
Clinical Utility of Optical Genome Mapping and 523-Gene Next Generation Sequencing Panel for Comprehensive Evaluation of Myeloid Cancers

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
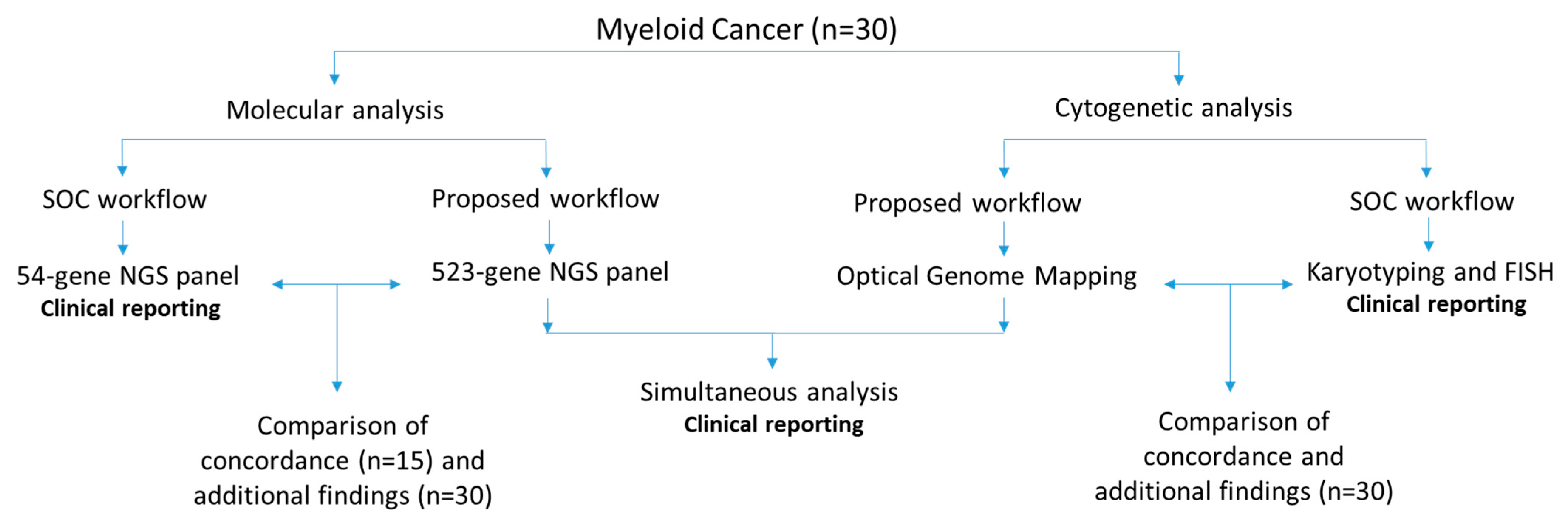
2. Materials and Methods
2.1. Sample Selection
2.2. Optical Genome Mapping
2.3. OGM Variant Calling and Data Analysis
2.4. Analytical Comparison between OGM and SOC Results
2.5. 523-Gene NGS Panel
2.6. NGS Variant Calling and Data Analysis
2.7. Analytical Comparison between 523-Gene NGS Panel and 54-Gene NGS Panel
2.8. OGM and Sequencing Data: Compound Heterozygous Events in NxClinical Software
3. Results
3.1. OGM Quality Control Metrics and Variant Filtering
3.2. OGM Results: Concordance, Higher Resolution/Resolving Identified Events, and Additional Findings
3.3. 523-Gene NGS Panel Quality Control Metrics
3.4. 523-Gene NGS Panel: Concordance and Additional Findings
3.5. OGM and 523-Gene NGS Panel: Compound Heterozygous Events
4. Risk Stratification
5. Discussion
5.1. Cytogenomics: OGM Compared to Standard-of-Care Technologies (Karyotype and FISH)
5.2. Molecular Profiling: 523-Gene NGS Panel Compared to 54-Gene NGS Panel
5.3. OGM and 523-Gene NGS Panel Compared to the Current Diagnostic Workflow (Karyotype, FISH, and 54-Gene NGS Panel)
5.4. OGM and 523-Gene NGS Panel Compared to GS for Myeloid Cancers
6. Limitations of the Study
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vardiman, J.W.; Harris, N.L.; Brunning, R.D. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 2002, 100, 2292–2302. [Google Scholar] [CrossRef] [Green Version]
- Tallman, M.S.; Wang, E.S.; Altman, J.K.; Appelbaum, F.R.; Bhatt, V.R.; Bixby, D.; Coutre, S.E.; De Lima, M.; Fathi, A.T.; Fiorella, M.; et al. Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 721–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Mikhail, F.M.; Heerema, N.A.; Rao, K.W.; Burnside, R.D.; Cherry, A.M.; Cooley, L.D. Section E6.1-6.4 of the ACMG technical standards and guidelines: Chromosome studies of neoplastic blood and bone marrow-acquired chromosomal abnormalities. Genet. Med. 2016, 18, 635–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.R.; Patel, K.P.; Routbort, M.J.; Reddy, N.G.; Barkoh, B.A.; Handal, B.; Kanagal-Shamanna, R.; Greaves, W.O.; Medeiros, L.J.; Aldape, K.D.; et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J. Mol. Diagn. 2013, 15, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Diaz, A.; Vazquez, I.; Ariceta, B.; Mañú, A.; Blasco-Iturri, Z.; Palomino-Echeverría, S.; Larrayoz, M.J.; García-Sanz, R.; Prieto-Conde, M.I.; del Carmen Chillón, M.; et al. Assessment of the clinical utility of four NGS panels in myeloid malignancies. Suggestions for NGS panel choice or design. PLoS ONE 2020, 15, e0227986. [Google Scholar] [CrossRef] [Green Version]
- Sahajpal, N.S.; Mondal, A.K.; Ananth, S.; Njau, A.; Ahluwalia, P.; Jones, K.; Ahluwalia, M.; Okechukwu, N.; Savage, N.M.; Kota, V.; et al. Clinical performance and utility of a comprehensive next-generation sequencing DNA panel for the simultaneous analysis of variants, TMB and MSI for myeloid neoplasms. PLoS ONE 2020, 15, e0240976. [Google Scholar] [CrossRef]
- Tsui, S.P.; Ip, H.W.; Saw, N.Y.; Zhang, C.; Cheung, A.K.; Ng, N.K.; Man, C.H.; Lam, S.S.; Tang, W.F.; Lin, C.H.; et al. Redefining prognostication of de novo cytogenetically normal acute myeloid leukemia in young adults. Blood Cancer J. 2020, 10, 104. [Google Scholar] [CrossRef]
- Nimer, S.D. Is it important to decipher the heterogeneity of “normal karyotype AML”? Best Pract. Res. Clin. Haematol. 2008, 21, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.; Marcucci, G. Molecular prognostic factors in cytogenetically normal acute myeloid leukemia. Expert Rev. Hematol. 2012, 5, 547–558. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahajpal, N.S.; Barseghyan, H.; Kolhe, R.; Hastie, A.; Chaubey, A. Optical Genome Mapping as a Next-Generation Cytogenomic Tool for Detection of Structural and Copy Number Variations for Prenatal Genomic Analyses. Genes 2021, 12, 398. [Google Scholar] [CrossRef]
- Mantere, T.; Neveling, K.; Pebrel-Richard, C.; Benoist, M.; van der Zande, G.; Kater-Baats, E.; Baatout, I.; van Beek, R.; Yammine, T.; Oorsprong, M.; et al. Optical genome mapping enables constitutional chromosomal aberration detection. Am. J. Hum. Genet. 2021, 108, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Shieh, J.T.; Penon-Portmann, M.; Wong, K.H.Y.; Levy-Sakin, M.; Verghese, M.; Slavotinek, A.; Gallagher, R.C.; Mendelsohn, B.A.; Tenney, J.; Beleford, D.; et al. Application of full-genome analysis to diagnose rare monogenic disorders. NPJ Genom. Med. 2021, 6, 77. [Google Scholar] [CrossRef]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next-generation cytogenetics: Comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am. J. Hum. Genet. 2021, 108, 1423–1435. [Google Scholar] [CrossRef]
- Levy, B.; Baughn, L.B.; Akkari, Y.; Chartrand, S.; LaBarge, B.; Claxton, D.; Lennon, P.A.; Cujar, C.; Kolhe, R.; Kroeger, K.; et al. Optical genome mapping in acute myeloid leukemia: A multicenter evaluation. Blood Adv. 2023, 7, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Garcia-Manero, G.; Rush, D.; Montalban-Bravo, G.; Mallampati, S.; Medeiros, L.J.; Levy, B.; Luthra, R.; Kanagal-Shamanna, R. Application of Optical Genome Mapping For Comprehensive Assessment of Chromosomal Structural Variants for Clinical Evaluation of Myelodysplastic Syndromes. Leukemia 2022, 36, 2306–2316. [Google Scholar] [CrossRef]
- Sahajpal, N.S.; Mondal, A.K.; Tvrdik, T.; Hauenstein, J.; Shi, H.; Deeb, K.K.; Saxe, D.; Hastie, A.; Chaubey, A.; Savage, N.M.; et al. Optical Genome Mapping: Clinical Validation and Diagnostic Utility for Enhanced Cytogenomic Analysis of Hematological Neoplasms. J. Mol. Diagn. 2022, 24, 1279–1291. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Schroeder, M.C.; O’Laughlin, M.; Wilson, R.; MacMillan, S.; Bohannon, A.; Kruchowski, S.; Garza, J.; Du, F.; Hughes, A.E.O.; et al. Genome Sequencing as an Alternative to Cytogenetic Analysis in Myeloid Cancers. N. Engl. J. Med. 2021, 384, 924–935. [Google Scholar] [CrossRef]
- Balducci, E.; Kaltenbach, S.; Villarese, P.; Duroyon, E.; Zalmai, L.; Friedrich, C.; Suarez, F.; Marcais, A.; Bouscary, D.; Decroocq, J.; et al. Optical genome mapping refines cytogenetic diagnostics, prognostic stratification and provides new molecular insights in adult MDS/AML patients. Blood Cancer J. 2022, 12, 126. [Google Scholar] [CrossRef] [PubMed]
- Nilius-Eliliwi, V.; Gerding, W.M.; Schroers, R.; Nguyen, H.P.; Vangala, D.B. Optical Genome Mapping for Cytogenetic Diagnostics in AML. Cancers 2023, 15, 1684. [Google Scholar] [CrossRef] [PubMed]
- Nilius-Eliliwi, V.; Tembrink, M.; Gerding, W.M.; Lubieniecki, K.P.; Lubieniecka, J.M.; Kankel, S.; Liehr, T.; Mika, T.; Dimopoulos, F.; Döhner, K.; et al. Broad genomic workup including optical genome mapping uncovers a DDX3X: MLLT10 gene fusion in acute myeloid leukemia. Front. Oncol. 2022, 12, 959243. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant Classification | Variant Types | Variant Classes | Current Diagnostic Workflow | Duncavage et al., 2021 [19] | Proposed Workflow | |||
|---|---|---|---|---|---|---|---|---|
| Karyotype | FISH | 54-Gene NGS Panel | Short-Read WGS | OGM | 523-Gene NGS Panel | |||
| Sequencing variants and coverage | SNVs | _ | _ | _ | >500x | 50x (probability of false negatives for variants detected at low VAF with 500x panels) | _ | >250x |
| Indels | _ | _ | _ | √* | √* | √# | √* | |
| Aneuploidy | Monosomy | Monosomy | √ | Targeted FISH probes | _ | √ | √ | _ |
| Trisomy | Trisomy | √ | Targeted FISH probes | _ | √ | √ | _ | |
| Triploidy | Triploidy | √ | Targeted FISH probes | _ | √ | no | _ | |
| Tetraploidy | Tetraploidy | √ | Targeted FISH probes | _ | √ | no | _ | |
| Ring chromosome | Ring chromosome | √ | No | _ | No | ≥500 kbp + fusion break | _ | |
| Copy Number Variants | Deletions/Duplication | Interstitial | 5 Mb or larger | Targeted FISH probes | _ | 5 Mb or larger | ≥5 kbp | _ |
| Terminal | 5 Mb or larger | Targeted FISH probes | _ | 5 Mb or larger | ≥5 kbp | _ | ||
| Insertion | Interstitial (unknown sequence) | 5 Mb or larger | No | _ | No | ~5 kbp | _ | |
| _ | _ | |||||||
| Structural variants | Translocations | Balanced translocations | √ | Dependent on FISH probes | _ | Only recurrent translocations were investigated with current bioinformatics processing | √ | _ |
| Unbalanced translocations | √ | Dependent on FISH probes | _ | Only recurrent translocations were investigated with current bioinformatics processing | √ | _ | ||
| Inversions | Pericentric | _ | No | √ | _ | |||
| Paracentric | 5 Mb or larger | No | _ | No | ≥30 kbp | _ | ||
| Homozygosity mapping | LOH | AOH/ROH/LOH | No | No | _ | √ | 25 mb ^ | _ |
| Microsatellite/Macrostaellite repeats | Repeats | Expansions/Contractions | No | No | _ | Limited to short repeats | ≥500 bp | _ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahajpal, N.S.; Mondal, A.K.; Singh, H.; Vashisht, A.; Ananth, S.; Saul, D.; Hastie, A.R.; Hilton, B.; DuPont, B.R.; Savage, N.M.; et al. Clinical Utility of Optical Genome Mapping and 523-Gene Next Generation Sequencing Panel for Comprehensive Evaluation of Myeloid Cancers. Cancers 2023, 15, 3214. https://doi.org/10.3390/cancers15123214
Sahajpal NS, Mondal AK, Singh H, Vashisht A, Ananth S, Saul D, Hastie AR, Hilton B, DuPont BR, Savage NM, et al. Clinical Utility of Optical Genome Mapping and 523-Gene Next Generation Sequencing Panel for Comprehensive Evaluation of Myeloid Cancers. Cancers. 2023; 15(12):3214. https://doi.org/10.3390/cancers15123214
Chicago/Turabian StyleSahajpal, Nikhil Shri, Ashis K. Mondal, Harmanpreet Singh, Ashutosh Vashisht, Sudha Ananth, Daniel Saul, Alex R. Hastie, Benjamin Hilton, Barbara R. DuPont, Natasha M. Savage, and et al. 2023. "Clinical Utility of Optical Genome Mapping and 523-Gene Next Generation Sequencing Panel for Comprehensive Evaluation of Myeloid Cancers" Cancers 15, no. 12: 3214. https://doi.org/10.3390/cancers15123214
APA StyleSahajpal, N. S., Mondal, A. K., Singh, H., Vashisht, A., Ananth, S., Saul, D., Hastie, A. R., Hilton, B., DuPont, B. R., Savage, N. M., Kota, V., Chaubey, A., Cortes, J. E., & Kolhe, R. (2023). Clinical Utility of Optical Genome Mapping and 523-Gene Next Generation Sequencing Panel for Comprehensive Evaluation of Myeloid Cancers. Cancers, 15(12), 3214. https://doi.org/10.3390/cancers15123214