
Multi-Omic Analysis of CIC’s Functional Networks Reveals Novel Interaction Partners and a Potential Role in Mitotic Fidelity

 , , , ,
, , , ,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Cell Culture
2.2. CIC Knockout Cell Line Generation and Expression Constructs
2.3. scRNA-Seq
2.4. IP-MS
2.5. Co-Essentiality Mapping and In Silico Genetic Screening Framework
2.6. Co-Expression Analysis
2.7. ChIP-Seq Analysis
2.8. Alternative Splicing Quantification
3. Results
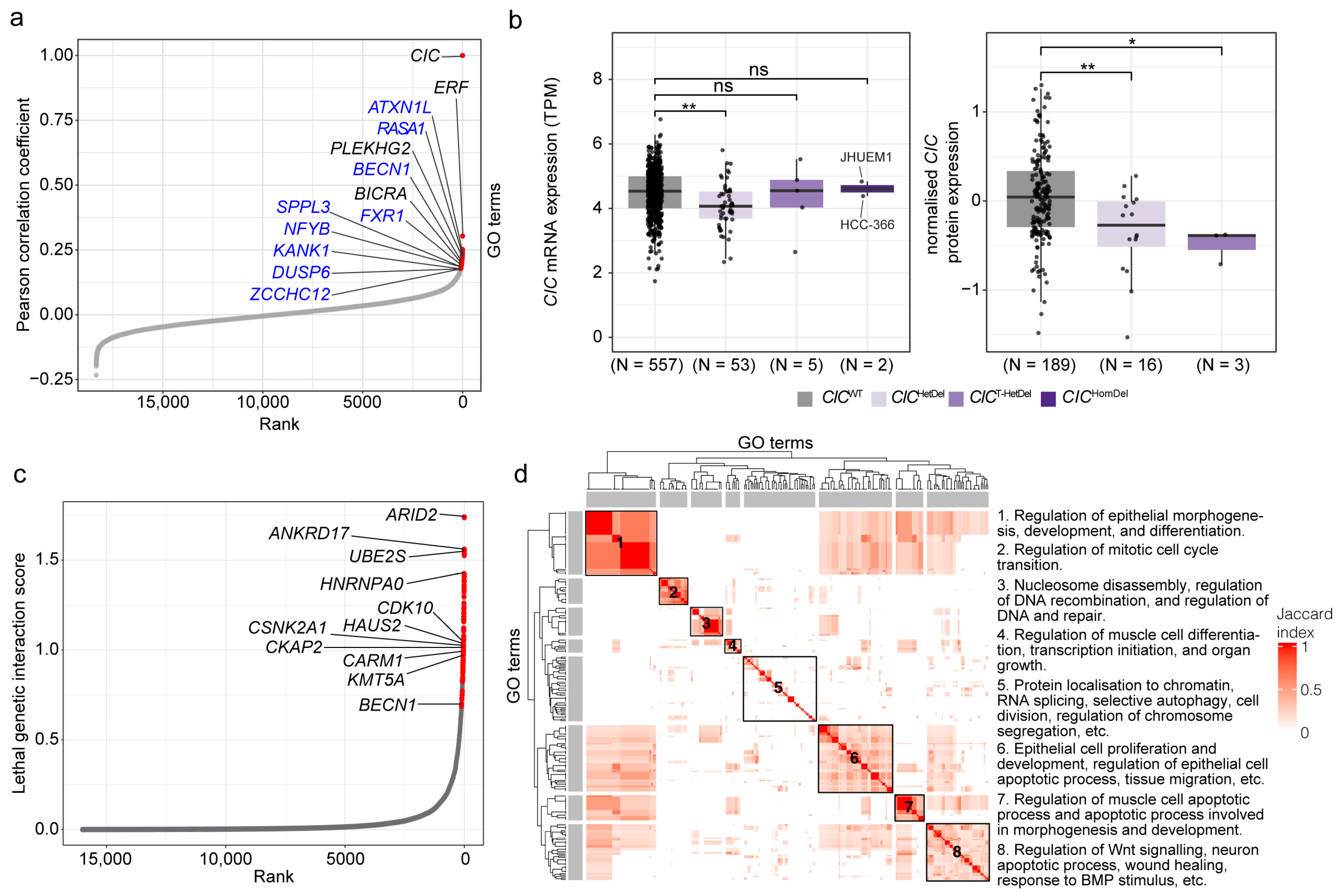
3.1. Co-Essential Network Analysis Reinforces CIC’s Role in MAPK Signalling and Identifies Candidate Additional Functions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Candidate Co-Essential Gene | Candidate Gene Function(s) | Related CIC Function(s) |
|---|---|---|
| ATXN1L | Interacts with and stabilises CIC for DNA binding [29]. | Interacts with and is stabilised by ATXN1L [29]. |
| BECN1 | Regulates apoptosis and cytoskeletal dynamics [76,77]. Mediator of autophagy [76]. | No known related function. |
| DUSP6 | Negative regulator of MAPK signalling and known target of CIC-mediated repression [21]. | Represses DUSP6 expression [21]. |
| FXR1 | RNA-binding protein, has been implicated in the stabilisation of multiple transcripts through binding to their 5′ or 3′ UTRs [78,79,80]. | No known related function. |
| KANK1 | Regulates apoptosis and cytoskeletal dynamics [81,82]. | No known related function. |
| NFYB | Regulates cell cycle progression, differentiation, and apoptosis [83,84]. | Regulates cell cycle progression and differentiation [30]. |
| RASA1 | Regulates MAPK signalling through suppression of ERK1/2 expression [85]. Regulates cell cycle progression, differentiation, and apoptosis [85]. | Regulates MAPK signalling through suppression of ERK1/2 expression [19]. Regulates cell cycle progression and differentiation [30]. |
| SPPL3 | Protease that participates in the regulation of T-cell responses by interfering with human leukocyte antigen detection [86]. | Plays a role in T-cell development [10,87,88,89,90]. |
| ZCCHC12 | Downstream effector of bone morphogenic protein signalling and has been shown to co-regulate cellular development with AP-1 and CREB [91]. | Implicated in cellular development in various contexts [30]. FOS and FOSL1, members of the AP-1 complex, are candidate targets of transcriptional regulation by CIC [22,24]. |
3.2. Lethal Genetic Interactors of CIC Are Associated with Regulation of Apoptotic Processes, Cell Division and Differentiation, and Chromatin Organisation
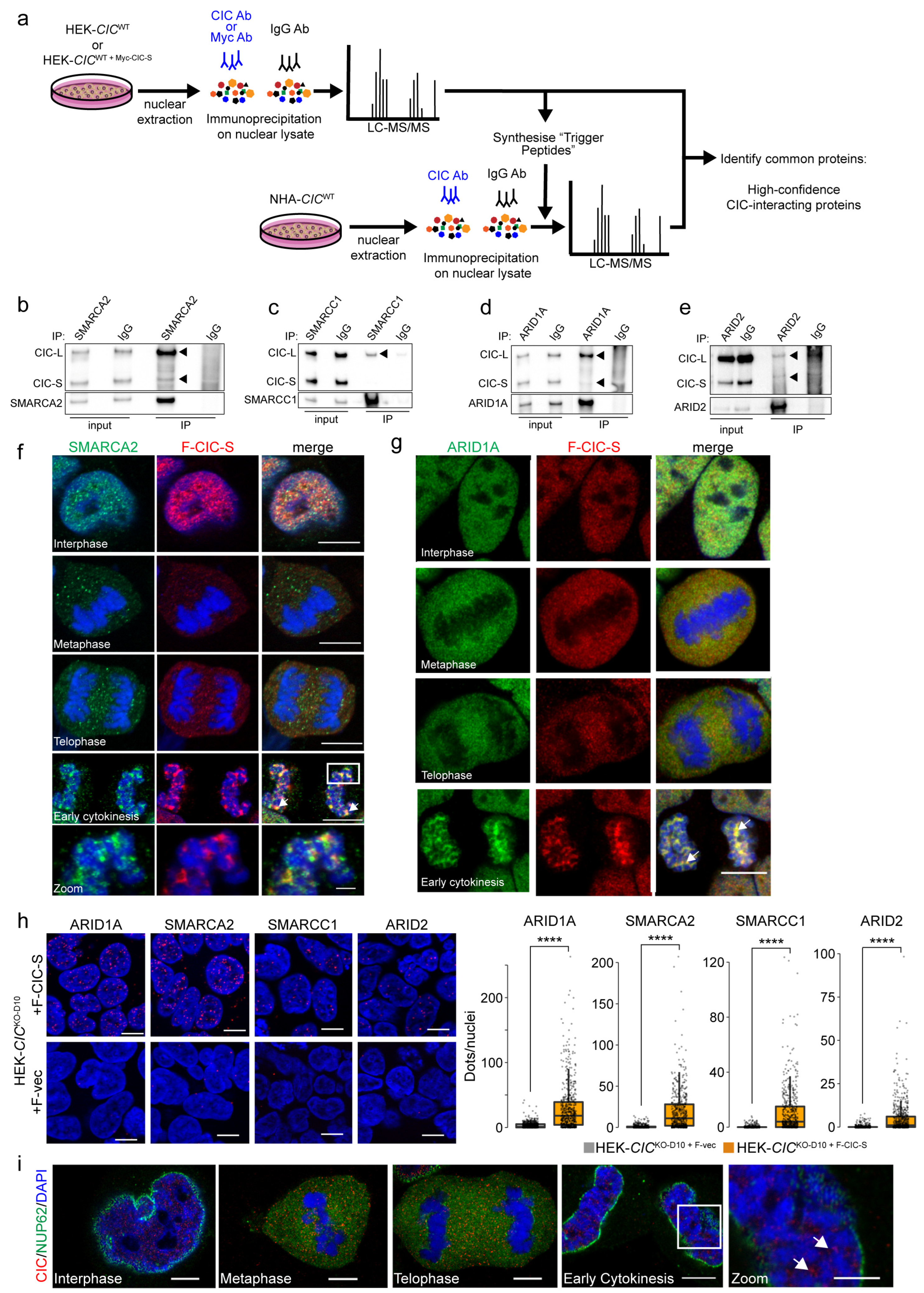
3.3. Nuclear CIC Interacts with Mitotic Regulators and SWI/SNF Complex Members
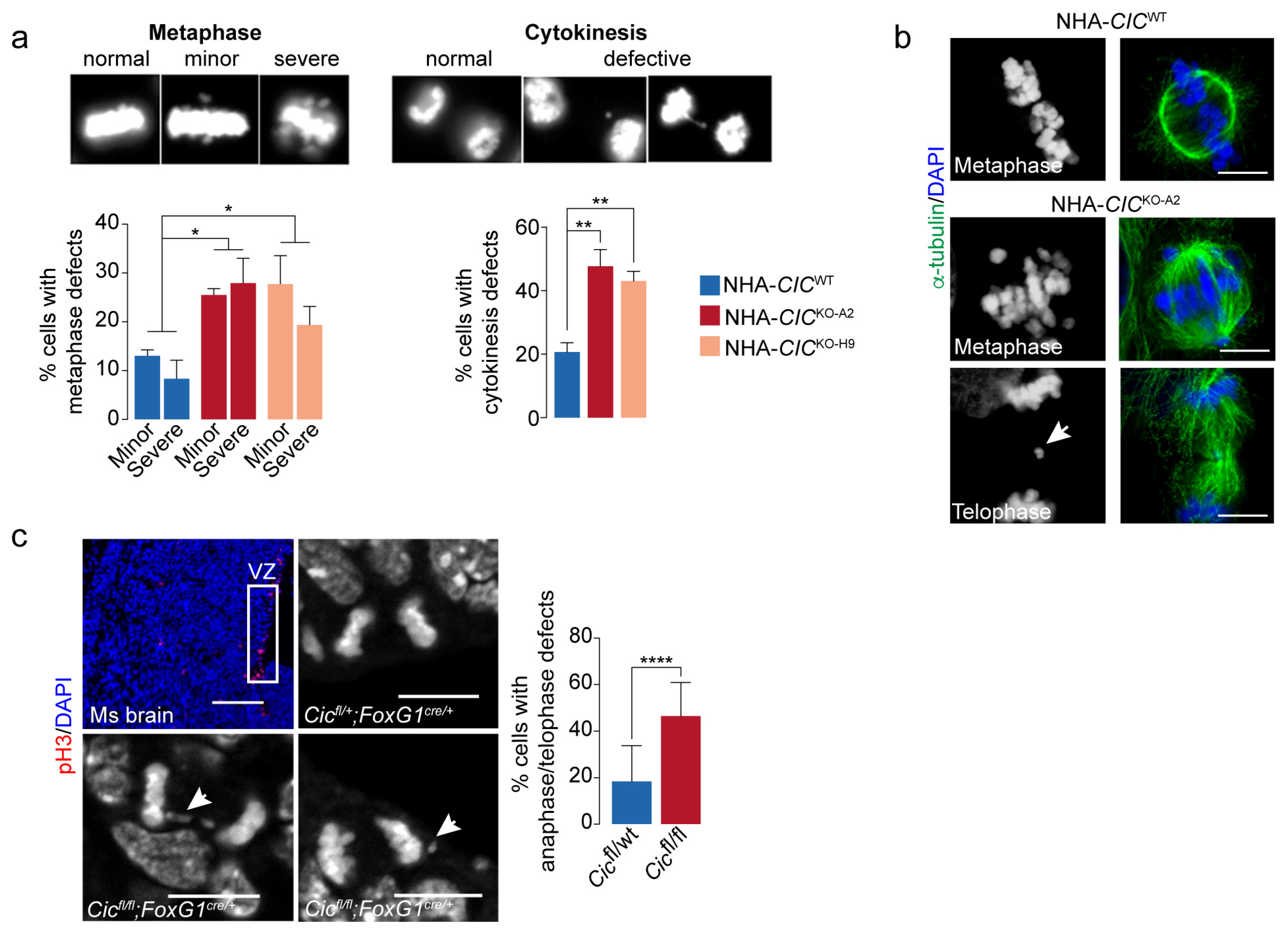
3.4. Loss of CIC Is Associated with Mitotic Defects in Mammalian Cells
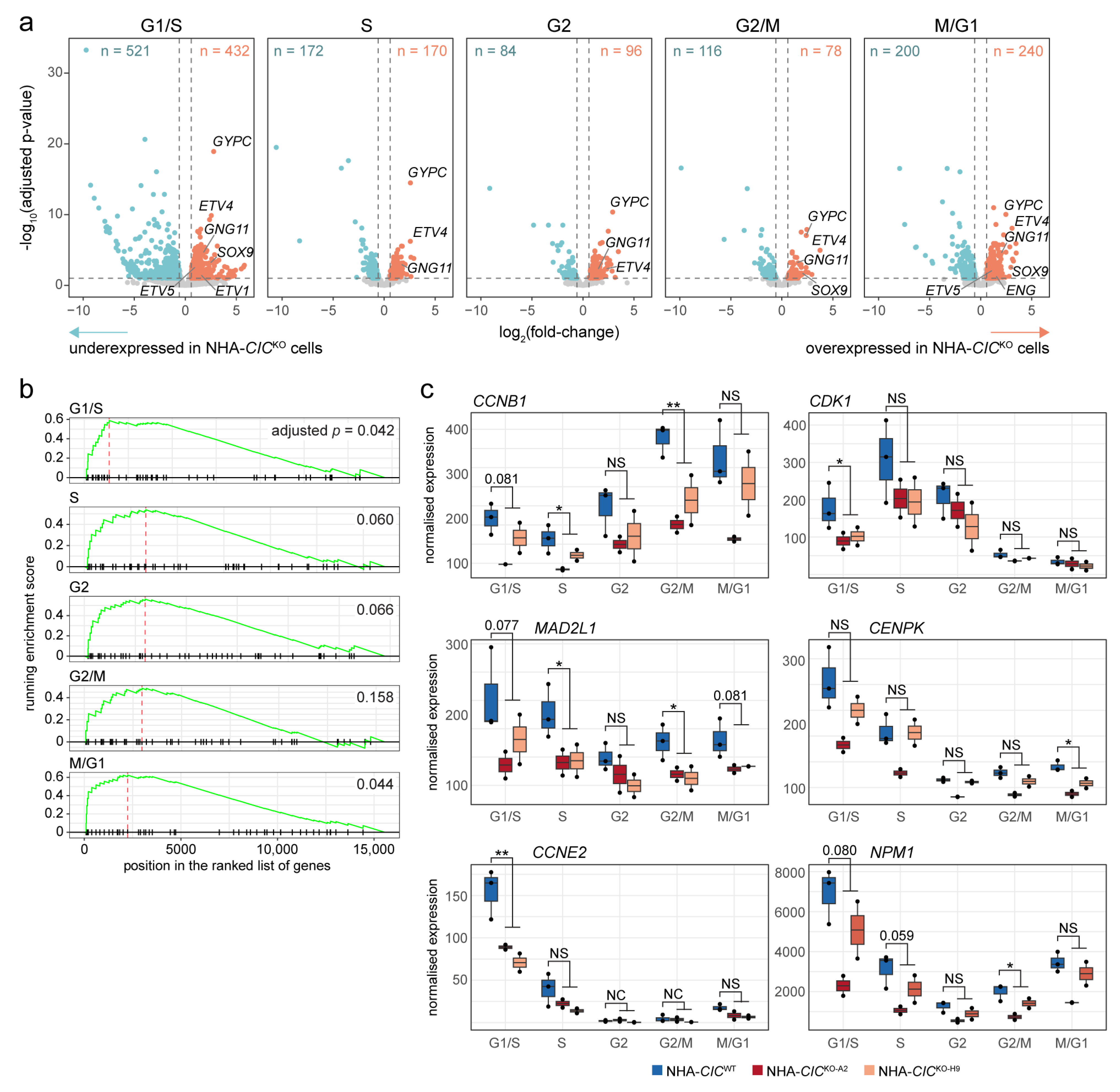
3.5. Loss of CIC Is Associated with Transcriptional Dysregulation across All Cell Cycle Phases
3.6. CIC Binding Sites Overlap with a Subset of SWI/SNF Binding Sites
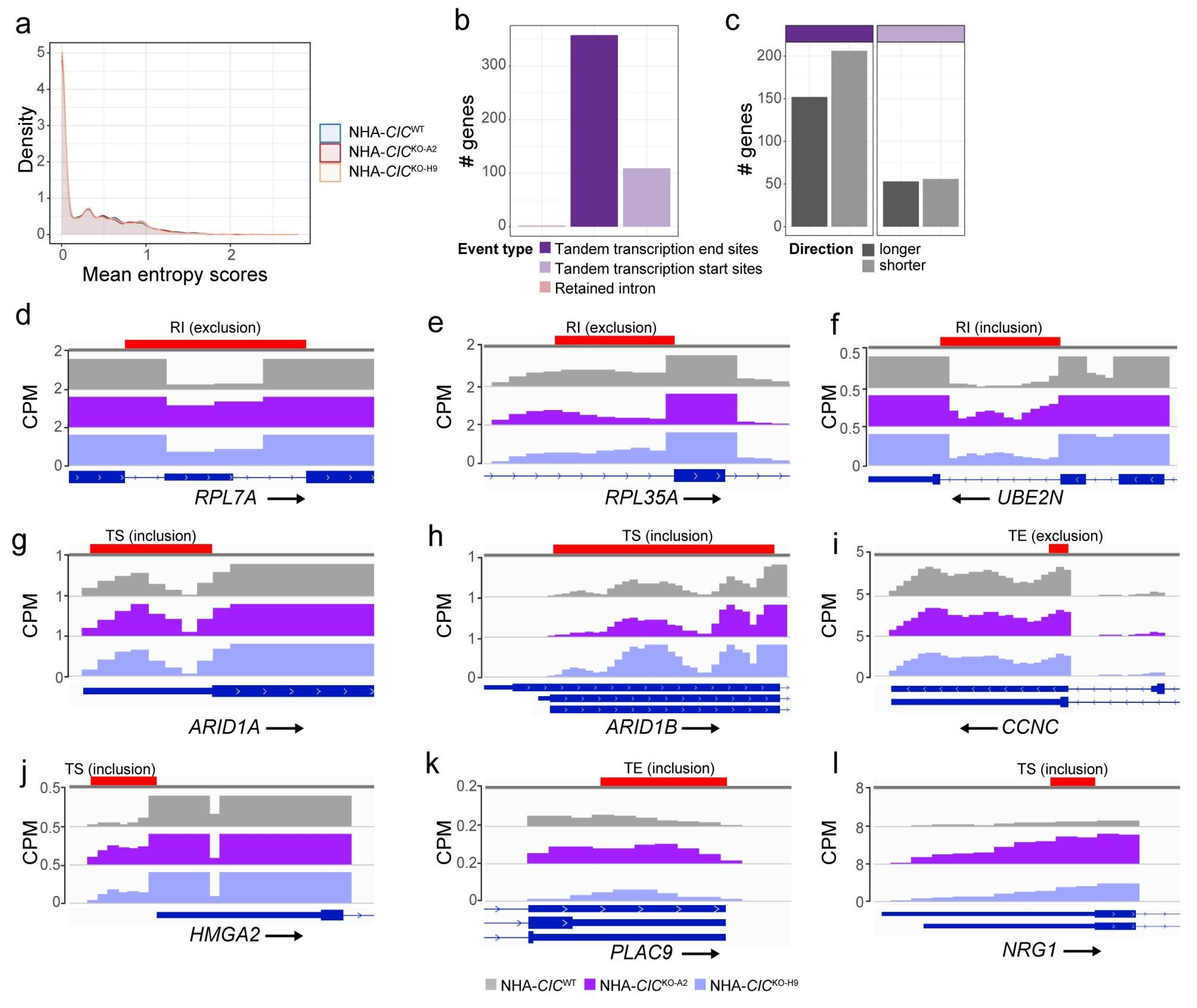
3.7. CIC Loss Destabilises Splicing at Untranslated Regions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haidar, A.; Arekapudi, S.; DeMattia, F.; Abu-Isa, E.; Kraut, M. High-Grade Undifferentiated Small Round Cell Sarcoma with t(4;19)(Q35;Q13.1) CIC-DUX4 Fusion: Emerging Entities of Soft Tissue Tumors with Unique Histopathologic Features—A Case Report and Literature Review. Am. J. Case Rep. 2015, 16, 87–94. [Google Scholar] [CrossRef]
- Bettegowda, C.; Agrawal, N.; Jiao, Y.; Sausen, M.; Wood, L.D.; Hruban, R.H.; Rodriguez, F.J.; Cahill, D.P.; McLendon, R.; Riggins, G.; et al. Mutations in CIC and FUBP1 Contribute to Human Oligodendroglioma. Science 2011, 333, 1453–1455. [Google Scholar] [CrossRef]
- Yip, S.; Butterfield, Y.S.; Morozova, O.; Chittaranjan, S.; Blough, M.D.; An, J.; Birol, I.; Chesnelong, C.; Chiu, R.; Chuah, E.; et al. Concurrent CIC Mutations, IDH Mutations, and 1p/19q Loss Distinguish Oligodendrogliomas from Other Cancers. J. Pathol. 2012, 226, 7–16. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Endometrial Carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Choi, N.; Park, J.; Lee, J.S.; Yoe, J.; Park, G.Y.; Kim, E.; Jeon, H.; Cho, Y.M.; Roh, T.Y.; Lee, Y. MiR-93/MiR-106b/MiR-375-CIC-CRABP1: A Novel Regulatory Axis in Prostate Cancer Progression. Oncotarget 2015, 6, 23533–23547. [Google Scholar] [CrossRef]
- Okimoto, R.A.; Breitenbuecher, F.; Olivas, V.R.; Wu, W.; Gini, B.; Hofree, M.; Asthana, S.; Hrustanovic, G.; Flanagan, J.; Tulpule, A.; et al. Inactivation of Capicua Drives Cancer Metastasis. Nat. Genet. 2017, 49, 87–96. [Google Scholar] [CrossRef]
- Simon-Carrasco, L.; Grana, O.; Salmon, M.; Jacob, H.K.C.; Gutierrez, A.; Jimenez, G.; Drosten, M.; Barbacid, M. Inactivation of Capicua in Adult Mice Causes T-Cell Lymphoblastic Lymphoma. Genes Dev. 2017, 31, 1456–1468. [Google Scholar] [CrossRef]
- Tan, Q.; Brunetti, L.; Rousseaux, M.W.C.; Lu, H.C.; Wan, Y.W.; Revelli, J.P.; Liu, Z.; Goodell, M.A.; Zoghbi, H.Y. Loss of Capicua Alters Early T Cell Development and Predisposes Mice to T Cell Lymphoblastic Leukemia/Lymphoma. Proc. Natl. Acad. Sci. USA 2018, 115, E1511–E1519. [Google Scholar] [CrossRef]
- Chan, A.K.; Pang, J.C.; Chung, N.Y.; Li, K.K.; Poon, W.S.; Chan, D.T.; Shi, Z.; Chen, L.; Zhou, L.; Ng, H.K. Loss of CIC and FUBP1 Expressions Are Potential Markers of Shorter Time to Recurrence in Oligodendroglial Tumors. Mod. Pathol. 2014, 27, 332–342. [Google Scholar] [CrossRef]
- Gleize, V.; Alentorn, A.; Connen de Kerillis, L.; Labussiere, M.; Nadaradjane, A.; Mundwiller, E.; Ottolenghi, C.; Mangesius, S.; Rahimian, A.; Ducray, F.; et al. CIC Inactivating Mutations Identify Aggressive Subset of 1p19q Codeleted Gliomas. Ann. Neurol. 2015, 78, 355–374. [Google Scholar] [CrossRef]
- Specht, K.; Sung, Y.S.; Zhang, L.; Richter, G.H.; Fletcher, C.D.; Antonescu, C.R. Distinct Transcriptional Signature and Immunoprofile of CIC-DUX4 Fusion-Positive Round Cell Tumors Compared to EWSR1-Rearranged Ewing Sarcomas: Further Evidence toward Distinct Pathologic Entities. Genes Chromosomes Cancer 2014, 53, 622–633. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Owosho, A.A.; Zhang, L.; Chen, S.; Deniz, K.; Huryn, J.M.; Kao, Y.C.; Huang, S.C.; Singer, S.; Tap, W.; et al. Sarcomas with CIC-Rearrangements Are a Distinct Pathologic Entity with Aggressive Outcome: A Clinicopathologic and Molecular Study of 115 Cases. Am. J. Surg. Pathol. 2017, 41, 941. [Google Scholar] [CrossRef]
- Liao, S.; Davoli, T.; Leng, Y.; Li, M.Z.; Xu, Q.; Elledge, S.J. A Genetic Interaction Analysis Identifies Cancer Drivers That Modify EGFR Dependency. Genes Dev. 2017, 31, 184–196. [Google Scholar] [CrossRef]
- Wang, B.; Krall, E.B.; Aguirre, A.J.; Kim, M.; Widlund, H.R.; Doshi, M.B.; Sicinska, E.; Sulahian, R.; Goodale, A.; Cowley, G.S.; et al. ATXN1L, CIC, and ETS Transcription Factors Modulate Sensitivity to MAPK Pathway Inhibition. Cell Rep. 2017, 18, 1543–1557. [Google Scholar] [CrossRef]
- Hashiba, T.; Yamashita, T.; Okada, H.; Nio, K.; Hayashi, T.; Asahina, Y.; Hayashi, T.; Terashima, T.; Iida, N.; Takatori, H.; et al. Inactivation of Transcriptional Repressor Capicua Confers Sorafenib Resistance in Human Hepatocellular Carcinoma. Cell Mol. Gastroenterol. Hepatol. 2020, 10, 269–285. [Google Scholar] [CrossRef]
- Simón-Carrasco, L.; Jiménez, G.; Barbacid, M.; Drosten, M. The Capicua Tumor Suppressor: A Gatekeeper of Ras Signaling in Development and Cancer. Cell Cycle 2018, 17, 702–711. [Google Scholar] [CrossRef]
- Padul, V.; Epari, S.; Moiyadi, A.; Shetty, P.; Shirsat, N.V. ETV/Pea3 Family Transcription Factor-Encoding Genes Are Overexpressed in CIC-Mutant Oligodendrogliomas. Genes Chromosomes Cancer 2015, 54, 725–733. [Google Scholar] [CrossRef]
- LeBlanc, V.G.; Firme, M.; Song, J.; Chan, S.Y.; Lee, M.H.; Yip, S.; Chittaranjan, S.; Marra, M.A. Comparative Transcriptome Analysis of Isogenic Cell Line Models and Primary Cancers Links CIC Loss to Activation of the MAPK Signalling Cascade. J. Pathol. 2017, 242, 206–220. [Google Scholar] [CrossRef]
- Weissmann, S.; Cloos, P.A.; Sidoli, S.; Jensen, O.N.; Pollard, S.; Helin, K. The Tumor Suppressor CIC Directly Regulates MAPK Pathway Genes via Histone Deacetylation. Cancer Res. 2018, 78, 4114–4125. [Google Scholar] [CrossRef]
- Ren, Y.; Ouyang, Z.; Hou, Z.; Yan, Y.; Zhi, Z.; Shi, M.; Du, M.; Liu, H.; Wen, Y.; Shao, Y. CIC Is a Mediator of the ERK1/2-DUSP6 Negative Feedback Loop. iScience 2020, 23, 101635. [Google Scholar] [CrossRef]
- Lee, S.D.; Song, J.; LeBlanc, V.G.; Marra, M.A. Integrative Multi-Omic Analysis Reveals Neurodevelopmental Gene Dysregulation in CIC-Knockout and IDH1-Mutant Cells. J. Pathol. 2022, 256, 297–309. [Google Scholar] [CrossRef]
- Jimenez, G.; Shvartsman, S.Y.; Paroush, Z. The Capicua Repressor—A General Sensor of RTK Signaling in Development and Disease. J. Cell Sci. 2012, 125, 1383–1391. [Google Scholar] [CrossRef]
- Tseng, A.S.; Tapon, N.; Kanda, H.; Cigizoglu, S.; Edelmann, L.; Pellock, B.; White, K.; Hariharan, I.K. Capicua Regulates Cell Proliferation Downstream of the Receptor Tyrosine Kinase/Ras Signaling Pathway. Curr. Biol. 2007, 17, 728–733. [Google Scholar] [CrossRef]
- Jin, Y.; Ha, N.; Fores, M.; Xiang, J.; Glasser, C.; Maldera, J.; Jimenez, G.; Edgar, B.A. EGFR/Ras Signaling Controls Drosophila Intestinal Stem Cell Proliferation via Capicua-Regulated Genes. PLoS Genet. 2015, 11, e1005634. [Google Scholar] [CrossRef]
- Yang, R.; Chen, L.H.; Hansen, L.J.; Carpenter, A.B.; Moure, C.J.; Liu, H.; Pirozzi, C.J.; Diplas, B.H.; Waitkus, M.S.; Greer, P.K.; et al. Cic Loss Promotes Gliomagenesis via Aberrant Neural Stem Cell Proliferation and Differentiation. Cancer Res. 2017, 77, 6097–6108. [Google Scholar] [CrossRef]
- Wong, D.; Lounsbury, K.; Lum, A.; Song, J.; Chan, S.; LeBlanc, V.; Chittaranjan, S.; Marra, M.; Yip, S. Transcriptomic Analysis of CIC and ATXN1L Reveal a Functional Relationship Exploited by Cancer. Oncogene 2019, 38, 273–290. [Google Scholar] [CrossRef]
- Lee, Y. Regulation and Function of Capicua in Mammals. Exp. Mol. Med. 2020, 52, 531–537. [Google Scholar] [CrossRef]
- Lam, Y.C.; Bowman, A.B.; Jafar-Nejad, P.; Lim, J.; Richman, R.; Fryer, J.D.; Hyun, E.D.; Duvick, L.A.; Orr, H.T.; Botas, J.; et al. ATAXIN-1 Interacts with the Repressor Capicua in Its Native Complex to Cause SCA1 Neuropathology. Cell 2006, 127, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Pan, H.; Yao, J.; Elemento, O.; Zheng, H.; Paik, J. CIC Is a Critical Regulator of Neuronal Differentiation. JCI Insight 2020, 5, 135826. [Google Scholar] [CrossRef] [PubMed]
- VanderSluis, B.; Costanzo, M.; Billmann, M.; Ward, H.N.; Myers, C.L.; Andrews, B.J.; Boone, C. Integrating Genetic and Protein-Protein Interaction Networks Maps a Functional Wiring Diagram of a Cell. Curr. Opin. Microbiol. 2018, 45, 170–179. [Google Scholar] [CrossRef]
- Takemon, Y.; Chick, J.M.; Gerdes Gyuricza, I.; Skelly, D.A.; Devuyst, O.; Gygi, S.P.; Churchill, G.A.; Korstanje, R. Proteomic and Transcriptomic Profiling Reveal Different Aspects of Aging in the Kidney. eLife 2021, 10, e62585. [Google Scholar] [CrossRef]
- Shan, G.; Zhang, H.; Bi, G.; Bian, Y.; Liang, J.; Valeria, B.; Zeng, D.; Yao, G.; Zhan, C.; Fan, H. Multi-Omics Analysis of Cancer Cell Lines with High/Low Ferroptosis Scores and Development of a Ferroptosis-Related Model for Multiple Cancer Types. Front. Cell Dev. Biol. 2021, 9, 794475. [Google Scholar] [CrossRef]
- Chittaranjan, S.; Chan, S.; Yang, C.; Yang, K.C.; Chen, V.; Moradian, A.; Firme, M.; Song, J.; Go, N.E.; Blough, M.D.; et al. Mutations in CIC and IDH1 Cooperatively Regulate 2-Hydroxyglutarate Levels and Cell Clonogenicity. Oncotarget 2014, 5, 7960–7979. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2020. [Google Scholar]
- Lun, A.T.; Bach, K.; Marioni, J.C. Pooling across Cells to Normalize Single-Cell RNA Sequencing Data with Many Zero Counts. Genome Biol. 2016, 17, 75. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-Wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Schwabe, D.; Formichetti, S.; Junker, J.P.; Falcke, M.; Rajewsky, N. The Transcriptome Dynamics of Single Cells during the Cell Cycle. Mol. Syst. Biol. 2020, 16, e9946. [Google Scholar] [CrossRef]
- Whitfield, M.L.; Sherlock, G.; Saldanha, A.J.; Murray, J.I.; Ball, C.A.; Alexander, K.E.; Matese, J.C.; Perou, C.M.; Hurt, M.M.; Brown, P.O.; et al. Identification of Genes Periodically Expressed in the Human Cell Cycle and Their Expression in Tumors. Mol. Biol. Cell 2002, 13, 1977–2000. [Google Scholar] [CrossRef]
- Zheng, S.C.; Stein-O’Brien, G.; Augustin, J.J.; Slosberg, J.; Carosso, G.A.; Winer, B.; Shin, G.; Bjornsson, H.T.; Goff, L.A.; Hansen, K.D. Universal Prediction of Cell-Cycle Position Using Transfer Learning. Genome Biol. 2022, 23, 41. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Takemon, Y.; Marra, M.A. GRETA: An R Package for Mapping in Silico Genetic Interaction and Essentiality Networks. bioRxiv 2022. bioRxiv:2022-09. [Google Scholar] [CrossRef]
- DepMap, Broad (2020): DepMap 20Q1 Public. Figshare. Dataset. Available online: https://figshare.com/articles/dataset/DepMap_20Q1_Public/11791698/3 (accessed on 10 February 2020).
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational Correction of Copy Number Effect Improves Specificity of CRISPR-Cas9 Essentiality Screens in Cancer Cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Dempster, J.M.; Rossen, J.; Kazachkova, M.; Pan, J.; Kugener, G.; Root, D.E.; Tsherniak, A. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv 2019, 720243. [Google Scholar] [CrossRef]
- Obayashi, T.; Kagaya, Y.; Aoki, Y.; Tadaka, S.; Kinoshita, K. COXPRESdb v7: A Gene Coexpression Database for 11 Animal Species Supported by 23 Coexpression Platforms for Technical Evaluation and Evolutionary Inference. Nucleic Acids Res. 2019, 47, D55–D62. [Google Scholar] [CrossRef]
- Pan, J.; Meyers, R.M.; Michel, B.C.; Mashtalir, N.; Sizemore, A.E.; Wells, J.N.; Cassel, S.H.; Vazquez, F.; Weir, B.A.; Hahn, W.C.; et al. Interrogation of Mammalian Protein Complex Structure, Function, and Membership Using Genome-Scale Fitness Screens. Cell Syst. 2018, 6, 555–568.e7. [Google Scholar] [CrossRef]
- Wainberg, M.; Kamber, R.A.; Balsubramani, A.; Meyers, R.M.; Sinnott-Armstrong, N.; Hornburg, D.; Jiang, L.; Chan, J.; Jian, R.; Gu, M.; et al. A Genome-Wide Atlas of Co-Essential Modules Assigns Function to Uncharacterized Genes. Nat. Genet. 2021, 53, 638–649. [Google Scholar] [CrossRef]
- Gatchalian, J.; Malik, S.; Ho, J.; Lee, D.-S.; Kelso, T.W.R.; Shokhirev, M.N.; Dixon, J.R.; Hargreaves, D.C. A Non-Canonical BRD9-Containing BAF Chromatin Remodeling Complex Regulates Naive Pluripotency in Mouse Embryonic Stem Cells. Nat. Commun. 2018, 9, 5139. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-Based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; He, Q.-Y. ChIPseeker: An R/Bioconductor Package for ChIP Peak Annotation, Comparison and Visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, M.; Wu, T.; Zhan, L.; Li, L.; Chen, M.; Xie, W.; Xie, Z.; Hu, E.; Xu, S.; et al. Exploring Epigenomic Datasets by ChIPseeker. Curr. Protoc. 2022, 2, e585. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pages, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.J.; Gazin, C.; Lawson, N.D.; Pagès, H.; Lin, S.M.; Lapointe, D.S.; Green, M.R. ChIPpeakAnno: A Bioconductor Package to Annotate ChIP-Seq and ChIP-Chip Data. BMC Bioinform. 2010, 11, 237. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.J. Integrative Analysis of ChIP-Chip and ChIP-Seq Dataset. In Tiling Arrays: Methods and Protocols; Lee, T.-L., Shui Luk, A.C., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 105–124. ISBN 9781627036078. [Google Scholar]
- Sterne-Weiler, T.; Weatheritt, R.J.; Best, A.J.; Ha, K.C.H.; Blencowe, B.J. Efficient and Accurate Quantitative Profiling of Alternative Splicing Patterns of Any Complexity on a Laptop. Mol. Cell 2018, 72, 187–200.e6. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-Performance Genomics Data Visualization and Exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. DeepTools2: A next Generation Web Server for Deep-Sequencing Data Analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Michel, B.C.; D’Avino, A.R.; Cassel, S.H.; Mashtalir, N.; McKenzie, Z.M.; McBride, M.J.; Valencia, A.M.; Zhou, Q.; Bocker, M.; Soares, L.M.M.; et al. A Non-Canonical SWI/SNF Complex Is a Synthetic Lethal Target in Cancers Driven by BAF Complex Perturbation. Nat. Cell Biol. 2018, 20, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- van Dam, S.; Võsa, U.; van der Graaf, A.; Franke, L.; de Magalhães, J.P. Gene Co-Expression Analysis for Functional Classification and Gene-Disease Predictions. Brief. Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, N.R.; Van Der Niet, S.; Chertkova, A.; Postma, M.; Hordijk, P.L.; Gadella, T.W.J., Jr.; Goedhart, J. Identification of Guanine Nucleotide Exchange Factors That Increase Cdc42 Activity in Primary Human Endothelial Cells. Small GTPases 2021, 12, 226–240. [Google Scholar] [CrossRef]
- Chou, M.M.; Masuda-Robens, J.M.; Gupta, M.L. Cdc42 Promotes G1 Progression through P70 S6 Kinase-Mediated Induction of Cyclin E Expression. J. Biol. Chem. 2003, 278, 35241–35247. [Google Scholar] [CrossRef]
- Yasuda, S.; Taniguchi, H.; Oceguera-Yanez, F.; Ando, Y.; Watanabe, S.; Monypenny, J.; Narumiya, S. An Essential Role of Cdc42-like GTPases in Mitosis of HeLa Cells. FEBS Lett. 2006, 580, 3375–3380. [Google Scholar] [CrossRef]
- Le Gallic, L.; Virgilio, L.; Cohen, P.; Biteau, B.; Mavrothalassitis, G. ERF Nuclear Shuttling, a Continuous Monitor of Erk Activity That Links It to Cell Cycle Progression. Mol. Cell. Biol. 2004, 24, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Twigg, S.R.F.; Vorgia, E.; McGowan, S.J.; Peraki, I.; Fenwick, A.L.; Sharma, V.P.; Allegra, M.; Zaragkoulias, A.; Akha, E.S.; Knight, S.J.L.; et al. Reduced Dosage of ERF Causes Complex Craniosynostosis in Humans and Mice and Links ERK1/2 Signaling to Regulation of Osteogenesis. Nat. Genet. 2013, 45, 308–313. [Google Scholar] [CrossRef]
- Singh, R.; Cohen, A.S.A.; Poulton, C.; Hjortshøj, T.D.; Akahira-Azuma, M.; Mendiratta, G.; Khan, W.A.; Azmanov, D.N.; Woodward, K.J.; Kirchhoff, M.; et al. Deletion of ERF and CIC Causes Abnormal Skull Morphology and Global Developmental Delay. Cold Spring Harb. Mol. Case Stud. 2021, 7, a005991. [Google Scholar] [CrossRef]
- Gupta, N.; Song, H.; Wu, W.; Ponce, R.K.; Lin, Y.K.; Kim, J.W.; Small, E.J.; Feng, F.Y.; Huang, F.W.; Okimoto, R.A. The CIC-ERF Co-Deletion Underlies Fusion-Independent Activation of ETS Family Member, ETV1, to Drive Prostate Cancer Progression. eLife 2022, 11, e77072. [Google Scholar] [CrossRef]
- Alpsoy, A.; Dykhuizen, E.C. Glioma Tumor Suppressor Candidate Region Gene 1 (GLTSCR1) and Its Paralog GLTSCR1-like Form SWI/SNF Chromatin Remodeling Subcomplexes. J. Biol. Chem. 2018, 293, 3892–3903. [Google Scholar] [CrossRef]
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-J.; Pan, W.-W.; Liu, S.-B.; Shen, Z.-F.; Xu, Y.; Hu, L.-L. ERK/MAPK Signalling Pathway and Tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 Network Regulates Autophagy and Apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Frémont, S.; Gérard, A.; Galloux, M.; Janvier, K.; Karess, R.E.; Berlioz-Torrent, C. Beclin-1 Is Required for Chromosome Congression and Proper Outer Kinetochore Assembly. EMBO Rep. 2013, 14, 364–372. [Google Scholar] [CrossRef]
- Deng, M.; Wang, N.; Li, Z.; Chen, R.; Duan, J.; Peng, Y.; Wu, Z.; Zhang, Z.; Jiang, L.; Zheng, X.; et al. FXR1 Can Bind with the CFIm25/CFIm68 Complex and Promote the Progression of Urothelial Carcinoma of the Bladder by Stabilizing TRAF1 MRNA. Cell Death Dis. 2022, 13, 170. [Google Scholar] [CrossRef]
- George, J.; Li, Y.; Kadamberi, I.P.; Parashar, D.; Tsaih, S.-W.; Gupta, P.; Geethadevi, A.; Chen, C.; Ghosh, C.; Sun, Y.; et al. RNA-Binding Protein FXR1 Drives CMYC Translation by Recruiting EIF4F Complex to the Translation Start Site. Cell Rep. 2021, 37, 109934. [Google Scholar] [CrossRef]
- Davidovic, L.; Durand, N.; Khalfallah, O.; Tabet, R.; Barbry, P.; Mari, B.; Sacconi, S.; Moine, H.; Bardoni, B. A Novel Role for the RNA-Binding Protein FXR1P in Myoblasts Cell-Cycle Progression by Modulating P21/Cdkn1a/Cip1/Waf1 MRNA Stability. PLoS Genet. 2013, 9, e1003367. [Google Scholar] [CrossRef]
- Cui, Z.; Shen, Y.; Chen, K.H.; Mittal, S.K.; Yang, J.-Y.; Zhang, G. KANK1 Inhibits Cell Growth by Inducing Apoptosis through Regulating CXXC5 in Human Malignant Peripheral Nerve Sheath Tumors. Sci. Rep. 2017, 7, 40325. [Google Scholar] [CrossRef]
- Chen, N.-P.; Sun, Z.; Fässler, R. The Kank Family Proteins in Adhesion Dynamics. Curr. Opin. Cell Biol. 2018, 54, 130–136. [Google Scholar] [CrossRef]
- Benatti, P.; Dolfini, D.; Viganò, A.; Ravo, M.; Weisz, A.; Imbriano, C. Specific Inhibition of NF-Y Subunits Triggers Different Cell Proliferation Defects. Nucleic Acids Res. 2011, 39, 5356–5368. [Google Scholar] [CrossRef]
- Moeinvaziri, F.; Shahhoseini, M. Epigenetic Role of CCAAT Box-Binding Transcription Factor NF-Y on ID Gene Family in Human Embryonic Carcinoma Cells. IUBMB Life 2015, 67, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Y.; Wang, Q.; Su, B.; Xu, H.; Sun, Y.; Sun, P.; Li, R.; Peng, X.; Cai, J. Role of RASA1 in Cancer: A Review and Update. Oncol. Rep. 2020, 44, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- Jongsma, M.L.M.; de Waard, A.A.; Raaben, M.; Zhang, T.; Cabukusta, B.; Platzer, R.; Blomen, V.A.; Xagara, A.; Verkerk, T.; Bliss, S.; et al. The SPPL3-Defined Glycosphingolipid Repertoire Orchestrates HLA Class I-Mediated Immune Responses. Immunity 2021, 54, 132–150.e9. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, S.; Lee, C.G.; Park, G.Y.; Hong, H.; Lee, J.S.; Kim, Y.M.; Lee, S.B.; Hwang, D.; Choi, Y.S.; et al. Capicua Deficiency Induces Autoimmunity and Promotes Follicular Helper T Cell Differentiation via Derepression of ETV5. Nat. Commun. 2017, 8, 16037. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Park, J.; Kim, E.; Lee, Y. The Capicua-ETV5 Axis Regulates Liver-Resident Memory CD8(+) T Cell Development and the Pathogenesis of Liver Injury. Hepatology 2019, 391, 806–811. [Google Scholar] [CrossRef]
- Park, G.-Y.; Lee, G.-W.; Kim, S.; Hong, H.; Park, J.S.; Cho, J.-H.; Lee, Y. Deletion Timing of Cic Alleles during Hematopoiesis Determines the Degree of Peripheral CD4+ T Cell Activation and Proliferation. Immune Netw. 2020, 20, e43. [Google Scholar] [CrossRef]
- Kim, S.; Park, G.-Y.; Park, J.S.; Park, J.; Hong, H.; Lee, Y. Regulation of Positive and Negative Selection and TCR Signaling during Thymic T Cell Development by Capicua. eLife 2021, 10, e71769. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Q.; Hu, X.; Feng, D.; Xiang, S.; He, Z.; Hu, X.; Zhou, J.; Ding, X.; Zhou, C.; et al. Human ZCCHC12 Activates AP-1 and CREB Signaling as a Transcriptional Co-Activator. Acta Biochim. Biophys. Sin. 2009, 41, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-Generation Characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Nusinow, D.P.; Szpyt, J.; Ghandi, M.; Rose, C.M.; McDonald, E.R., 3rd; Kalocsay, M.; Jané-Valbuena, J.; Gelfand, E.; Schweppe, D.K.; Jedrychowski, M.; et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 2020, 180, 387–402.e16. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of Cancer Therapeutic Targets Using CRISPR–Cas9 Screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M. Synthetic Lethality: General Principles, Utility and Detection Using Genetic Screens in Human Cells. FEBS Lett. 2011, 585, 1–6. [Google Scholar] [CrossRef]
- Van Leeuwen, J.; Boone, C.; Andrews, B.J. Mapping a Diversity of Genetic Interactions in Yeast. Curr. Opin. Syst. Biol. 2017, 6, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Mashtalir, N.; D’Avino, A.R.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; St Pierre, R.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288.e20. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, Y.; Ozawa, T.; Hirose, Y.; Aldape, K.D.; McMahon, M.; Berger, M.S.; Pieper, R.O. Formation of Intracranial Tumors by Genetically Modified Human Astrocytes Defines Four Pathways Critical in the Development of Human Anaplastic Astrocytoma. Cancer Res. 2001, 61, 4956–4960. [Google Scholar]
- Erickson, B.K.; Rose, C.M.; Braun, C.R.; Erickson, A.R.; Knott, J.; McAlister, G.C.; Wühr, M.; Paulo, J.A.; Everley, R.A.; Gygi, S.P. A Strategy to Combine Sample Multiplexing with Targeted Proteomics Assays for High-Throughput Protein Signature Characterization. Mol. Cell 2017, 65, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Hasskamp, P.; Schmedding, I.; Morandell, S.; van Vugt, M.A.T.M.; Wang, X.; Linding, R.; Ong, S.-E.; Weaver, D.; Carr, S.A.; et al. DNA Damage Activates a Spatially Distinct Late Cytoplasmic Cell-Cycle Checkpoint Network Controlled by MK2-Mediated RNA Stabilization. Mol. Cell 2010, 40, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Mittal, P.; Roberts, C.W.M. The SWI/SNF Complex in Cancer—Biology, Biomarkers and Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef]
- Gañez-Zapater, A.; Mackowiak, S.D.; Guo, Y.; Tarbier, M.; Jordán-Pla, A.; Friedländer, M.R.; Visa, N.; Östlund Farrants, A.-K. The SWI/SNF Subunit BRG1 Affects Alternative Splicing by Changing RNA Binding Factor Interactions with Nascent RNA. Mol. Genet. Genom. 2022, 297, 463–484. [Google Scholar] [CrossRef]
- Lin, J.-S.; Lai, E.-M. Protein–Protein Interactions: Co-Immunoprecipitation. In Bacterial Protein Secretion Systems: Methods and Protocols; Journet, L., Cascales, E., Eds.; Springer: New York, NY, USA, 2017; pp. 211–219. ISBN 9781493970339. [Google Scholar]
- Söderberg, O.; Gullberg, M.; Jarvius, M.; Ridderstråle, K.; Leuchowius, K.-J.; Jarvius, J.; Wester, K.; Hydbring, P.; Bahram, F.; Larsson, L.-G.; et al. Direct Observation of Individual Endogenous Protein Complexes in Situ by Proximity Ligation. Nat. Methods 2006, 3, 995–1000. [Google Scholar] [CrossRef]
- Post, G.R.; Dawson, G. Characterization of a Cell Line Derived from a Human Oligodendroglioma. Mol. Chem. Neuropathol. 1992, 16, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.T.; Rogers, A.D.; Chen, M.J.; Dixit, R.; Adnani, L.; Frankiw, L.S.; Lawn, S.O.; Blough, M.D.; Alshehri, M.; Wu, W.; et al. Capicua Regulates Neural Stem Cell Proliferation and Lineage Specification through Control of Ets Factors. Nat. Commun. 2019, 10, 2000. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S. Rethinking Synchronization of Mammalian Cells for Cell Cycle Analysis. Cell. Mol. Life Sci. 2003, 60, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.J.; Tung, P.; Blischak, J.D.; Burnett, J.E.; Barr, K.A.; Dey, K.K.; Stephens, M.; Gilad, Y. Characterizing and Inferring Quantitative Cell Cycle Phase in Single-Cell RNA-Seq Data Analysis. Genome Res. 2020, 30, 611–621. [Google Scholar] [CrossRef]
- Conte, A.; Valente, V.; Paladino, S.; Pierantoni, G.M. HIPK2 in Cancer Biology and Therapy: Recent Findings and Future Perspectives. Cell. Signal. 2023, 101, 110491. [Google Scholar] [CrossRef]
- Meister, G.; Landthaler, M.; Peters, L.; Chen, P.Y.; Urlaub, H.; Lührmann, R.; Tuschl, T. Identification of Novel Argonaute-Associated Proteins. Curr. Biol. 2005, 15, 2149–2155. [Google Scholar] [CrossRef]
- Lazzaretti, D.; Tournier, I.; Izaurralde, E. The C-Terminal Domains of Human TNRC6A, TNRC6B, and TNRC6C Silence Bound Transcripts Independently of Argonaute Proteins. RNA 2009, 15, 1059–1066. [Google Scholar] [CrossRef]
- Pong, S.K.; Gullerova, M. Noncanonical Functions of MicroRNA Pathway Enzymes—Drosha, DGCR8, Dicer and Ago Proteins. FEBS Lett. 2018, 592, 2973–2986. [Google Scholar] [CrossRef]
- Park, J.-W.; Kilic, O.; Deo, M.; Jimenez-Cowell, K.; Demirdizen, E.; Kim, H.; Turcan, Ş. CIC Reduces XCT/SLC7A11 Expression and Glutamate Release in Glioma. Acta Neuropathol. Commun. 2023, 11, 13. [Google Scholar] [CrossRef]
- Dissanayake, K.; Toth, R.; Blakey, J.; Olsson, O.; Campbell, D.G.; Prescott, A.R.; MacKintosh, C. ERK/P90(RSK)/14-3-3 Signalling Has an Impact on Expression of PEA3 Ets Transcription Factors via the Transcriptional Repressor Capicua. Biochem. J. 2011, 433, 515–525. [Google Scholar] [CrossRef]
- Gardino, A.K.; Yaffe, M.B. 14-3-3 Proteins as Signaling Integration Points for Cell Cycle Control and Apoptosis. Semin. Cell Dev. Biol. 2011, 22, 688–695. [Google Scholar] [CrossRef]
- Heath, E.M.; Chan, S.M.; Minden, M.D.; Murphy, T.; Shlush, L.I.; Schimmer, A.D. Biological and Clinical Consequences of NPM1 Mutations in AML. Leukemia 2017, 31, 798–807. [Google Scholar] [CrossRef]
- Owens, D.M.; Keyse, S.M. Differential Regulation of MAP Kinase Signalling by Dual-Specificity Protein Phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef]
- Sobolev, V.V.; Khashukoeva, A.Z.; Evina, O.E.; Geppe, N.A.; Chebysheva, S.N.; Korsunskaya, I.M.; Tchepourina, E.; Mezentsev, A. Role of the Transcription Factor FOSL1 in Organ Development and Tumorigenesis. Int. J. Mol. Sci. 2022, 23, 1521. [Google Scholar] [CrossRef]
- Chaudhari, H.G.; Cohen, B.A. Local Sequence Features That Influence AP-1 Cis-Regulatory Activity. Genome Res. 2018, 28, 171–181. [Google Scholar] [CrossRef]
- Kannan, M.B.; Solovieva, V.; Blank, V. The Small MAF Transcription Factors MAFF, MAFG and MAFK: Current Knowledge and Perspectives. Biochim. Biophys. Acta 2012, 1823, 1841–1846. [Google Scholar] [CrossRef]
- Katsuoka, F.; Yamamoto, M. Small Maf Proteins (MafF, MafG, MafK): History, Structure and Function. Gene 2016, 586, 197–205. [Google Scholar] [CrossRef]
- Amit, I.; Citri, A.; Shay, T.; Lu, Y.; Katz, M.; Zhang, F.; Tarcic, G.; Siwak, D.; Lahad, J.; Jacob-Hirsch, J.; et al. A Module of Negative Feedback Regulators Defines Growth Factor Signaling. Nat. Genet. 2007, 39, 503–512. [Google Scholar] [CrossRef]
- Hu, J.; Yao, H.; Gan, F.; Tokarski, A.; Wang, Y. Interaction of OKL38 and P53 in Regulating Mitochondrial Structure and Function. PLoS ONE 2012, 7, e43362. [Google Scholar] [CrossRef]
- Melé, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. Human Genomics. The Human Transcriptome across Tissues and Individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef]
- Georgakopoulos-Soares, I.; Parada, G.E.; Wong, H.Y.; Medhi, R.; Furlan, G.; Munita, R.; Miska, E.A.; Kwok, C.K.; Hemberg, M. Alternative Splicing Modulation by G-Quadruplexes. Nat. Commun. 2022, 13, 2404. [Google Scholar] [CrossRef]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-Damage Response by the RNF8 Ubiquitin Ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef]
- Hughes, R.; Gilley, J.; Kristiansen, M.; Ham, J. The MEK-ERK Pathway Negatively Regulates Bim Expression through the 3′ UTR in Sympathetic Neurons. BMC Neurosci. 2011, 12, 69. [Google Scholar] [CrossRef]
- Moore, M.J. From Birth to Death: The Complex Lives of Eukaryotic MRNAs. Science 2005, 309, 1514–1518. [Google Scholar] [CrossRef]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating Cells Express MRNAs with Shortened 3′ Untranslated Regions and Fewer MicroRNA Target Sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef]
- Cenik, C.; Derti, A.; Mellor, J.C.; Berriz, G.F.; Roth, F.P. Genome-Wide Functional Analysis of Human 5′ Untranslated Region Introns. Genome Biol. 2010, 11, R29. [Google Scholar] [CrossRef]
- Grabski, D.F.; Broseus, L.; Kumari, B.; Rekosh, D.; Hammarskjold, M.-L.; Ritchie, W. Intron Retention and Its Impact on Gene Expression and Protein Diversity: A Review and a Practical Guide. Wiley Interdiscip. Rev. RNA 2021, 12, e1631. [Google Scholar] [CrossRef]
- Li, N.; Fassl, A.; Chick, J.; Inuzuka, H.; Li, X.; Mansour, M.R.; Liu, L.; Wang, H.; King, B.; Shaik, S.; et al. Cyclin C Is a Haploinsufficient Tumour Suppressor. Nat. Cell Biol. 2014, 16, 1080–1091. [Google Scholar] [CrossRef]
- Chen, Q.; Fu, Q.; Pu, L.; Liu, X.; Liu, Y. Effects of HMGA2 Gene Silencing on Cell Cycle and Apoptosis in the Metastatic Renal Carcinoma Cell Line ACHN. J. Int. Med. Res. 2022, 50, 3000605221075511. [Google Scholar] [CrossRef]
- Wang, H.-X.; Qin, X.-H.; Shen, J.; Liu, Q.-H.; Shi, Y.-B.; Xue, L. Proteomic Analysis Reveals That Placenta-Specific Protein 9 Inhibits Proliferation and Stimulates Motility of Human Bronchial Epithelial Cells. Front. Oncol. 2021, 11, 628480. [Google Scholar] [CrossRef]
- Fernandez-Cuesta, L.; Thomas, R.K. Molecular Pathways: Targeting NRG1 Fusions in Lung Cancer. Clin. Cancer Res. 2015, 21, 1989–1994. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Xiong, W.-C. Neuregulin 1 in Neural Development, Synaptic Plasticity and Schizophrenia. Nat. Rev. Neurosci. 2008, 9, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Nave, K.-A. Neuregulin-ERBB Signaling in the Nervous System and Neuropsychiatric Diseases. Neuron 2014, 83, 27–49. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 Extraterminal Domain Confers Transcription Activation Independent of PTEFb by Recruiting Multiple Proteins, Including NSD3. Mol. Cell. Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [PubMed]
- Innis, S.M.; Cabot, B. GBAF, a Small BAF Sub-Complex with Big Implications: A Systematic Review. Epigen. Chromatin 2020, 13, 48. [Google Scholar] [CrossRef]
- Coisy-Quivy, M.; Disson, O.; Roure, V.; Muchardt, C.; Blanchard, J.-M.; Dantonel, J.-C. Role for Brm in Cell Growth Control. Cancer Res. 2006, 66, 5069–5076. [Google Scholar] [CrossRef]
- Dykhuizen, E.C.; Hargreaves, D.C.; Miller, E.L.; Cui, K.; Korshunov, A.; Kool, M.; Pfister, S.; Cho, Y.J.; Zhao, K.; Crabtree, G.R. BAF Complexes Facilitate Decatenation of DNA by Topoisomerase IIalpha. Nature 2013, 497, 624–627. [Google Scholar] [CrossRef]
- Brownlee, P.M.; Chambers, A.L.; Cloney, R.; Bianchi, A.; Downs, J.A. BAF180 Promotes Cohesion and Prevents Genome Instability and Aneuploidy. Cell Rep. 2014, 6, 973–981. [Google Scholar] [CrossRef]
- Xue, Y.; Canman, J.C.; Lee, C.S.; Nie, Z.; Yang, D.; Moreno, G.T.; Young, M.K.; Salmon, E.D.; Wang, W. The Human SWI/SNF-B Chromatin-Remodeling Complex Is Related to Yeast Rsc and Localizes at Kinetochores of Mitotic Chromosomes. Proc. Natl. Acad. Sci. USA 2000, 97, 13015–13020. [Google Scholar] [CrossRef]
- You, S.Y.; Park, Y.S.; Jeon, H.-J.; Cho, D.-H.; Jeon, H.B.; Kim, S.H.; Chang, J.W.; Kim, J.-S.; Oh, J.S. Beclin-1 Knockdown Shows Abscission Failure but Not Autophagy Defect during Oocyte Meiotic Maturation. Cell Cycle 2016, 15, 1611–1619. [Google Scholar] [CrossRef]
- Henkel, L.; Rauscher, B.; Boutros, M. Context-Dependent Genetic Interactions in Cancer. Curr. Opin. Genet. Dev. 2019, 54, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Zhao, J.; Wang, Y.; Lu, W.; Liu, Z.; Zhou, Y.; Martin, W.R.; Wang, R.; Huang, J.; Hao, T.; et al. Comprehensive Characterization of Protein–Protein Interactions Perturbed by Disease Mutations. Nat. Genet. 2021, 53, 166. [Google Scholar] [CrossRef] [PubMed]
- Zinman, G.E.; Zhong, S.; Bar-Joseph, Z. Biological Interaction Networks Are Conserved at the Module Level. BMC Syst. Biol. 2011, 5, 134. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-Based Protein Identification by Searching Sequence Databases Using Mass Spectrometry Data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT Protein Sequence Database and Its Supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Hughes, C.S.; Foehr, S.; Garfield, D.A.; Furlong, E.E.; Steinmetz, L.M.; Krijgsveld, J. Ultrasensitive Proteome Analysis Using Paramagnetic Bead Technology. Mol. Syst. Biol. 2014, 10, 757. [Google Scholar] [CrossRef]
- Hughes, C.S.; McConechy, M.K.; Cochrane, D.R.; Nazeran, T.; Karnezis, A.N.; Huntsman, D.G.; Morin, G.B. Quantitative Profiling of Single Formalin Fixed Tumour Sections: Proteomics for Translational Research. Sci. Rep. 2016, 6, 34949. [Google Scholar] [CrossRef]
- Zolg, D.P.; Wilhelm, M.; Schnatbaum, K.; Zerweck, J.; Knaute, T.; Delanghe, B.; Bailey, D.J.; Gessulat, S.; Ehrlich, H.-C.; Weininger, M.; et al. Building ProteomeTools Based on a Complete Synthetic Human Proteome. Nat. Methods 2017, 14, 259–262. [Google Scholar] [CrossRef]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and Go Extraction Tips for Matrix-Assisted Laser Desorption/Ionization, Nanoelectrospray, and LC/MS Sample Pretreatment in Proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef]
- Käll, L.; Canterbury, J.D.; Weston, J.; Noble, W.S.; MacCoss, M.J. Semi-Supervised Learning for Peptide Identification from Shotgun Proteomics Datasets. Nat. Methods 2007, 4, 923–925. [Google Scholar] [CrossRef]
- Spivak, M.; Weston, J.; Bottou, L.; Käll, L.; Noble, W.S. Improvements to the Percolator Algorithm for Peptide Identification from Shotgun Proteomics Data Sets. J. Proteome Res. 2009, 8, 3737–3745. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]





| Enriched Functions | Selected SL Interactors in Relevant Term(s) |
|---|---|
| Regulation of epithelial cell function (morphogenesis, differentiation, proliferation, and migration) | RREB1, GATA3, PAX8, CEBPB, and SP1 |
| Regulation of cell cycle processes and cell division | ANKRD17, CARM1, CSNK2A1, HAUS2, CDK10, BECN1, UBE2S, CKAP2, and KMT5A |
| Chromatin/nucleosome disassembly | ARID2, and NFE2 |
| Histone modification and chromatin organisation | BECN1, CARM1, GATA3, KMT5A, and RIF1 |
| Regulation of double-stranded break repair | ARID2, RIF1, and ATP23 |
| RNA splicing | LUC7L2, HNRNPA0, and RBM23 |
| Apoptotic processes | CEBPB, GATA3, ILK, NKX2-5, PAX8, and PRKC1 |
| Wnt signalling | FERMT2, ILK, GATA3, GNAQ, CSNK2A1, NKX2-5, and CALCOCO1 |
| Enriched Functions | High-Confidence Interactors |
|---|---|
| RNA splicing and ribosomal and messenger RNA (rRNA and mRNA, respectively) metabolism and processing | >30 interactors |
| DNA conformation change and duplex unwinding | DDX3X, G3BP1, HNRNPA2B1, HP1BP3, NPM1, RUVBL1, TOP1, TTN, and XRCC5/6 |
| Chromatin remodelling and nucleosome organisation | ACTB, HNRNPC, HP1BP3, NPM1, PBRM1, RUVBL1, SMARCA2/A4/A5/C1/C2, TOP1, and TRIM28 |
| Chromatin organisation and epigenetic regulation of gene expression | ACTB, DDX21, FMR1, GLYR1, HNRNPU, MECP2, MKI67, SIN3A, SMARCA5, SNW1, TRIM28, and UPF1 |
| Telomere organisation and maintenance | DKC1, GAR1, GNL3, HNRNPA1/A2B1/C/D/U, NAT10, UPF1, XRCC5/6, and YLPM1 |
| Intrinsic apoptotic signalling | DDX3X, DDX5, DNM1L, HNRNPK, NONO, PRKDC, RPL11, SFPQ, and SNW1 |
| Gene(s) | Function(s) |
|---|---|
| ETV4 and ETV5 | Effectors of MAPK signalling. Known targets of CIC-mediated transcriptional regulation in multiple cellular contexts [19] |
| DUSP4 and DUSP5 | Negative feedback regulators of MAPK signalling [117]. Proposed direct targets of CIC in HEK cells, mouse embryonic stem cells, and developing mouse brains [21,22,32]. |
| FOSL1 | Proto-oncogene and member of the FOS gene family, which encodes a group of proteins that can dimerize with proteins of the JUN family to form the transcription factor AP-1. Activated by the RAS/MAPK signalling cascade through phosphorylation by ERK2, ERK5, and RSK2 [118]. Proposed direct target of CIC in mouse embryonic stem cells [22]. |
| MAFF | Member of the basic leucine zipper (bZIP) family of transcription factors. Can also participate in the formation and binding activity of the AP-1 complex [119]. Has been associated with a variety of functions, mostly related to the stress response. Its activity has been implicated in cancer [120,121]. Has been implicated as a feedback regulator of the MAPK signalling cascade, potentially downstream of AP-1 activity [122]. Proposed direct target of CIC in mouse embryonic stem cells [22]. |
| OSGIN1 | Involved in oxidative stress and DNA damage responses. Tumour suppressor that acts downstream of and in concert with p53 to induce mitochondrial cytochrome c release and apoptosis [123]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takemon, Y.; LeBlanc, V.G.; Song, J.; Chan, S.Y.; Lee, S.D.; Trinh, D.L.; Ahmad, S.T.; Brothers, W.R.; Corbett, R.D.; Gagliardi, A.; et al. Multi-Omic Analysis of CIC’s Functional Networks Reveals Novel Interaction Partners and a Potential Role in Mitotic Fidelity. Cancers 2023, 15, 2805. https://doi.org/10.3390/cancers15102805
Takemon Y, LeBlanc VG, Song J, Chan SY, Lee SD, Trinh DL, Ahmad ST, Brothers WR, Corbett RD, Gagliardi A, et al. Multi-Omic Analysis of CIC’s Functional Networks Reveals Novel Interaction Partners and a Potential Role in Mitotic Fidelity. Cancers. 2023; 15(10):2805. https://doi.org/10.3390/cancers15102805
Chicago/Turabian StyleTakemon, Yuka, Véronique G. LeBlanc, Jungeun Song, Susanna Y. Chan, Stephen Dongsoo Lee, Diane L. Trinh, Shiekh Tanveer Ahmad, William R. Brothers, Richard D. Corbett, Alessia Gagliardi, and et al. 2023. "Multi-Omic Analysis of CIC’s Functional Networks Reveals Novel Interaction Partners and a Potential Role in Mitotic Fidelity" Cancers 15, no. 10: 2805. https://doi.org/10.3390/cancers15102805
APA StyleTakemon, Y., LeBlanc, V. G., Song, J., Chan, S. Y., Lee, S. D., Trinh, D. L., Ahmad, S. T., Brothers, W. R., Corbett, R. D., Gagliardi, A., Moradian, A., Cairncross, J. G., Yip, S., Aparicio, S. A. J. R., Chan, J. A., Hughes, C. S., Morin, G. B., Gorski, S. M., Chittaranjan, S., & Marra, M. A. (2023). Multi-Omic Analysis of CIC’s Functional Networks Reveals Novel Interaction Partners and a Potential Role in Mitotic Fidelity. Cancers, 15(10), 2805. https://doi.org/10.3390/cancers15102805