- Nasmall, U.A.; Merhi, M.; Inchakalody, V.; Fernandes, Q.; Mestiri, S.; Prabhu, K.S.; Uddin, S.; Dermime, S. The role of PAK4 in the immune system and its potential implication in cancer immunotherapy. Cell Immunol. 2021, 367, 104408. [Google Scholar] [CrossRef]
- Abril-Rodriguez, G.; Torrejon, D.Y.; Liu, W.; Zaretsky, J.M.; Nowicki, T.S.; Tsoi, J.; Puig-Saus, C.; Baselga-Carretero, I.; Medina, E.; Quist, M.J.; et al. PAK4 inhibition improves PD-1 blockade immunotherapy. Nat. Cancer 2020, 1, 46–58. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug Name | ClinicalTrials.gov Identifier (NCT) | Phase | Combined with | Targets |
|---|---|---|---|---|
| CI-8993 [48] | NCT04475523 | Phase 1 | - | Solid tumors |
| CA-170 [49] | NCT02812875 | Phase 1 completed | - | Advance solid tumors and lymphomas |
| HMBD-002 [50] | NCT05082610 | Phase 1 | Pembrolizumab (anti-PD-1) | Advance solid malignancy |
| Drug Name | ClinicalTrials.gov Identifier (NCT) | Phase | Combined with | Targets |
|---|---|---|---|---|
| JS006 [71] | NCT05061628 | Phase 1 | Toripalimab | Advanced tumors |
| IBI939 [72] | NCT04672369 | Phase 1 | Sintilimab | Advanced malignancies |
| COM902 [73] | NCT04354246 | Phase 1 | - | Advanced malignancies |
| M6223 [74] | NCT04457778 | Phase 1 | Bintrafusp alfa | Metastatic solid tumors |
| BAT6021 [75] | NCT05073484 | Phase 1 | BAT1308 (anti-PD1) | Advanced solid tumors |
| Domvanalimab [76] | NCT05502237 | Phase 1 | Zimberelimab (anti-PD1) | Solid tumors |
| COM701 [77] | NCT04570839 | Phase 1 | Nivolumab (anti-PD1) | Advanced cancers |
| NTX-1088 [78] | NCT05378425 | Phase 1 | Pembrolizumab (anti-PD1) | Cancer |
| OMP-313M32 [79] | NCT03119428 | Phase 1 | Nivolumab (anti-PD1) | Metastatic cancer |
| BMS-986207 [80] | NCT02913313 | Phase 2 | Nivolumab Ipilimumab | Broad solid tumors |
| Tiragolumab [81] | NCT04294810 | Phase 1 | Atezolizumab Nab-paclitaxel Carboplatin | TNBC |
| Drug Name | ClinicalTrials.gov Identifier (NCT) | Phase | Combined with | Targets |
|---|---|---|---|---|
| BI754111 [97] | NCT03156114 | Phase 1 | BI754091 (anti-PD1) | Carcinoma |
| LAG525 [98] | NCT03365791 | Phase 2 | PDR001 (anti-PD1) | TNBC |
| Drug Name | ClinicalTrials.gov Identifier (NCT) | Phase | Combined with | Targets |
|---|---|---|---|---|
| Sym023 [123] | NCT03489343 | Phase 1 | - | Metastatic cancers |
| MBG453 [124] | NCT04266301 | Phase 2 | PDR001 (anti-PD1) Decitabine | Advance malignancies |
| LY3321367 [125] | NCT03099109 | Phase 1 | - | Solid tumor |
| RO7121661 [126] | NCT04785820 | Phase 1 | - | Solid tumor |
| LB1410 [127] | NCT05357651 | Phase 1 | - | Solid tumor |
| TSR-022 [128] | NCT03680508. | Phase 1 | TSR-042 (anti-PD1) | Advance solid tumor |
| BGBA425 [129] | NCT03744468 | Phase 1 | Tislelizumab (anti-PD1) | Metastatic tumors |
| INCAGN02390 [130] | NCT03652077 | Phase 1 | - | Solid tumors |
| BMS-986258 [131] | NCT03446040 | Phase 1 | Nivolumab | Advanced cancer |
| Drug | Class | First FDA Approval Date |
|---|---|---|
| Nivolumab | Anti-PD-1 | 22 December 2014 |
| Pembrolizumab | 4 September 2014 | |
| Cemiplimab | 28 September 2018 | |
| Atezolizumab | Anti-PD-L1 | 18 May 2016 |
| Avelumab | 23 March 2017 | |
| Durvalumab | 1 May 2017 | |
| Ipilimumab | Anti-CTLA-4 | 28 March 2011 |
| Tremelimumab | 24 October 2022 | |
| Relatlimab | Anti-LAG-3 | 18 March 2022 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dulal, D.; Boring, A.; Terrero, D.; Johnson, T.; Tiwari, A.K.; Raman, D. Tackling of Immunorefractory Tumors by Targeting Alternative Immune Checkpoints. Cancers 2023, 15, 2774. https://doi.org/10.3390/cancers15102774
Dulal D, Boring A, Terrero D, Johnson T, Tiwari AK, Raman D. Tackling of Immunorefractory Tumors by Targeting Alternative Immune Checkpoints. Cancers. 2023; 15(10):2774. https://doi.org/10.3390/cancers15102774
Chicago/Turabian StyleDulal, Dharmindra, Andrew Boring, David Terrero, Tiffany Johnson, Amit K. Tiwari, and Dayanidhi Raman. 2023. "Tackling of Immunorefractory Tumors by Targeting Alternative Immune Checkpoints" Cancers 15, no. 10: 2774. https://doi.org/10.3390/cancers15102774
APA StyleDulal, D., Boring, A., Terrero, D., Johnson, T., Tiwari, A. K., & Raman, D. (2023). Tackling of Immunorefractory Tumors by Targeting Alternative Immune Checkpoints. Cancers, 15(10), 2774. https://doi.org/10.3390/cancers15102774