
Voltage-Gated Sodium Channel NaV1.7 Inhibitors with Potent Anticancer Activities in Medullary Thyroid Cancer Cells

 ,
,  , ,
, ,  , , ,
, , , 
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Materials and Methods
2.1. Cell Culture
2.2. Human Tissue Samples
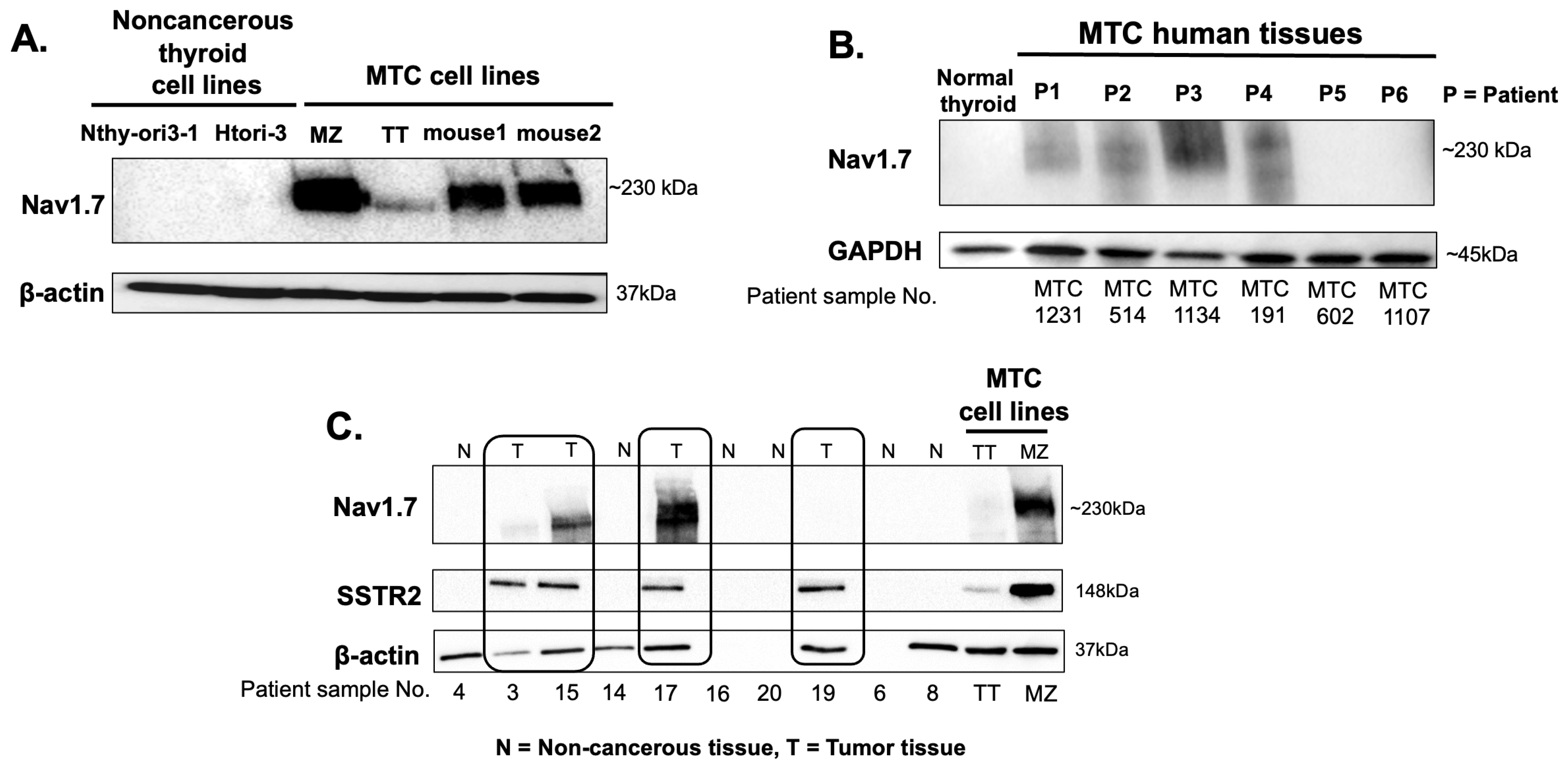
2.3. Western Blot Analysis
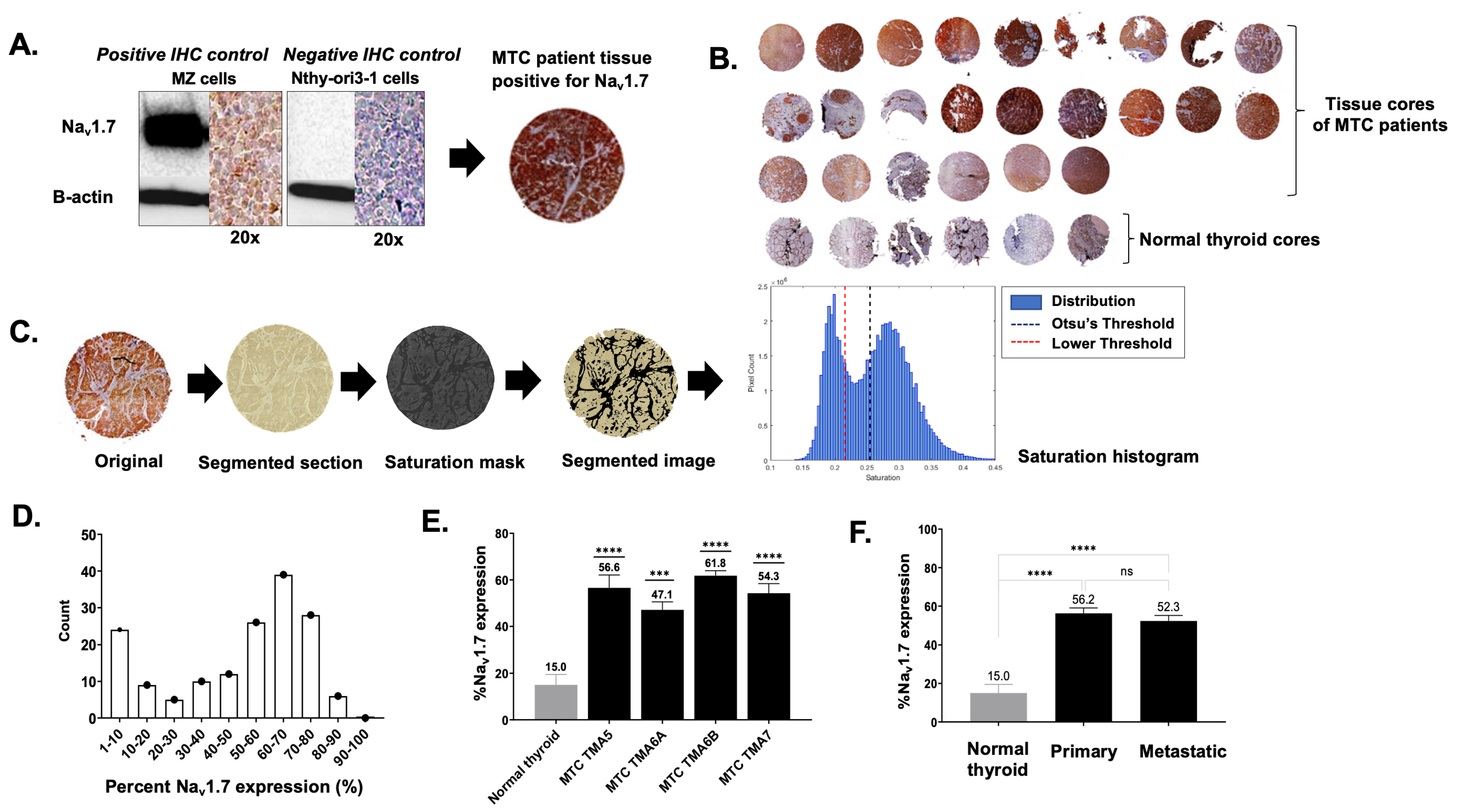
2.4. TMA Staining, Quantification, and Evaluation
2.5. Real-Time Quantitative PCR (RT-qPCR)
2.6. NaV1.7 Transfection
2.7. Electrophysiology
2.8. Cell Viability Assay
2.9. Motility Assays (Migration/Invasion)
2.10. Cell Cycle Analysis Flow Cytometry
2.11. Statistical Analysis
2.12. General Methods for Compound Synthesis and Characterization
2.12.1. 4,4-Diphenylbutyric Acid (2)
2.12.2. 3-(Piperidin-1-yl)propan-1-amine (5)
2.12.3. 4,4-Diphenyl-N-[3-(piperidin-1-yl)propyl]butanamide (6)
2.12.4. 4,4-Diphenylbutyl [3-(piperidin-1-yl)propyl]amine Hydrochloride (SV188)
2.12.5. 4,4-Diphenyl-N-(3-phenylpropyl)butanamide (8)
2.12.6. 4,4-Diphenylbutyl(3-phenylpropyl)amine Hydrochloride (WJB-133)
2.12.7. 4-(4-Fluorophenyl)butyl][3-(piperidin-1-yl)propyl Amine Hydrochloride (Compound 4)
3. Results and Discussion
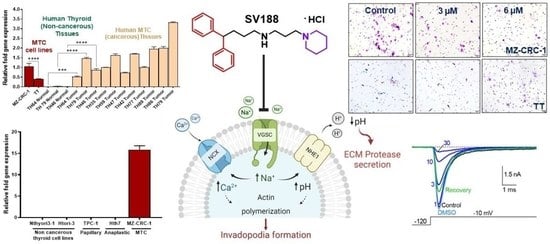
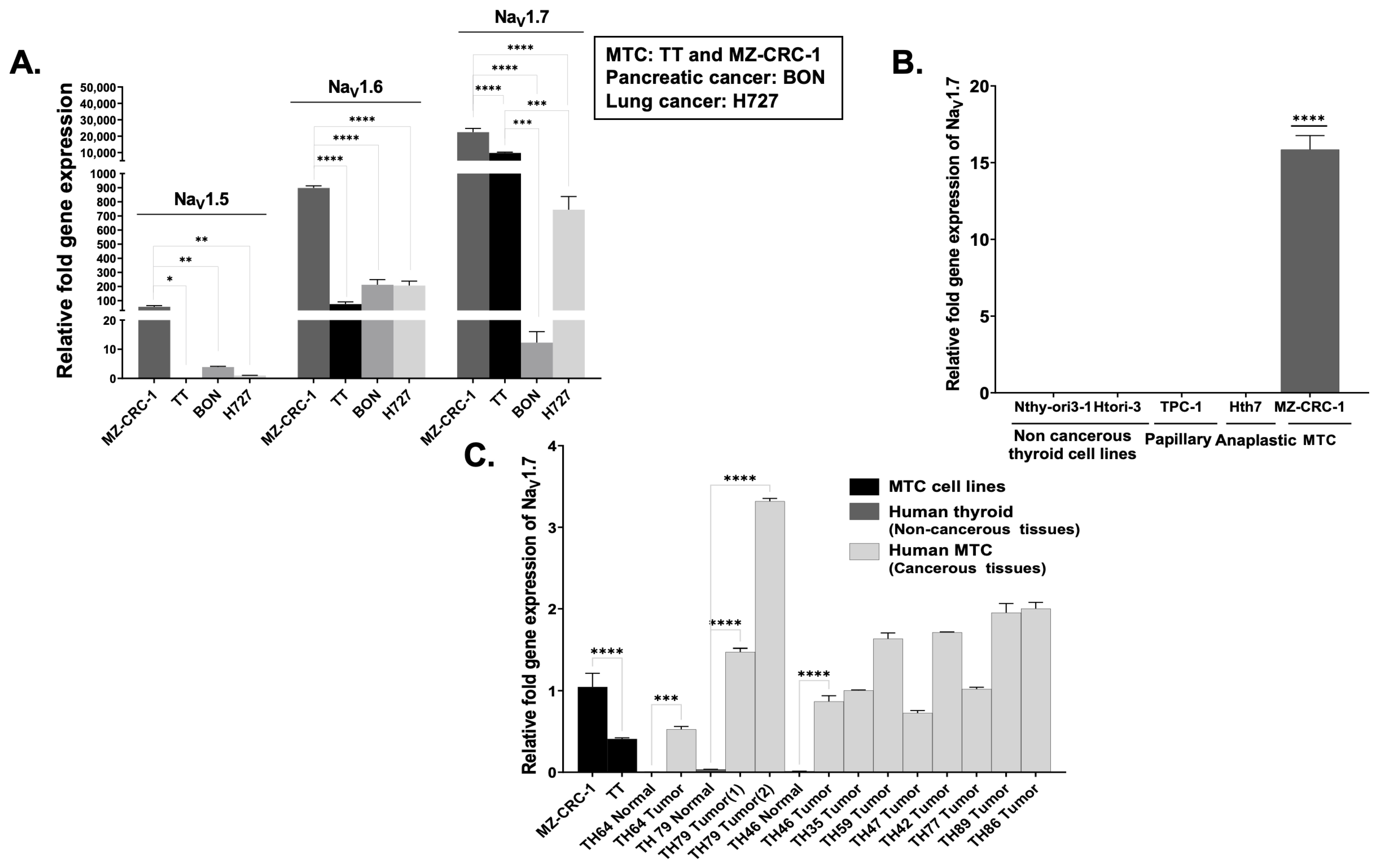
3.1. VGSC Expression in Neuroendocrine Tumors (NETs)
3.2. High-Throughput Analysis of NaV1.7 Expression in Human MTC
3.3. Identification of NaV1.7 Inhibitors
3.4. Synthesis of Compound 4, SV188 and WJB-133
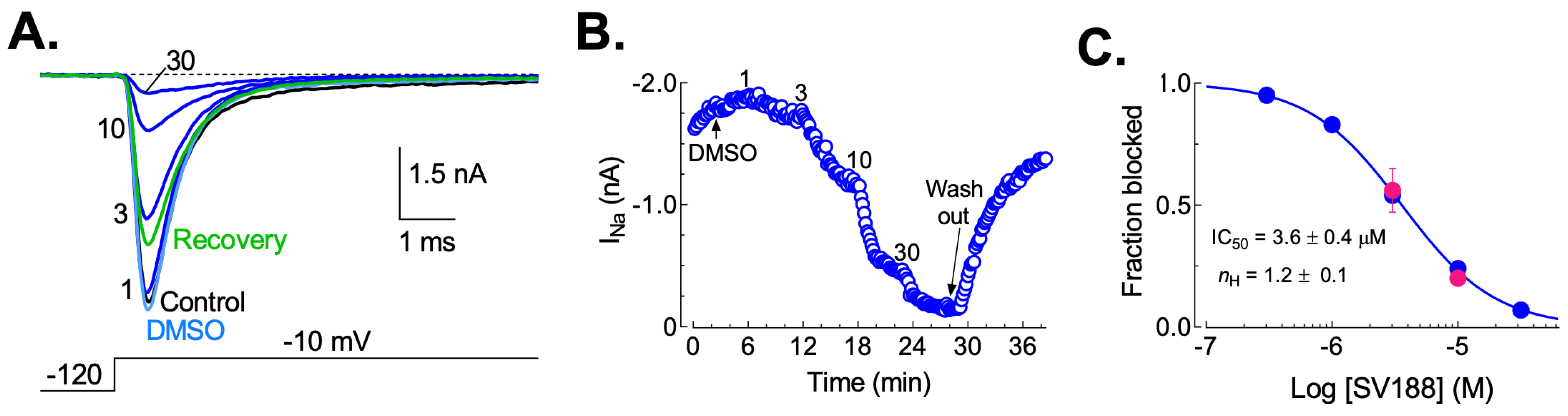
3.5. Dose-Dependent Inhibition of NaV1.7 Currents (INa) by SV188
3.6. Effects of SV188 on NaV1.7 Channels Gating
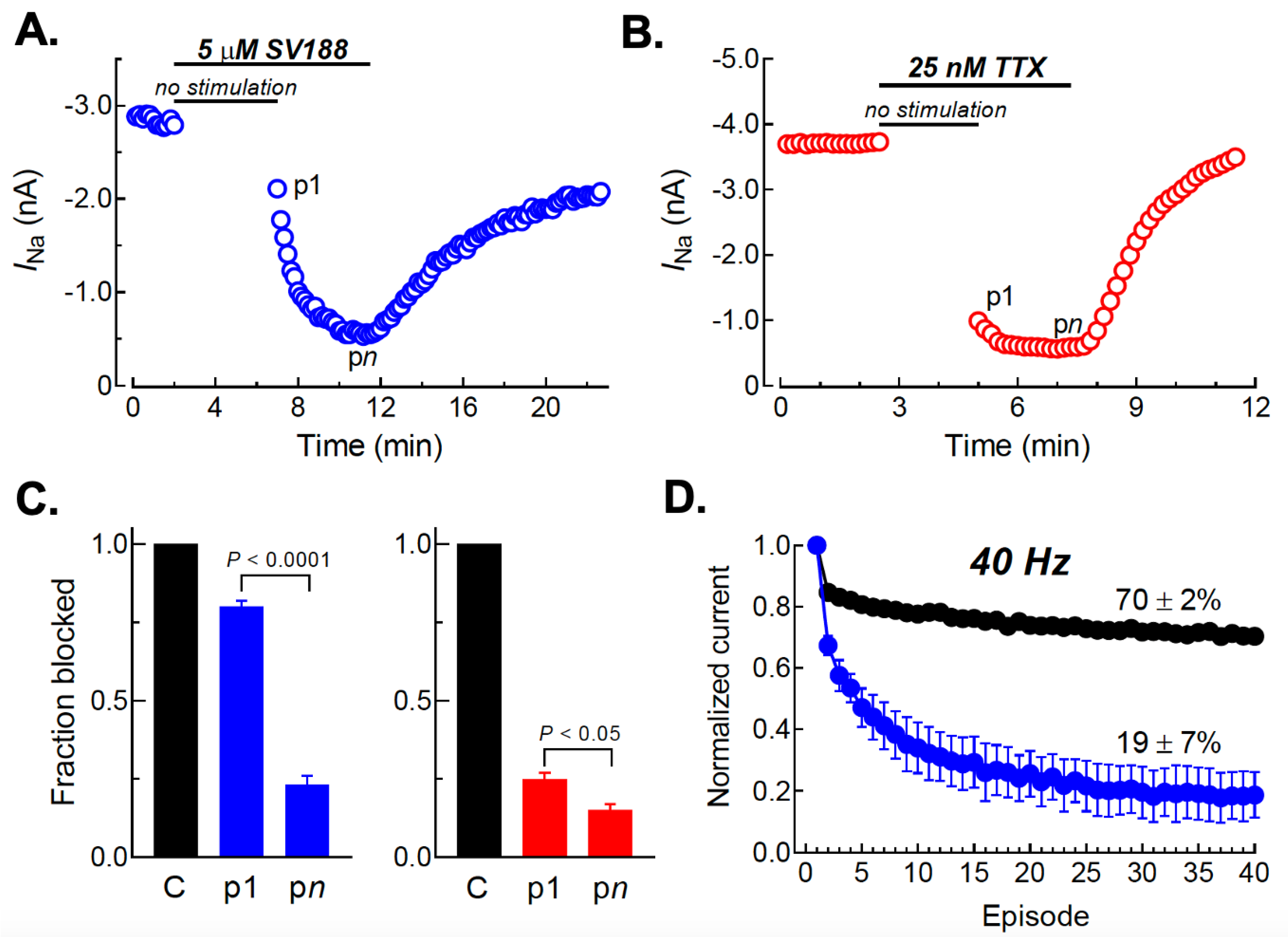
3.7. Use-Dependent Blockade of NaV1.7 Channels by SV188
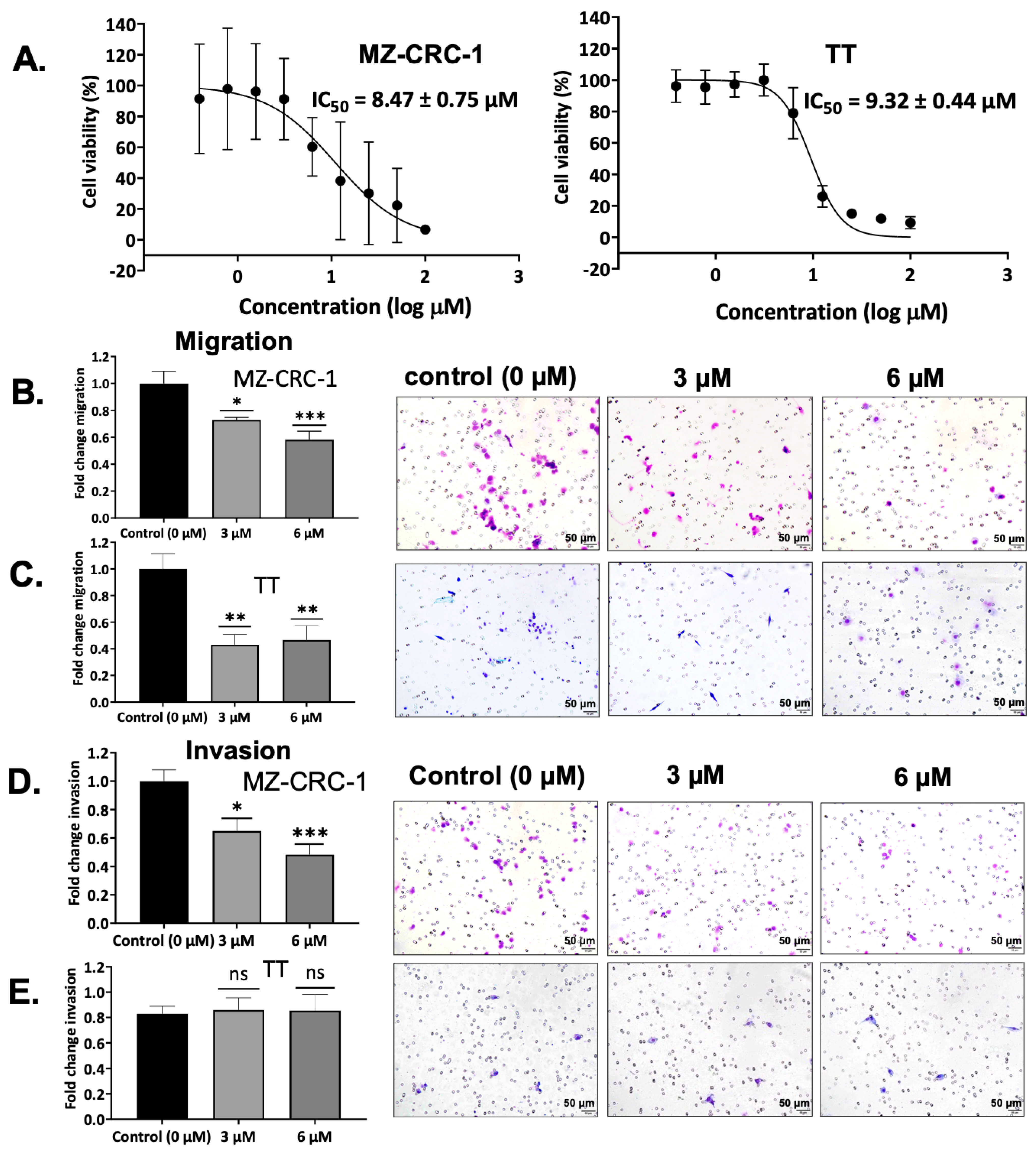
3.8. Effect on MTC Cell Viability by SV188
3.9. Effect on Cell Migration by SV188
3.10. Effect on Cell Invasion by SV188
3.11. Cell Cycle Analysis in Response to SV188 Treatment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Greenblatt, D.Y.; Elson, D.; Mack, E.; Chen, H. Initial lymph node dissection increases cure rates in patients with medullary thyroid cancer. Asian J. Surg. 2007, 30, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Sippel, R.S.; Kunnimalaiyaan, M.; Chen, H. Current management of medullary thyroid cancer. Oncologist 2008, 13, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sippel, R.S.; O’Dorisio, M.S.; Vinik, A.I.; Lloyd, R.V.; Pacak, K. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: Pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas 2010, 39, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Okafor, C.; Hogan, J.; Raygada, M.; Thomas, B.J.; Akshintala, S.; Glod, J.W.; Del Rivero, J. Update on Targeted Therapy in Medullary Thyroid Cancer. Front. Endocrinol. 2021, 12, 708949. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Chen, H.; Sippel, R.S. Current understanding and management of medullary thyroid cancer. Oncologist 2013, 18, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Carlomagno, F.; Baudin, E.; Bidart, J.M.; Santoro, M. New therapeutic approaches to treat medullary thyroid carcinoma. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 22–32. [Google Scholar] [CrossRef]
- Sandilos, G.; Lou, J.; Butchy, M.V.; Gaughan, J.P.; Reid, L.; Spitz, F.R.; Beninato, T.; Moore, M.D. Features of mixed medullary thyroid tumors: An NCDB analysis of clinicopathologic characteristics and survival. Am. J. Surg. 2023, in press. [Google Scholar] [CrossRef]
- Shepet, K.; Alhefdhi, A.; Lai, N.; Mazeh, H.; Sippel, R.; Chen, H. Hereditary medullary thyroid cancer: Age-appropriate thyroidectomy improves disease-free survival. Ann. Surg. Oncol. 2013, 20, 1451–1455. [Google Scholar] [CrossRef]
- Pajak, C.; Cadili, L.; Nabata, K.; Wiseman, S.M. 68Ga-DOTATATE-PET shows promise for diagnosis of recurrent or persistent medullary thyroid cancer: A systematic review. Am. J. Surg. 2022, 224, 670–675. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Habra, M.A. Lenvatinib: Role in thyroid cancer and other solid tumors. Cancer Treat. Rev. 2016, 42, 47–55. [Google Scholar] [CrossRef]
- Priya, S.R.; Dravid, C.S.; Digumarti, R.; Dandekar, M. Targeted Therapy for Medullary Thyroid Cancer: A Review. Front. Oncol. 2017, 7, 238. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cheng, L.; Sa, R.; Qiu, X.; Chen, L. Real-world insights into the efficacy and safety of tyrosine kinase inhibitors against thyroid cancers. Crit. Rev. Oncol. Hematol. 2022, 172, 103624. [Google Scholar] [CrossRef] [PubMed]
- Efstathiadou, Z.A.; Tsentidis, C.; Bargiota, A.; Daraki, V.; Kotsa, K.; Ntali, G.; Papanastasiou, L.; Tigas, S.; Toulis, K.; Pazaitou-Panayiotou, K.; et al. Benefits and Limitations of TKIs in Patients with Medullary Thyroid Cancer: A Systematic Review and Meta-Analysis. Eur. Thyroid J. 2021, 10, 125–139. [Google Scholar] [CrossRef]
- Roger, S.; Gillet, L.; Le Guennec, J.-Y.; Besson, P. Voltage-gated sodium channels and cancer: Is excitability their primary role? Front. Pharmacol. 2015, 6, 152. [Google Scholar] [CrossRef]
- Roger, S.; Potier, M.; Vandier, C.; Besson, P.; Le Guennec, J.Y. Voltage-gated sodium channels: New targets in cancer therapy? Curr. Pharm. Des. 2006, 12, 3681–3695. [Google Scholar] [CrossRef]
- Brackenbury, W.J. Voltage-gated sodium channels and metastatic disease. Channels 2012, 6, 352–361. [Google Scholar] [CrossRef]
- Lastraioli, E.; Iorio, J.; Arcangeli, A. Ion channel expression as promising cancer biomarker. Biochim. Biophys. Acta 2015, 1848, 2685–2702. [Google Scholar] [CrossRef] [PubMed]
- Hodgkin, A.L.; Huxley, A.F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952, 117, 500–544. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef]
- de Lera Ruiz, M.; Kraus, R.L. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 2015, 58, 7093–7118. [Google Scholar] [CrossRef]
- Quicke, P.; Sun, Y.; Arias-Garcia, M.; Beykou, M.; Acker, C.D.; Djamgoz, M.B.A.; Bakal, C.; Foust, A.J. Voltage imaging reveals the dynamic electrical signatures of human breast cancer cells. Commun. Biol. 2022, 5, 1178. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.; Elghajiji, A.; Fraser, S.P.; Burke, Z.D.; Tosh, D.; Djamgoz, M.B.A.; Rocha, P.R.F. Human Breast Cancer Cells Demonstrate Electrical Excitability. Front. Neurosci. 2020, 14, 404. [Google Scholar] [CrossRef] [PubMed]
- Miura, R.M. Analysis of excitable cell models. J. Comput. Appl. Math. 2002, 144, 29–47. [Google Scholar] [CrossRef]
- Sundelacruz, S.; Levin, M.; Kaplan, D.L. Role of Membrane Potential in the Regulation of Cell Proliferation and Differentiation. Stem Cell Rev. Rep. 2009, 5, 231–246. [Google Scholar] [CrossRef]
- Brackenbury, W.J.; Chioni, A.-M.; Diss, J.K.J.; Djamgoz, M.B.A. The neonatal splice variant of Nav1.5 potentiates in vitro invasive behaviour of MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 2007, 101, 149–160. [Google Scholar] [CrossRef]
- Yang, M.; Brackenbury, W.J. Membrane potential and cancer progression. Front. Physiol. 2013, 4, 185. [Google Scholar] [CrossRef]
- Nuccitelli, R. Endogenous electric fields in embryos during development, regeneration and wound healing. Radiat. Prot. Dosim. 2003, 106, 375–383. [Google Scholar] [CrossRef]
- Tokuoka, S.; Morioka, H. The membrane potential of the human cancer and related cells. Gann 1957, 48, 353–354. [Google Scholar]
- Djamgoz, M.B.A. Hyponatremia and Cancer Progression: Possible Association with Sodium-Transporting Proteins. Bioelectricity 2020, 2, 14–20. [Google Scholar] [CrossRef]
- Yu, H. Depolarization or hyperpolarization: Emerging role of altered bioelectricity in breast cancer metastasis. EBioMedicine 2022, 76, 103853. [Google Scholar] [CrossRef]
- James, A.D.; Leslie, T.K.; Kaggie, J.D.; Wiggins, L.; Patten, L.; Murphy O’Duinn, J.; Langer, S.; Labarthe, M.-C.; Riemer, F.; Baxter, G.; et al. Sodium accumulation in breast cancer predicts malignancy and treatment response. Br. J. Cancer 2022, 127, 337–349. [Google Scholar] [CrossRef]
- Lopez-Charcas, O.; Pukkanasut, P.; Velu, S.E.; Brackenbury, W.J.; Hales, T.G.; Besson, P.; Gomora, J.C.; Roger, S. Pharmacological and nutritional targeting of voltage-gated sodium channels in the treatment of cancers. iScience 2021, 24, 102270. [Google Scholar] [CrossRef] [PubMed]
- Angus, M.; Ruben, P. Voltage gated sodium channels in cancer and their potential mechanisms of action. Channels 2019, 13, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Huang, N.; Huang, H.; Sun, L.; Dong, S.; Su, J.; Zhang, J.; Wang, L.; Lin, L.; Shi, M.; et al. Voltage-gated sodium channel Nav1.7 promotes gastric cancer progression through MACC1-mediated upregulation of NHE1. Int. J. Cancer 2016, 139, 2553–2569. [Google Scholar] [CrossRef]
- Hossain, M.M.; Sonsalla, P.K.; Richardson, J.R. Coordinated role of voltage-gated sodium channels and the Na+/H+ exchanger in sustaining microglial activation during inflammation. Toxicol. Appl. Pharmacol. 2013, 273, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Besson, P.; Driffort, V.; Bon, É.; Gradek, F.; Chevalier, S.; Roger, S. How do voltage-gated sodium channels enhance migration and invasiveness in cancer cells? Biochim. Biophys. Acta 2015, 1848, 2493–2501. [Google Scholar] [CrossRef]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Gillet, L.; Roger, S.; Besson, P.; Lecaille, F.; Gore, J.; Bougnoux, P.; Lalmanach, G.; Le Guennec, J.-Y. Voltage-gated Sodium Channel Activity Promotes Cysteine Cathepsin-dependent Invasiveness and Colony Growth of Human Cancer Cells. J. Biol. Chem. 2009, 284, 8680–8691. [Google Scholar] [CrossRef]
- Pal, R.; Kumar, B.; Akhtar, M.J.; Chawla, P.A. Voltage gated sodium channel inhibitors as anticonvulsant drugs: A systematic review on recent developments and structure activity relationship studies. Bioorg. Chem. 2021, 115, 105230. [Google Scholar] [CrossRef]
- Dutta, S.; Lopez Charcas, O.; Tanner, S.; Gradek, F.; Driffort, V.; Roger, S.; Selander, K.; Velu, S.E.; Brouillette, W. Discovery and evaluation of nNav1.5 sodium channel blockers with potent cell invasion inhibitory activity in breast cancer cells. Bioorg. Med. Chem. 2018, 26, 2428–2436. [Google Scholar] [CrossRef]
- Djamgoz, M.B.A.; Fraser, S.P.; Brackenbury, W.J. In Vivo Evidence for Voltage-Gated Sodium Channel Expression in Carcinomas and Potentiation of Metastasis. Cancers 2019, 11, 1675. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Liu, D.; Wu, K.; Lei, J.; Yan, N. Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science 2019, 363, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Lenkey, N.; Karoly, R.; Lukacs, P.; Vizi, E.S.; Sunesen, M.; Fodor, L.; Mike, A. Classification of Drugs Based on Properties of Sodium Channel Inhibition: A Comparative Automated Patch-Clamp Study. PLoS ONE 2010, 5, e15568. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, D.S.; McPhee, J.C.; Scheuer, T.; Catterall, W.A. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 1994, 265, 1724–1728. [Google Scholar] [CrossRef]
- Benjamin, E.R.; Pruthi, F.; Olanrewaju, S.; Ilyin, V.I.; Crumley, G.; Kutlina, E.; Valenzano, K.J.; Woodward, R.M. State-dependent compound inhibition of Nav1.2 sodium channels using the FLIPR Vm dye: On-target and off-target effects of diverse pharmacological agents. J. Biomol. Screen 2006, 11, 29–39. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-Gated Sodium Channels: Structure, Function, and Pathophysiology. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Waltham, MA, USA, 2013; pp. 564–569. [Google Scholar]
- Chevrier, P.; Vijayaragavan, K.; Chahine, M. Differential modulation of Nav1.7 and Nav1.8 peripheral nerve sodium channels by the local anesthetic lidocaine. Br. J. Pharmacol. 2004, 142, 576–584. [Google Scholar] [CrossRef]
- Ragsdale, D.S.; McPhee, J.C.; Scheuer, T.; Catterall, W.A. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. USA 1996, 93, 9270–9275. [Google Scholar] [CrossRef]
- House, C.D.; Vaske, C.J.; Schwartz, A.M.; Obias, V.; Frank, B.; Luu, T.; Sarvazyan, N.; Irby, R.; Strausberg, R.L.; Hales, T.G.; et al. Voltage-gated Na+ channel SCN5A is a key regulator of a gene transcriptional network that controls colon cancer invasion. Cancer Res. 2010, 70, 6957–6967. [Google Scholar] [CrossRef]
- Fairhurst, C.; Martin, F.; Watt, I.; Doran, T.; Bland, M.; Brackenbury, W.J. Sodium channel-inhibiting drugs and cancer survival: Protocol for a cohort study using the CPRD primary care database. BMJ Open 2016, 6, e011661. [Google Scholar] [CrossRef]
- Driffort, V.; Gillet, L.; Bon, E.; Marionneau-Lambot, S.; Oullier, T.; Joulin, V.; Collin, C.; Pagès, J.C.; Jourdan, M.L.; Chevalier, S.; et al. Ranolazine inhibits NaV1.5-mediated breast cancer cell invasiveness and lung colonization. Mol. Cancer 2014, 13, 264. [Google Scholar] [CrossRef]
- Martin, F.; Ufodiama, C.; Watt, I.; Bland, M.; Brackenbury, W.J. Therapeutic Value of Voltage-Gated Sodium Channel Inhibitors in Breast, Colorectal, and Prostate Cancer: A Systematic Review. Front. Pharmacol. 2015, 6, 273. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, J.; Fan, N.; Wang, H.; Thomas, A.M.; Yan, Q.; Li, S.; Qin, H. Nav1.6 promotes the progression of human follicular thyroid carcinoma cells via JAK-STAT signaling pathway. Pathol. Res. Pract. 2022, 236, 153984. [Google Scholar] [CrossRef] [PubMed]
- Klugbauer, N.; Lacinova, L.; Flockerzi, V.; Hofmann, F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995, 14, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Charcas, O.; Poisson, L.; Benouna, O.; Lemoine, R.; Chadet, S.; Pétereau, A.; Lahlou, W.; Guyétant, S.; Ouaissi, M.; Pukkanasut, P.; et al. Voltage-Gated Sodium Channel NaV1.5 Controls NHE-1-Dependent Invasive Properties in Colon Cancer Cells. Cancers 2023, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Jaskula-Sztul, R.; Eide, J.; Tesfazghi, S.; Dammalapati, A.; Harrison, A.D.; Yu, X.M.; Scheinebeck, C.; Winston-McPherson, G.; Kupcho, K.R.; Robers, M.B.; et al. Tumor-suppressor role of Notch3 in medullary thyroid carcinoma revealed by genetic and pharmacological induction. Mol. Cancer Ther. 2015, 14, 499–512. [Google Scholar] [CrossRef]
- Aburjania, Z.; Whitt, J.D.; Jang, S.; Nadkarni, D.H.; Chen, H.; Rose, J.B.; Velu, S.E.; Jaskula-Sztul, R. Synthetic Makaluvamine Analogs Decrease c-Kit Expression and Are Cytotoxic to Neuroendocrine Tumor Cells. Molecules 2020, 25, 4940. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Hamill, O.P.; Marty, A.; Neher, E.; Sakmann, B.; Sigworth, F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981, 391, 85–100. [Google Scholar] [CrossRef]
- Marty, A.; Neher, E. Tight-Seal Whole-Cell Recording. In Single-Channel Recording; Sakmann, B., Neher, E., Eds.; Springer: Boston, MA, USA, 1995; pp. 31–52. [Google Scholar]
- GraphPad Prism Version 9.3.1 for Windows, Version 9.3.1; GraphPad Software: San Diego, CA, USA, 2021.
- Ashland, O.B. FlowJo™ Software for Windows Version 10.8.1, Version 10.8.1; Dickinson and Company: Franklin Lakes, NJ, USA, 2021.
- Kirkpatrick, L.A. A Simple Guide to IBM SPSS Statistics for Versions 20.0 & 21.0, Versions 20.0 & 21.0; Wadsworth Publishing: Belmont, CA, USA, 2013. [Google Scholar]
- Liu, C.; Yu, M.; Li, Y.; Wang, H.; Xu, C.; Zhang, X.; Li, M.; Guo, H.; Ma, D.; Guo, X. Lidocaine inhibits the metastatic potential of ovarian cancer by blocking NaV1.5-mediated EMT and FAK/Paxillin signaling pathway. Cancer Med. 2020, 10, 337–349. [Google Scholar] [CrossRef]
- Lopez-Charcas, O.; Espinosa, A.M.; Alfaro, A.; Herrera-Carrillo, Z.; Ramirez-Cordero, B.E.; Cortes-Reynosa, P.; Perez Salazar, E.; Berumen, J.; Gomora, J.C. The invasiveness of human cervical cancer associated to the function of NaV1.6 channels is mediated by MMP-2 activity. Sci. Rep. 2018, 8, 12995. [Google Scholar] [CrossRef]
- Diss, J.K.; Archer, S.N.; Hirano, J.; Fraser, S.P.; Djamgoz, M.B. Expression profiles of voltage-gated Na+ channel alpha-subunit genes in rat and human prostate cancer cell lines. Prostate 2001, 48, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Diss, J.K.J.; Stewart, D.; Pani, F.; Foster, C.S.; Walker, M.M.; Patel, A.; Djamgoz, M.B.A. A potential novel marker for human prostate cancer: Voltage-gated sodium channel expression in vivo. Prostate Cancer Prostatic Dis. 2005, 8, 266–273. [Google Scholar] [CrossRef]
- Campbell, T.M.; Main, M.J.; Fitzgerald, E.M. Functional expression of the voltage-gated Na⁺-channel Nav1.7 is necessary for EGF-mediated invasion in human non-small cell lung cancer cells. J. Cell Sci. 2013, 126, 4939–4949. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tan, H.; Yang, W.; Yao, S.; Hong, L. The voltage-gated sodium channel Nav1.7 associated with endometrial cancer. J. Cancer 2019, 10, 4954–4960. [Google Scholar] [CrossRef] [PubMed]
- Horne, J.; Mansur, S.; Bao, Y. Sodium ion channels as potential therapeutic targets for cancer metastasis. Drug Discov. Today 2021, 26, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- Pacini, F.; Elisei, R.; Anelli, S.; Basolo, F.; Cola, A.; Pinchera, A. Somatostatin in medullary thyroid cancer. In vitro and in vivo studies. Cancer 1989, 63, 1189–1195. [Google Scholar] [CrossRef]
- Gould, H.J., 3rd; Norleans, J.; Ward, T.D.; Reid, C.; Paul, D. Selective lysis of breast carcinomas by simultaneous stimulation of sodium channels and blockade of sodium pumps. Oncotarget 2018, 9, 15606–15615. [Google Scholar] [CrossRef]
- Yang, M.; Kozminski, D.J.; Wold, L.A.; Modak, R.; Calhoun, J.D.; Isom, L.L.; Brackenbury, W.J. Therapeutic potential for phenytoin: Targeting Nav1.5 sodium channels to reduce migration and invasion in metastatic breast cancer. Breast Cancer Res. Treat. 2012, 134, 603–615. [Google Scholar] [CrossRef]
- Simon, R.; Mirlacher, M.; Sauter, G. Immunohistochemical analysis of tissue microarrays. Methods Mol. Biol. 2010, 664, 113–126. [Google Scholar] [CrossRef]
- Wu, Y.; Zou, B.; Liang, L.; Li, M.; Tao, Y.X.; Yu, H.; Wang, X.; Li, M. Loperamide inhibits sodium channels to alleviate inflammatory hyperalgesia. Neuropharmacology 2017, 117, 282–291. [Google Scholar] [CrossRef]
- Bankar, G.; Goodchild, S.J.; Howard, S.; Nelkenbrecher, K.; Waldbrook, M.; Dourado, M.; Shuart, N.G.; Lin, S.; Young, C.; Xie, Z.; et al. Selective NaV1.7 Antagonists with Long Residence Time Show Improved Efficacy against Inflammatory and Neuropathic Pain. Cell Rep. 2018, 24, 3133–3145. [Google Scholar] [CrossRef]
- Wang, G.K.; Russell, C.; Wang, S.Y. Mexiletine block of wild-type and inactivation-deficient human skeletal muscle hNav1.4 Na+ channels. J. Physiol. 2004, 554, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Panigel, J.; Cook, S.P. A point mutation at F1737 of the human Nav1.7 sodium channel decreases inhibition by local anesthetics. J. Neurogenet. 2011, 25, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.K.; Pesti, K.; Lukacs, P.; Földi, M.C.; Gerevich, Z.; Sperlagh, B.; Mike, A. Non-blocking modulation as the major mechanism of sodium channel inhibition by riluzole. bioRxiv 2017. [Google Scholar] [CrossRef]
- Gamal El-Din, T.M.; Lenaeus, M.J.; Zheng, N.; Catterall, W.A. Fenestrations control resting-state block of a voltage-gated sodium channel. Proc. Natl. Acad. Sci. USA 2018, 115, 13111–13116. [Google Scholar] [CrossRef] [PubMed]
- Hille, B. Local anesthetics: Hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 1977, 69, 497–515. [Google Scholar] [CrossRef]
- Huang, C.J.; Schild, L.; Moczydlowski, E.G. Use-dependent block of the voltage-gated Na+ channel by tetrodotoxin and saxitoxin: Effect of pore mutations that change ionic selectivity. J. Gen. Physiol. 2012, 140, 435–454. [Google Scholar] [CrossRef]
- Narahashi, T. Tetrodotoxin: A brief history. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2008, 84, 147–154. [Google Scholar] [CrossRef]
- Van Meerloo, J.; Kaspers, G.J.; Cloos, J. Cell sensitivity assays: The MTT assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar] [CrossRef]
- Chen, H.C. Boyden chamber assay. Methods Mol. Biol. 2005, 294, 15–22. [Google Scholar] [CrossRef]
- Edenfield, S.; Sims, A.M.; Porretta, C.; Gould, H.J., 3rd; Paul, D. Effect of Cell Cycle on Cell Surface Expression of Voltage-Gated Sodium Channels and Na+,K+-ATPase. Cells 2022, 11, 3240. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.R.; Perez-Neut, M.; Kaja, S.; Gentile, S. Voltage-gated ion channels in cancer cell proliferation. Cancers 2015, 7, 849–875. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 1995, 80, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Abdul Kadir, L.; Stacey, M.; Barrett-Jolley, R. Emerging Roles of the Membrane Potential: Action Beyond the Action Potential. Front. Physiol. 2018, 9, 1661. [Google Scholar] [CrossRef]
- Li, R.; Xiao, C.; Liu, H.; Huang, Y.; Dilger, J.P.; Lin, J. Effects of local anesthetics on breast cancer cell viability and migration. BMC Cancer 2018, 18, 666. [Google Scholar] [CrossRef]
- Haraguchi-Suzuki, K.; Kawabata-Iwakawa, R.; Suzuki, T.; Suto, T.; Takazawa, T.; Saito, S. Local anesthetic lidocaine induces growth suppression of HeLa cells by decreasing and changing the cellular localization of the proliferation marker Ki-67. Genes Cells 2022, 27, 675–684. [Google Scholar] [CrossRef]
- Piegeler, T.; Votta-Velis, E.G.; Liu, G.; Place, A.T.; Schwartz, D.E.; Beck-Schimmer, B.; Minshall, R.D.; Borgeat, A. Antimetastatic Potential of Amide-linked Local Anesthetics: Inhibition of Lung Adenocarcinoma Cell Migration and Inflammatory Src Signaling Independent of Sodium Channel Blockade. Anesthesiology 2012, 117, 548–559. [Google Scholar] [CrossRef]
- Le Gac, G.; Angenard, G.; Clément, B.; Laviolle, B.; Coulouarn, C.; Beloeil, H. Local Anesthetics Inhibit the Growth of Human Hepatocellular Carcinoma Cells. Anesth. Analg. 2017, 125, 1600–1609. [Google Scholar] [CrossRef]
- Xu, X.; Dai, Y.; Feng, L.; Zhang, H.; Hu, Y.; Xu, L.; Zhu, X.; Jiang, Y. Knockdown of Nav1.5 inhibits cell proliferation, migration and invasion via Wnt/-catenin signaling pathway in oral squamous cell carcinoma. Acta Biochim. Biophys. Sin. 2020, 52, 527–535. [Google Scholar] [CrossRef]
- Li, K.; Zhou, Z.Y.; Ji, P.P.; Luo, H.S. Knockdown of β-catenin by siRNA influences proliferation, apoptosis and invasion of the colon cancer cell line SW480. Oncol. Lett. 2016, 11, 3896–3900. [Google Scholar] [CrossRef]
- Iwai, S.; Yonekawa, A.; Harada, C.; Hamada, M.; Katagiri, W.; Nakazawa, M.; Yura, Y. Involvement of the Wnt-β-catenin pathway in invasion and migration of oral squamous carcinoma cells. Int. J. Oncol. 2010, 37, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ou, S.W.; Wang, Y.J. Distribution and function of voltage-gated sodium channels in the nervous system. Channels 2017, 11, 534–554. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Wang, J.; Ou, S.; Wang, Y.; Qiu, B.; Ding, D.; Guo, F.; Gao, Q. Expression of neonatal Nav1.5 in human brain astrocytoma and its effect on proliferation, invasion and apoptosis of astrocytoma cells. Oncol. Rep. 2014, 31, 2692–2700. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef]
- Brisson, L.; Gillet, L.; Calaghan, S.; Besson, P.; Le Guennec, J.Y.; Roger, S.; Gore, J. NaV1.5 enhances breast cancer cell invasiveness by increasing NHE1-dependent H+ efflux in caveolae. Oncogene 2011, 30, 2070–2076. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Smallbone, K.; Maini, P.K.; Rose, F.; Averill, J.; Nagle, R.B.; Worrall, L.; Gillies, R.J. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br. J. Cancer 2007, 97, 646–653. [Google Scholar] [CrossRef]
- Brisson, L.; Driffort, V.; Benoist, L.; Poet, M.; Counillon, L.; Antelmi, E.; Rubino, R.; Besson, P.; Labbal, F.; Chevalier, S.; et al. NaV1.5 Na⁺ channels allosterically regulate the NHE-1 exchanger and promote the activity of breast cancer cell invadopodia. J. Cell Sci. 2013, 126, 4835–4842. [Google Scholar] [CrossRef]
- Roger, S.; Besson, P.; Le Guennec, J.Y. Influence of the whole-cell patch-clamp configuration on electrophysiological properties of the voltage-dependent sodium current expressed in MDA-MB-231 breast cancer cells. Eur. Biophys. J. 2004, 33, 274–279. [Google Scholar] [CrossRef]
- Wang, G.; Cao, R.; Wang, Y.; Qian, G.; Dan, H.C.; Jiang, W.; Ju, L.; Wu, M.; Xiao, Y.; Wang, X. Simvastatin induces cell cycle arrest and inhibits proliferation of bladder cancer cells via PPARγ signalling pathway. Sci. Rep. 2016, 6, 35783. [Google Scholar] [CrossRef] [PubMed]
- Eidizade, F.; Soukhtanloo, M.; Zhiani, R.; Mehrzad, J.; Mirzavi, F. Inhibition of glioblastoma proliferation, invasion, and migration by Urolithin B through inducing G0/G1 arrest and targeting MMP-2/-9 expression and activity. Biofactors 2022, 49, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Li, L.; Yang, L.; Duan, G.; Ma, T.; Li, N.; Liu, Y.; Yao, J.; Liu, J.Y.; Zhang, X. Novel SCN9A missense mutations contribute to congenital insensitivity to pain: Unexpected correlation between electrophysiological characterization and clinical phenotype. Mol. Pain 2020, 16, 1744806920923881. [Google Scholar] [CrossRef]
- Marchi, M.; D’Amato, I.; Andelic, M.; Cartelli, D.; Salvi, E.; Lombardi, R.; Gumus, E.; Lauria, G. Congenital insensitivity to pain: A novel mutation affecting a U12-type intron causes multiple aberrant splicing of SCN9A. Pain 2022, 163, e882–e887. [Google Scholar] [CrossRef]
- Trombetti, G.A.; Mezzelani, A.; Orro, A. A Drug Discovery Approach for an Effective Pain Therapy through Selective Inhibition of Nav1.7. Int. J. Mol. Sci. 2022, 23, 6793. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pukkanasut, P.; Whitt, J.; Guenter, R.; Lynch, S.E.; Gallegos, C.; Rosendo-Pineda, M.J.; Gomora, J.C.; Chen, H.; Lin, D.; Sorace, A.; et al. Voltage-Gated Sodium Channel NaV1.7 Inhibitors with Potent Anticancer Activities in Medullary Thyroid Cancer Cells. Cancers 2023, 15, 2806. https://doi.org/10.3390/cancers15102806
Pukkanasut P, Whitt J, Guenter R, Lynch SE, Gallegos C, Rosendo-Pineda MJ, Gomora JC, Chen H, Lin D, Sorace A, et al. Voltage-Gated Sodium Channel NaV1.7 Inhibitors with Potent Anticancer Activities in Medullary Thyroid Cancer Cells. Cancers. 2023; 15(10):2806. https://doi.org/10.3390/cancers15102806
Chicago/Turabian StylePukkanasut, Piyasuda, Jason Whitt, Rachael Guenter, Shannon E. Lynch, Carlos Gallegos, Margarita Jacaranda Rosendo-Pineda, Juan Carlos Gomora, Herbert Chen, Diana Lin, Anna Sorace, and et al. 2023. "Voltage-Gated Sodium Channel NaV1.7 Inhibitors with Potent Anticancer Activities in Medullary Thyroid Cancer Cells" Cancers 15, no. 10: 2806. https://doi.org/10.3390/cancers15102806
APA StylePukkanasut, P., Whitt, J., Guenter, R., Lynch, S. E., Gallegos, C., Rosendo-Pineda, M. J., Gomora, J. C., Chen, H., Lin, D., Sorace, A., Jaskula-Sztul, R., & Velu, S. E. (2023). Voltage-Gated Sodium Channel NaV1.7 Inhibitors with Potent Anticancer Activities in Medullary Thyroid Cancer Cells. Cancers, 15(10), 2806. https://doi.org/10.3390/cancers15102806