
KRAS and NRAS Translation Is Increased upon MEK Inhibitors-Induced Processing Bodies Dissolution

 , , , , , , , , , , , ,
, , , , , , , , , , , ,  and
and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. DDX6 GFP Stable Cell Lines
2.3. siRNA Transient Transfection
2.4. Cancer Cell Line Encyclopedia (CCLE) Expression Analyses
2.5. Immunoblotting
2.6. Isolation of RNA and Quantitative Real-Time RT-PCR Analysis
2.7. Immunofluorescence and Confocal Microscopy
2.8. Polysome Gradient
2.9. Statistical Analysis
3. Results
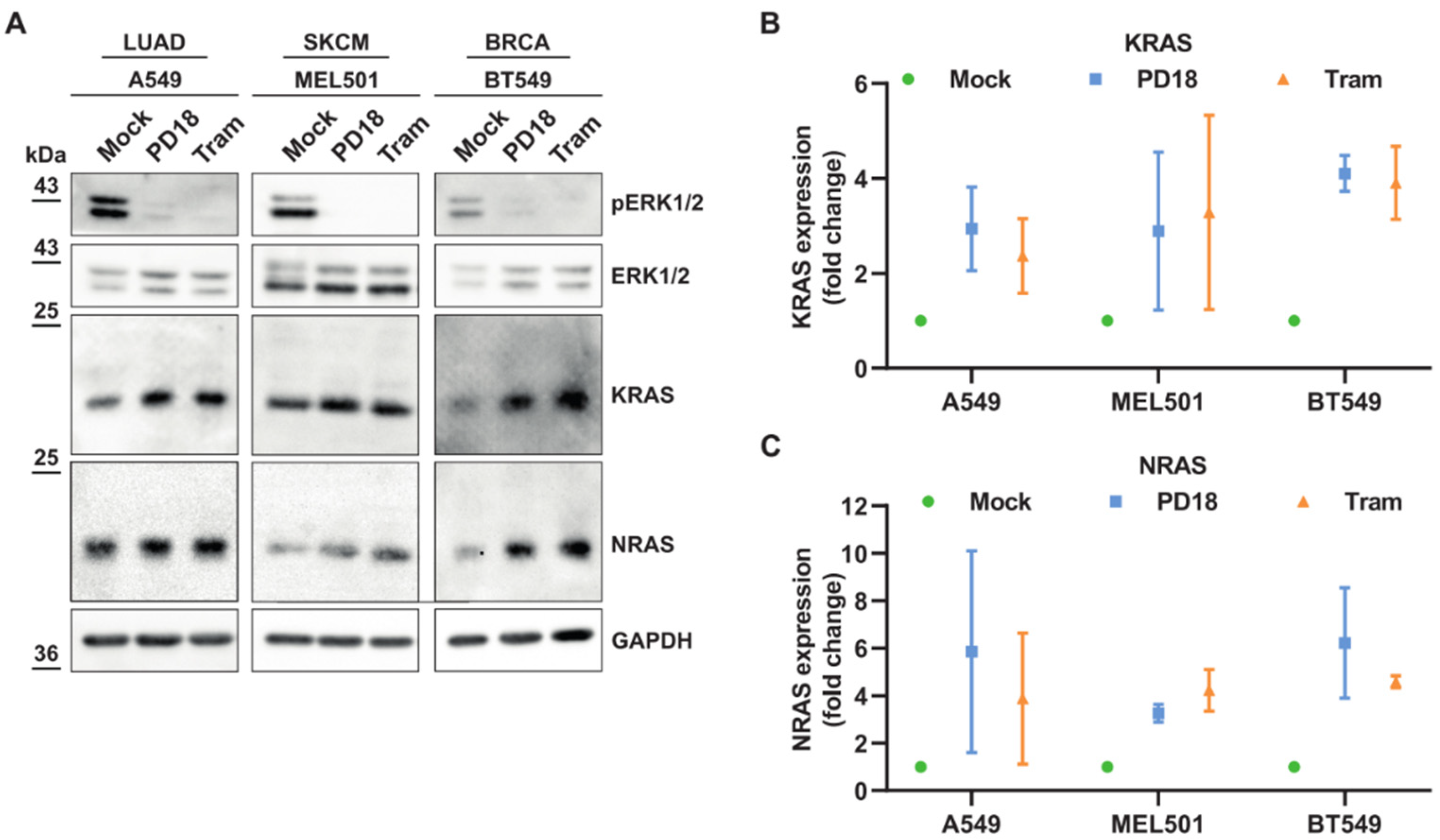
3.1. MEKi Response Promotes KRAS and NRAS Translation
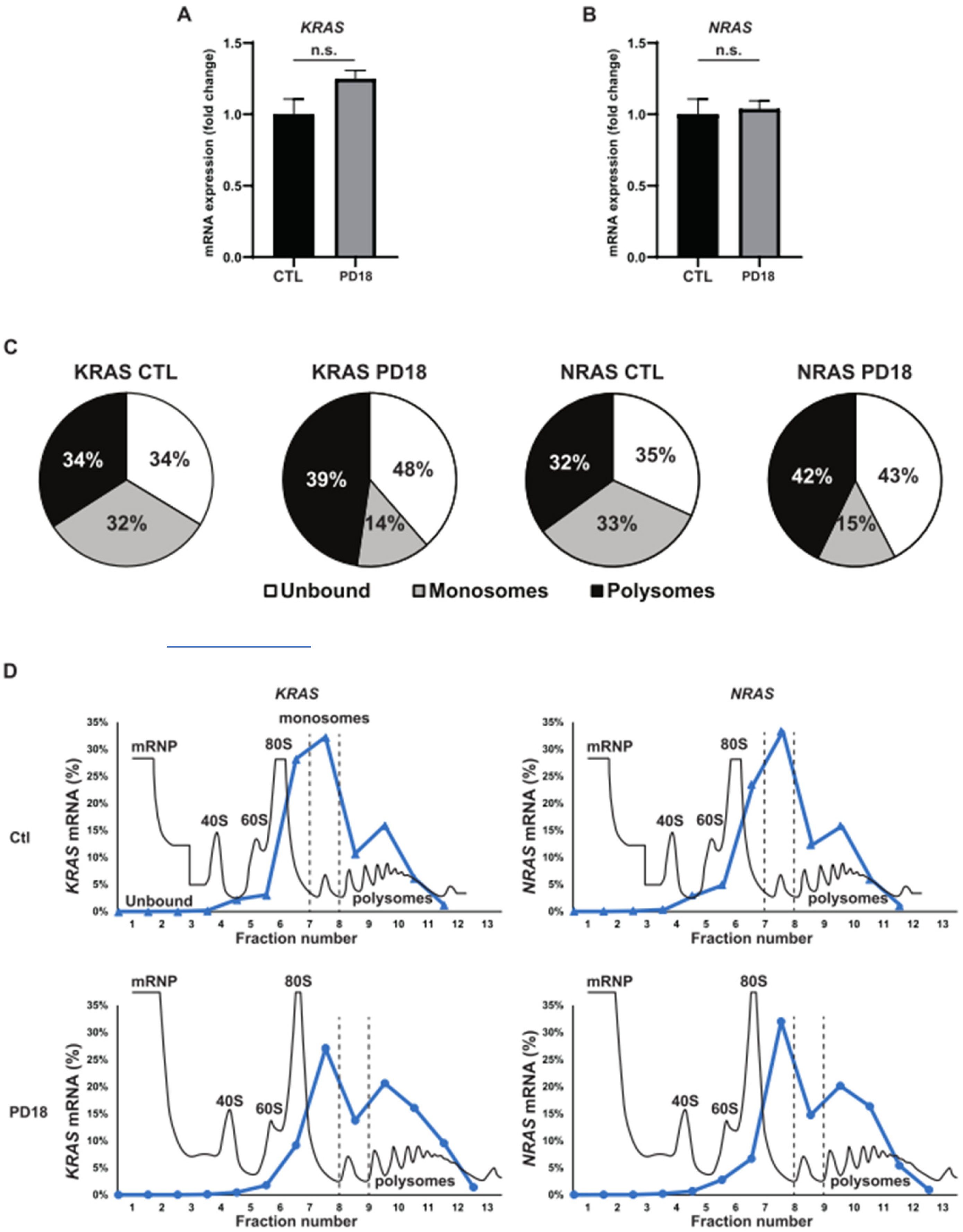
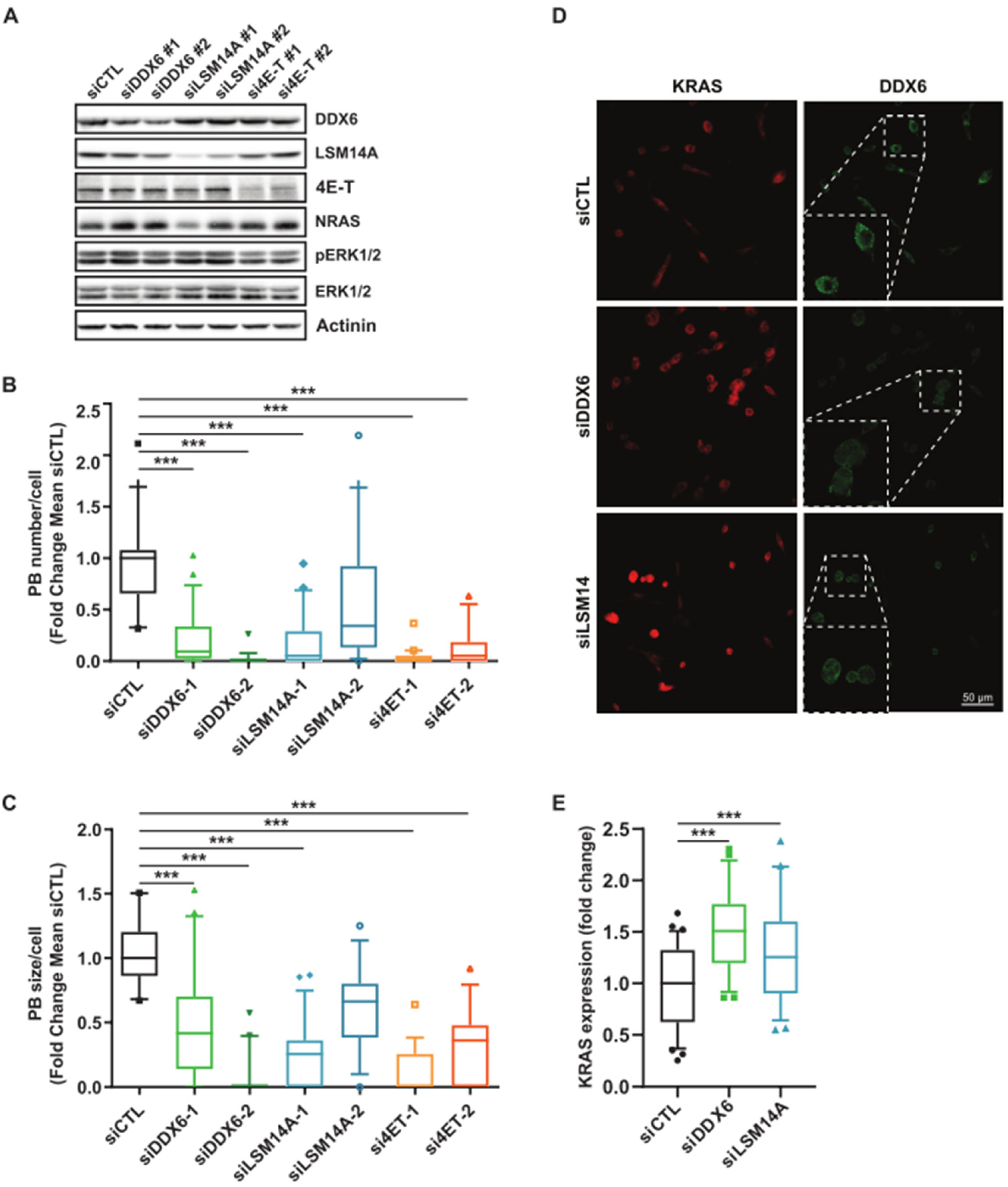
3.2. MEK Inhibition Promotes P-body Dissolution
3.3. Relationship between KRAS and NRAS Expression and P-body Components
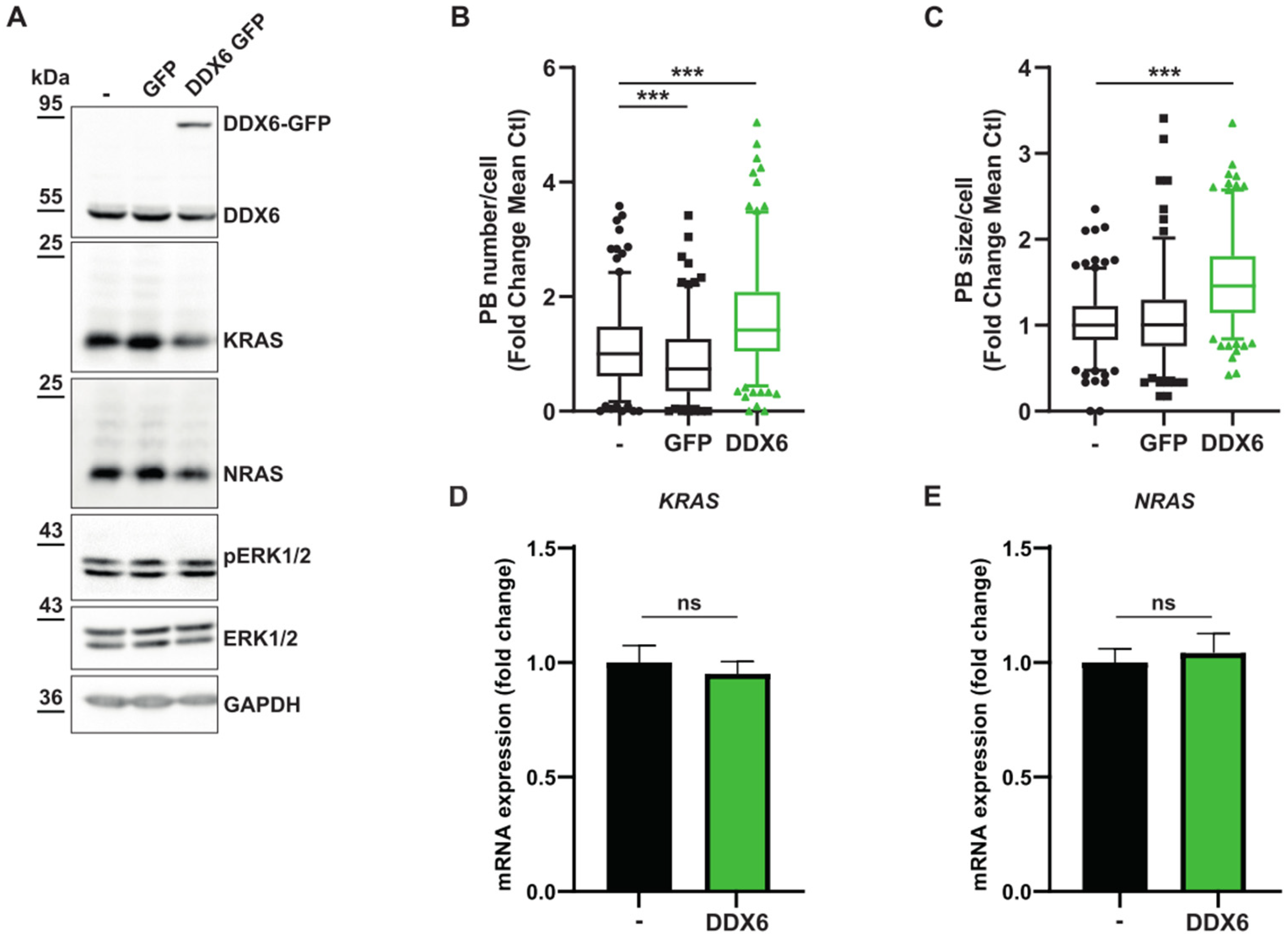
3.4. MAPK Signaling Promotes P-body Formation
3.5. Expression of KRAS or NRAS Is Sufficient to Induce Phosphorylation of BRAF
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2669–2974. [Google Scholar] [CrossRef] [Green Version]
- Anguera, G.; Majem, M. BRAF Inhibitors in Metastatic Non-Small Cell Lung Cancer. J. Thorac. Dis. 2018, 10, 589–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRAS G12C Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRAS G12C Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS–MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1923. [Google Scholar] [CrossRef]
- Smith, L.K.; Sheppard, K.E.; McArthur, G.A. Is Resistance to Targeted Therapy in Cancer Inevitable? Cancer Cell 2021, 39, 1047–1049. [Google Scholar] [CrossRef]
- Aldea, M.; Andre, F.; Marabelle, A.; Dogan, S.; Barlesi, F.; Soria, J.C. Overcoming Resistance to Tumor-Targeted and Immune-Targeted Therapies. Cancer Discov. 2021, 11, 874–899. [Google Scholar] [CrossRef]
- Wang, Y.; Van Becelaere, K.; Jiang, P.; Przybranowski, S.; Omer, C.; Sebolt-Leopold, J. A Role for K-Ras in Conferring Resistance to the MEK Inhibitor, CI-1040. Neoplasia 2005, 7, 336–347. [Google Scholar] [CrossRef] [Green Version]
- Ambrosini, G.; Khanin, R.; Carvajal, R.D.; Schwartz, G.K. Overexpression of DDX43 Mediates MEK Inhibitor Resistance through RAS Upregulation in Uveal Melanoma Cells. Mol. Cancer Ther. 2014, 13, 2073–2080. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.S.; Woodcock, M.G.; Azam, S.H.; Thorne, L.B.; Kanchi, K.L.; Parker, J.S.; Vincent, B.G.; Pecot, C. V Rapid Idiosyncratic Mechanisms of Clinical Resistance to KRAS G12C Inhibition. J. Clin. Investig. 2022, 132, e155523. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Kedersha, N.; Ivanov, P. Stress Granules, P-Bodies and Cancer. Biochim. Biophys. Acta-Gene Regul. Mech. 2015, 1849, 861–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, K.; Kuwano, Y. Diverse Roles of RNA-Binding Proteins in Cancer Traits and Their Implications in Gastrointestinal Cancers. Wiley Interdiscip. Rev. RNA 2019, 10, e1520. [Google Scholar] [CrossRef] [PubMed]
- Hubstenberger, A.; Courel, M.; Bénard, M.; Souquere, S.; Ernoult-Lange, M.; Chouaib, R.; Yi, Z.; Morlot, J.B.; Munier, A.; Fradet, M.; et al. P-Body Purification Reveals the Condensation of Repressed MRNA Regulons. Mol. Cell 2017, 68, 144–157.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khong, A.; Matheny, T.; Jain, S.; Mitchell, S.F.; Wheeler, J.R.; Parker, R. The Stress Granule Transcriptome Reveals Principles of MRNA Accumulation in Stress Granules. Mol. Cell 2017, 68, 808–820.e5. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Valencia-Sanchez, M.A.; Hannon, G.J.; Parker, R. MicroRNA-Dependent Localization of Targeted MRNAs to Mammalian P-Bodies. Nat. Cell Biol. 2005, 7, 719–723. [Google Scholar] [CrossRef] [Green Version]
- Pitchiaya, S.; Mourao, M.D.A.A.; Jalihal, A.P.; Xiao, L.; Jiang, X.; Chinnaiyan, A.M.; Schnell, S.; Walter, N.G. Dynamic Recruitment of Single RNAs to Processing Bodies Depends on RNA Functionality. Mol. Cell 2019, 74, 521–533. [Google Scholar] [CrossRef]
- Hardy, S.D.; Shinde, A.; Wang, W.H.; Wendt, M.K.; Geahlen, R.L. Regulation of Epithelial-Mesenchymal Transition and Metastasis by TGF-β, P-Bodies, and Autophagy. Oncotarget 2017, 8, 103302–103314. [Google Scholar] [CrossRef] [Green Version]
- Feillet, C.; Krusche, P.; Tamanini, F.; Janssens, R.C.; Downey, M.J.; Martin, P.; Teboul, M.; Saito, S.; Lévi, F.A.; Bretschneider, T.; et al. Phase Locking and Multiple Oscillating Attractors for the Coupled Mammalian Clock and Cell Cycle. Proc. Natl. Acad. Sci. USA 2014, 111, 9828–9833. [Google Scholar] [CrossRef] [Green Version]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bairoch, A. The Cellosaurus, a Cell-Line Knowledge Resource. J. Biomol. Tech. 2018, 29, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Ayache, J.; Bénard, M.; Ernoult-Lange, M.; Minshall, N.; Standart, N.; Kress, M.; Weil, D. P-Body Assembly Requires DDX6 Repression Complexes Rather than Decay or Ataxin2/2L Complexes. Mol. Biol. Cell 2015, 26, 2579–2595. [Google Scholar] [CrossRef] [PubMed]
- Kamenska, A.; Simpson, C.; Vindry, C.; Broomhead, H.; Bénard, M.; Ernoult-Lange, M.; Lee, B.P.; Harries, L.W.; Weil, D.; Standart, N. The DDX6-4E-T Interaction Mediates Translational Repression and P-Body Assembly. Nucleic Acids Res. 2016, 44, 6318–6334. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.-K. Non-Small-Cell Lung Cancers: A Heterogeneous Set of Diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- De Conti, G.; Dias, M.H.; Bernards, R. Fighting Drug Resistance through the Targeting of Drug-tolerant Persister Cells. Cancers 2021, 13, 1118. [Google Scholar] [CrossRef]
- Marin-Bejar, O.; Rogiers, A.; Dewaele, M.; Femel, J.; Karras, P.; Pozniak, J.; Bervoets, G.; Van Raemdonck, N.; Pedri, D.; Swings, T.; et al. Evolutionary Predictability of Genetic versus Nongenetic Resistance to Anticancer Drugs in Melanoma. Cancer Cell 2021, 39, 1135–1149.e8. [Google Scholar] [CrossRef]
- Marine, J.C.; Dawson, S.J.; Dawson, M.A. Non-Genetic Mechanisms of Therapeutic Resistance in Cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef]
- Meyer, M.; Paquet, A.; Arguel, M.-J.; Peyre, L.; Gomes-Pereira, L.C.; Lebrigand, K.; Mograbi, B.; Brest, P.; Waldmann, R.; Barbry, P.; et al. Profiling the Non-Genetic Origins of Cancer Drug Resistance with a Single-Cell Functional Genomics Approach Using Predictive Cell Dynamics. Cell Syst. 2020, 11, 367–374.e5. [Google Scholar] [CrossRef]
- Vendramin, R.; Litchfield, K.; Swanton, C. Cancer Evolution: Darwin and Beyond. EMBO J. 2021, 40, e108389. [Google Scholar] [CrossRef]
- Algazi, A.P.; Othus, M.; Daud, A.I.; Lo, R.S.; Mehnert, J.M.; Truong, T.G.; Conry, R.; Kendra, K.; Doolittle, G.C.; Clark, J.I.; et al. Continuous versus Intermittent BRAF and MEK Inhibition in Patients with BRAF-Mutated Melanoma: A Randomized Phase 2 Trial. Nat. Med. 2020, 26, 1564–1568. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hütter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling Cancer Persister Cells Arise from Lineages with Distinct Programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Bontoux, C.; Hofman, V.; Brest, P.; Ilié, M.; Mograbi, B.; Hofman, P. Daily Practice Assessment of KRAS Status in NSCLC Patients: A New Challenge for the Thoracic Pathologist Is Right around the Corner. Cancers 2022, 14, 1628. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. Bi-3406, a Potent and Selective Sos1–Kras Interaction Inhibitor, Is Effective in Kras-Driven Cancers through Combined Mek Inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, D.; Battich, N.; Pelkmans, L. A Systems-Level Study Reveals Regulators of Membrane-Less Organelles in Human Cells. Mol. Cell 2018, 72, 1035–1049.e5. [Google Scholar] [CrossRef] [Green Version]
- Ünal, E.B.; Uhlitz, F.; Blüthgen, N. A Compendium of ERK Targets. FEBS Lett. 2017, 591, 2607–2615. [Google Scholar] [CrossRef] [Green Version]
- Courel, M.; Clément, Y.; Bossevain, C.; Foretek, D.; Vidal Cruchez, O.; Yi, Z.; Bénard, M.; Benassy, M.-N.M.-N.; Kress, M.; Vindry, C.; et al. GC Content Shapes MRNA Storage and Decay in Human Cells. Elife 2019, 8, 1689–1699. [Google Scholar] [CrossRef]
- Lampson, B.L.; Pershing, N.L.K.; Prinz, J.A.; Lacsina, J.R.; Marzluff, W.F.; Nicchitta, C.V.; MacAlpine, D.M.; Counter, C.M. Rare Codons Regulate KRas Oncogenesis. Curr. Biol. 2013, 23, 70–75. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Dang, Y.; Counter, C.; Liu, Y. Codon Usage Regulates Human KRAS Expression at Both Transcriptional and Translational Levels. J. Biol. Chem. 2018, 293, 17929–17940. [Google Scholar] [CrossRef] [Green Version]
- Stopfer, L.E.; Rettko, N.J.; Leddy, O.; Mesfin, J.M.; Brown, E.; Winski, S.; Bryson, B.; Wells, J.A.; White, F.M. MEK Inhibition Enhances Presentation of Targetable MHC-I Tumor Antigens in Mutant Melanomas. Proc. Natl. Acad. Sci. USA 2022, 119, 2017. [Google Scholar] [CrossRef]
- Shen, S.; Faouzi, S.; Bastide, A.; Martineau, S.; Malka-Mahieu, H.; Fu, Y.; Sun, X.; Mateus, C.; Routier, E.; Roy, S.; et al. An Epitranscriptomic Mechanism Underlies Selective MRNA Translation Remodelling in Melanoma Persister Cells. Nat. Commun. 2019, 10, 5713. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidal-Cruchez, O.; Nicolini, V.J.; Rete, T.; Jacquet, K.; Rezzonico, R.; Lacoux, C.; Domdom, M.-A.; Roméo, B.; Roux, J.; Hubstenberger, A.; et al. KRAS and NRAS Translation Is Increased upon MEK Inhibitors-Induced Processing Bodies Dissolution. Cancers 2023, 15, 3078. https://doi.org/10.3390/cancers15123078
Vidal-Cruchez O, Nicolini VJ, Rete T, Jacquet K, Rezzonico R, Lacoux C, Domdom M-A, Roméo B, Roux J, Hubstenberger A, et al. KRAS and NRAS Translation Is Increased upon MEK Inhibitors-Induced Processing Bodies Dissolution. Cancers. 2023; 15(12):3078. https://doi.org/10.3390/cancers15123078
Chicago/Turabian StyleVidal-Cruchez, Olivia, Victoria J. Nicolini, Tifenn Rete, Karine Jacquet, Roger Rezzonico, Caroline Lacoux, Marie-Angela Domdom, Barnabé Roméo, Jérémie Roux, Arnaud Hubstenberger, and et al. 2023. "KRAS and NRAS Translation Is Increased upon MEK Inhibitors-Induced Processing Bodies Dissolution" Cancers 15, no. 12: 3078. https://doi.org/10.3390/cancers15123078
APA StyleVidal-Cruchez, O., Nicolini, V. J., Rete, T., Jacquet, K., Rezzonico, R., Lacoux, C., Domdom, M. -A., Roméo, B., Roux, J., Hubstenberger, A., Mari, B., Mograbi, B., Hofman, P., & Brest, P. (2023). KRAS and NRAS Translation Is Increased upon MEK Inhibitors-Induced Processing Bodies Dissolution. Cancers, 15(12), 3078. https://doi.org/10.3390/cancers15123078