
Evaluation of the Elements of Short Hairpin RNAs in Developing shRNA-Containing CAR T Cells
 , , ,
, , ,
Abstract
:Simple Summary
Abstract
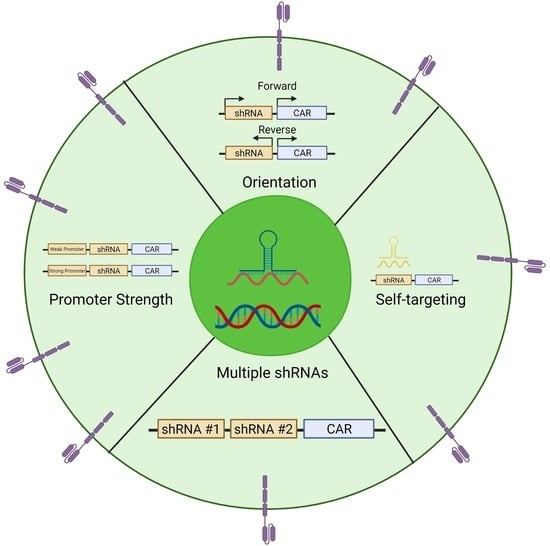
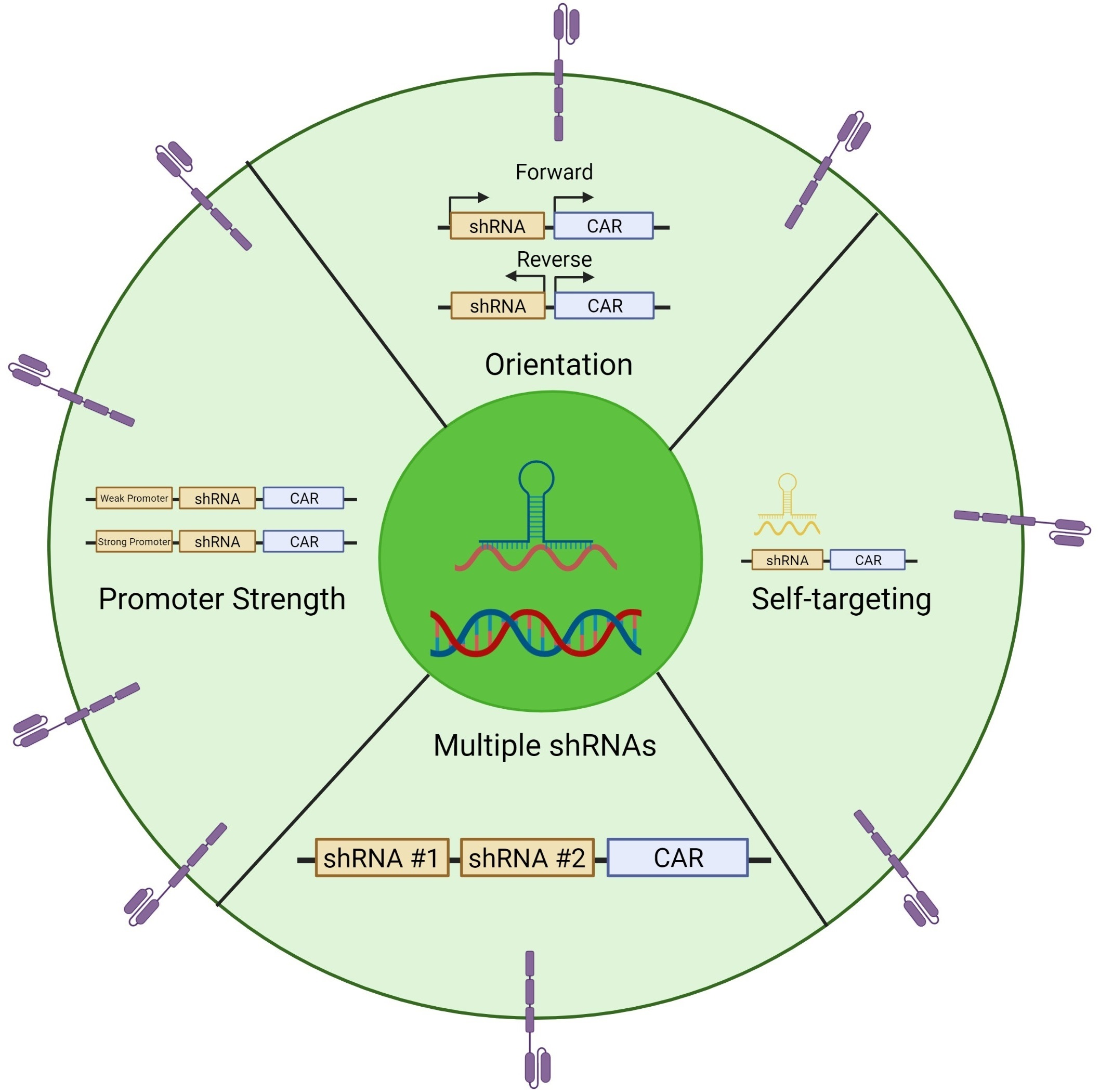
1. Introduction
2. Materials and Methods
2.1. DNA Constructs
2.2. Staining and Flow Cytometry
2.3. Cell Lines
2.4. Lentiviral and HIV Production
2.5. Generation of CAR T Cells
2.6. Knockdown Assay
2.7. Protection Assay
2.8. Activation Assay
2.9. T cell Cytotoxicity Assay
2.10. Degranulation Assay
2.11. Cytokine Production Assay
2.12. Acute Lymphoblastic Leukemia Xenograft Model
2.13. Statistics
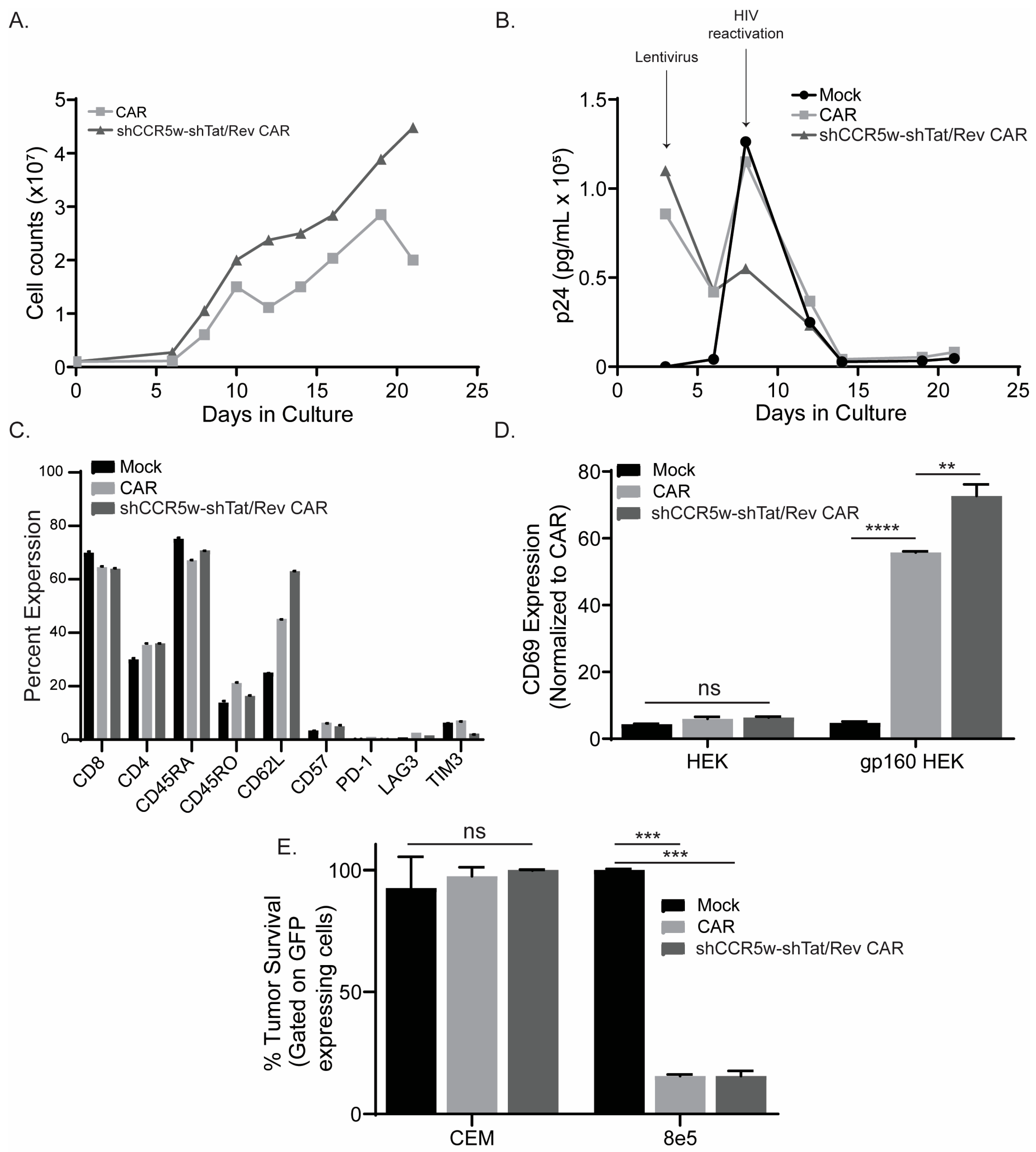
3. Results
3.1. Comparing the Orientation of the Polymerase III Promoter in CAR T cells during shRNA Expression
3.2. Wobble Bases Decrease shRNA Self-Targeting
3.3. Optimizing Multi-shRNA Containing CAR
3.4. Comparing Promoters in the Construction of shRNA-containing CAR
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Noopur, R.; Yi, L.; David, S.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. New Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Roschewski, M.; Longo, D.L.; Wilson, W.H. CAR T-Cell Therapy for Large B-Cell Lymphoma—Who, When, and How? N. Engl. J. Med. 2021, 386, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Zhu, L.; Chen, J. Current advances and challenges in CAR T-Cell therapy for solid tumors: Tumor-associated antigens and the tumor microenvironment. Exp. Hematol. Oncol. 2023, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Safarzadeh, K.P.; Rahbarizadeh, F. CAR-T cell therapy in T-cell malignancies: Is success a low-hanging fruit? Stem Cell Res. Ther. 2021, 12, 527. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, N.; Feng, K.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J.; Tang, N.; Zhang, X.; et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell. Mol. Immunol. 2021, 18, 2188–2198. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J., Jr.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef]
- Kouro, T.; Himuro, H.; Sasada, T. Exhaustion of CAR T cells: Potential causes and solutions. J. Transl. Med. 2022, 20, 239. [Google Scholar] [CrossRef]
- Stüber, T.; Monjezi, R.; Wallstabe, L.; Kühnemundt, J.; Nietzer, S.L.; Dandekar, G.; Wöckel, A.; Einsele, H.; Wischhusen, J.; Hudecek, M. Inhibition of TGF-β-receptor signaling augments the antitumor function of ROR1-specific CAR T-cells against triple-negative breast cancer. J. Immunother. Cancer 2020, 8, e000676. [Google Scholar] [CrossRef]
- Harris, E.; Elmer, J.J. Optimization of electroporation and other non-viral gene delivery strategies for T cells. Biotechnol. Prog. 2021, 37, e3066. [Google Scholar] [CrossRef]
- Zhang, Z.; Qiu, S.; Zhang, X.; Chen, W. Optimized DNA electroporation for primary human T cell engineering. BMC Biotechnol. 2018, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Seki, A.; Rutz, S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J. Exp. Med. 2018, 215, 985–997. [Google Scholar] [CrossRef]
- Li, C.; Mei, H.; Hu, Y. Applications and explorations of CRISPR/Cas9 in CAR T-cell therapy. Briefings Funct. Genom. 2020, 19, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.B.; Guthrie, E.H.; Huang, M.T.; Taxman, D.J. Short hairpin RNA (shRNA): Design, delivery, and assessment of gene knockdown. Methods Mol. Biol. 2010, 629, 141–158. [Google Scholar] [PubMed]
- Zhang, J.; Zhu, J.; Zheng, G.; Wang, Q.; Li, X.; Feng, Y.; Shang, F.; He, S.; Jiang, Q.; Shi, B.; et al. Co-Expression of miR155 or LSD1 shRNA Increases the Anti-Tumor Functions of CD19 CAR-T Cells. Front. Immunol. 2021, 12, 811364. [Google Scholar] [CrossRef]
- Zhou, J.E.; Yu, J.; Wang, Y.; Wang, H.; Wang, J.; Wang, Y.; Yu, L.; Yan, Z. ShRNA-mediated silencing of PD-1 augments the efficacy of chimeric antigen receptor T cells on subcutaneous prostate and leukemia xenograft. Biomed. Pharmacother. 2021, 137, 111339. [Google Scholar] [CrossRef]
- Steklov, M.; Lecalve, B.; Marijse, J.; Huberty, F.; Ramelot, N.; Jacques-Hespel, C.; Iserentant, H.; Breman, E.; Gilham, D. 146 Evolving Mutliplexed shRNA to generate tailored CAR T cell therapy. J. Immunother. Cancer 2021, 9 (Suppl. 2), A154. [Google Scholar] [CrossRef]
- An, D.S.; Qin, F.X.-F.; Auyeung, V.; Mao, S.H.; Kung, S.K.; Baltimore, D.; Chen, I.S. Optimization and Functional Effects of Stable Short Hairpin RNA Expression in Primary Human Lymphocytes via Lentiviral Vectors. Mol. Ther. 2006, 14, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Massengill, M.T.; Young, B.M.; Lewin, A.S.; Ildefonso, C.J. Co-Delivery of a Short-Hairpin RNA and a shRNA-Resistant Replacement Gene with Adeno-Associated Virus: An Allele-Independent Strategy for Autosomal-Dominant Retinal Disorders. Methods Mol. Biol. 2019, 1937, 235–258. [Google Scholar]
- Kang, L.; Tang, X.; Zhang, J.; Li, M.; Xu, N.; Qi, W.; Tan, J.; Lou, X.; Yu, Z.; Sun, J.; et al. Interleukin-6-knockdown of chimeric antigen receptor-modified T cells significantly reduces IL-6 release from monocytes. Exp. Hematol. Oncol. 2020, 9, 11. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Lee, H.J.; Kim, H.C.; Lee, Y.; Nam, S.K.; Hupperetz, C.; Ma, J.S.; Wang, X.; Singer, O.; Kim, W.S.; et al. PD-1 and TIGIT downregulation distinctly affect the effector and early memory phenotypes of CD19-targeting CAR T cells. Mol. Ther. 2022, 30, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.; Lim, L.; Holguin, L.; Han, T.; Vyas, V.; Urak, R.; Miller, A.; Browning, D.L.; Echavarria, L.; Li, S.; et al. Pre-clinical data supporting immunotherapy for HIV using CMV-HIV-specific CAR T cells with CMV vaccine. Mol. Ther. Methods Clin. Dev. 2022, 25, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Urak, R.; Walter, M.; Lim, L.; Wong, C.W.; Budde, L.E.; Thomas, S.; Forman, S.J.; Wang, X. Ex vivo Akt inhibition promotes the generation of potent CD19CAR T cells for adoptive immunotherapy. J. Immunother. Cancer 2017, 5, 26. [Google Scholar] [CrossRef]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986, 59, 284–291. [Google Scholar] [CrossRef]
- Herzig, E.; Kim, K.C.; Packard, T.A.; Vardi, N.; Schwarzer, R.; Gramatica, A.; Deeks, S.G.; Wiliams, S.R.; Landgraf, K.; Killeen, N.; et al. Attacking Latent HIV with convertibleCAR-T Cells, a Highly Adaptable Killing Platform. Cell 2019, 179, 880–894.e10. [Google Scholar] [CrossRef]
- Wang, X.; Huynh, C.; Urak, R.; Weng, L.; Walter, M.; Lim, L.; Vyas, V.; Chang, W.-C.; Aguilar, B.; Brito, A.; et al. The Cerebroventricular Environment Modifies CAR T Cells for Potent Activity against Both Central Nervous System and Systemic Lymphoma. Cancer Immunol. Res. 2021, 9, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Aguilar, B.; Starr, R.; Alizadeh, D.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; Forman, S.J.; Brown, C.E. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight 2018, 3, e99048. [Google Scholar] [CrossRef]
- McCutchan, F.E.; Salminen, M.O.; Carr, J.K.; Burke, D.S. HIV-1 genetic diversity. AIDS 1996, 10 (Suppl. 3), S13–S20. [Google Scholar]
- Robertson, D.L.; Anderson, J.P.; Bradac, J.A.; Carr, J.K.; Foley, B.; Funkhouser, R.K.; Gao, F.; Hahn, B.H.; Kalish, M.L.; Kuiken, C.; et al. HIV-1 Nomenclature Proposal. Science 2000, 288, 55. [Google Scholar] [CrossRef]
- Johnson, V.; Byington, R. Infectivity Assay (Virus Yield Assay) In Techniques in HIV Research; Aldovini, A., Walker, B.D., Eds.; Stockton Press: New York, NY, USA, 1990. [Google Scholar]
- Nie, L.; Das Thakur, M.; Wang, Y.; Su, Q.; Zhao, Y.; Feng, Y. Regulation of U6 promoter activity by transcriptional interference in viral vector-based RNAi. Genom. Proteom. Bioinform. 2010, 8, 170–179. [Google Scholar] [CrossRef]
- Liu, Y.P.; Vink, M.A.; Westerink, J.-T.; de Arellano, E.R.; Konstantinova, P.; Ter Brake, O.; Berkhout, B. Titers of lentiviral vectors encoding shRNAs and miRNAs are reduced by different mechanisms that require distinct repair strategies. RNA 2010, 16, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Poluri, A.; Sutton, R. Titers of HIV-based Vectors Encoding shRNAs are Reduced by a Dicer-dependent Mechanism. Mol. Ther. 2008, 16, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H. Codon—Anticodon pairing: The wobble hypothesis. J. Mol. Biol. 1966, 19, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Lopalco, L. CCR5: From Natural Resistance to a New Anti-HIV Strategy. Viruses 2010, 2, 574–600. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, H.; Gurrola, T.; Berman, R.; Collins, M.; Sariyer, I.K.; Nonnemacher, M.R.; Wigdahl, B. Targeting CCR5 as a Component of an HIV-1 Therapeutic Strategy. Front. Immunol. 2021, 12, 816515. [Google Scholar] [CrossRef] [PubMed]
- Urak, R.Z.; Soemardy, C.; Ray, R.; Li, S.; Shevchenko, G.; Scott, T.; Lim, L.; Wang, X.; Morris, K.V. Conditionally Replicating Vectors Mobilize Chimeric Antigen Receptors against HIV. Mol. Ther.-Methods Clin. Dev. 2020, 19, 285–294. [Google Scholar] [CrossRef]
- Wang, X.; Urak, R.; Walter, M.; Guan, M.; Han, T.; Vyas, V.; Chien, S.-H.; Gittins, B.; Clark, M.C.; Mokhtari, S.; et al. Large-scale manufacturing and characterization of CMV-CD19CAR T cells. J. Immunother. Cancer 2022, 10, e003461. [Google Scholar] [CrossRef]
- Ding, Y.; Kong, D.; Li, D.; Zhang, Y.; Hong, K.; Liang, H.; Ma, L. Characterization of antibody-dependent cellular cytotoxicity induced by the plasma from persons living with HIV-1 based on target cells with or without CD4 molecules. Microbes Infect. 2021, 23, 104805. [Google Scholar] [CrossRef]
- Quillent, C.; Dumey, N.; Dauguet, C.; Clavel, F. Reversion of a Polymerase-Defective Integrated HIV-1 Genome. AIDS Res. Hum. Retrovir. 1993, 9, 1031–1037. [Google Scholar] [CrossRef]
- Chang, Z.; Westaway, S.; Li, S.; Zaia, J.A.; Rossi, J.J.; Scherer, L.J. Enhanced expression and HIV-1 inhibition of chimeric tRNA(Lys3)-ribozymes under dual U6 snRNA and tRNA promoters. Mol. Ther. J. Am. Soc. Gene Ther. 2002, 6, 481–489. [Google Scholar] [CrossRef]
- Kleiman, L. tRNA(Lys3): The primer tRNA for reverse transcription in HIV-1. IUBMB Life 2002, 53, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Tammaro, M.; Yan, H. Enriching CRISPR-Cas9 targeted cells by co-targeting the HPRT gene. Nucleic Acids Res. 2015, 43, e134. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Fang, H.; Wang, D.; Chen, Y.; Zhai, Y.; Zhou, B.-B.S.; Li, H. HPRT1 activity loss is associated with resistance to thiopurine in ALL. Oncotarget 2018, 9, 2268–2278. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ramos, A.A.; Marchetti-Laurent, C.; Poindessous, V.; Antonio, S.; Laurent-Puig, P.; Bortoli, S.; Loriot, M.-A.; Pallet, N. 6-mercaptopurine promotes energetic failure in proliferating T cells. Oncotarget 2017, 8, 43048–43060. [Google Scholar] [CrossRef] [PubMed]
- Razeghian, E.; Nasution, M.K.M.; Rahman, H.S.; Gardanova, Z.R.; Abdelbasset, W.K.; Aravindhan, S.; Bokov, D.O.; Suksatan, W.; Nakhaei, P.; Shariatzadeh, S.; et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res. Ther. 2021, 12, 428. [Google Scholar] [CrossRef]
- Agarwal, S.; Wellhausen, N.; Levine, B.L.; June, C.H. Production of Human CRISPR-Engineered CAR-T Cells. J. Vis. Exp. 2021, 169, e62299. [Google Scholar]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef]
- Sheng, W.; LaFleur, M.W.; Nguyen, T.H.; Chen, S.; Chakravarthy, A.; Conway, J.R.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.-H.; et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018, 174, 549–563.e19. [Google Scholar] [CrossRef]
- Giering, J.C.; Grimm, D.; Storm, T.A.; Kay, M.A. Expression of shRNA from a tissue-specific pol II promoter is an effective and safe RNAi therapeutic. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1630–1636. [Google Scholar] [CrossRef]
- Lee, S.-K.; Dykxhoorn, D.M.; Kumar, P.; Ranjbar, S.; Song, E.; Maliszewski, L.E.; François-Bongarçon, V.; Goldfeld, A.; Swamy, N.M.; Lieberman, J.; et al. Lentiviral delivery of short hairpin RNAs protects CD4 T cells from multiple clades and primary isolates of HIV. Blood 2005, 106, 818–826. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Short Hairpin RNA | Sequence |
|---|---|
| shCCR5 | AGTGTCAAGTCCAATCTATGATTCAAGAGATCATAGATTGGACTTGACACTTTTTTT |
| shCCR5w | AAAAAAAGTGTCAAGTCCAATCTATGATCTCTTGAACCACAAATTGGACTTGACACT |
| shTat/Rev | GCGGAGACAGCGACGAAGAGCTTTGTGTAGGCTCTTCGTCGCTGTCTCCGCTTTTTT |
| shHPRT | GCACTGAATAGAAATGGTGGTTCAAGAGATCACTATTTCTATTCAGTGCTTTTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urak, R.; Gittins, B.; Soemardy, C.; Grepo, N.; Goldberg, L.; Maker, M.; Shevchenko, G.; Davis, A.; Li, S.; Scott, T.; et al. Evaluation of the Elements of Short Hairpin RNAs in Developing shRNA-Containing CAR T Cells. Cancers 2023, 15, 2848. https://doi.org/10.3390/cancers15102848
Urak R, Gittins B, Soemardy C, Grepo N, Goldberg L, Maker M, Shevchenko G, Davis A, Li S, Scott T, et al. Evaluation of the Elements of Short Hairpin RNAs in Developing shRNA-Containing CAR T Cells. Cancers. 2023; 15(10):2848. https://doi.org/10.3390/cancers15102848
Chicago/Turabian StyleUrak, Ryan, Brenna Gittins, Citradewi Soemardy, Nicole Grepo, Lior Goldberg, Madeleine Maker, Galina Shevchenko, Alicia Davis, Shirley Li, Tristan Scott, and et al. 2023. "Evaluation of the Elements of Short Hairpin RNAs in Developing shRNA-Containing CAR T Cells" Cancers 15, no. 10: 2848. https://doi.org/10.3390/cancers15102848
APA StyleUrak, R., Gittins, B., Soemardy, C., Grepo, N., Goldberg, L., Maker, M., Shevchenko, G., Davis, A., Li, S., Scott, T., Morris, K. V., Forman, S. J., & Wang, X. (2023). Evaluation of the Elements of Short Hairpin RNAs in Developing shRNA-Containing CAR T Cells. Cancers, 15(10), 2848. https://doi.org/10.3390/cancers15102848