


Immunotherapy for Recurrent Glioma—From Bench to Bedside

Abstract
:Simple Summary
Abstract
1. Introduction
2. Recurrent Glioma Features
3. Immune Checkpoint Blockade
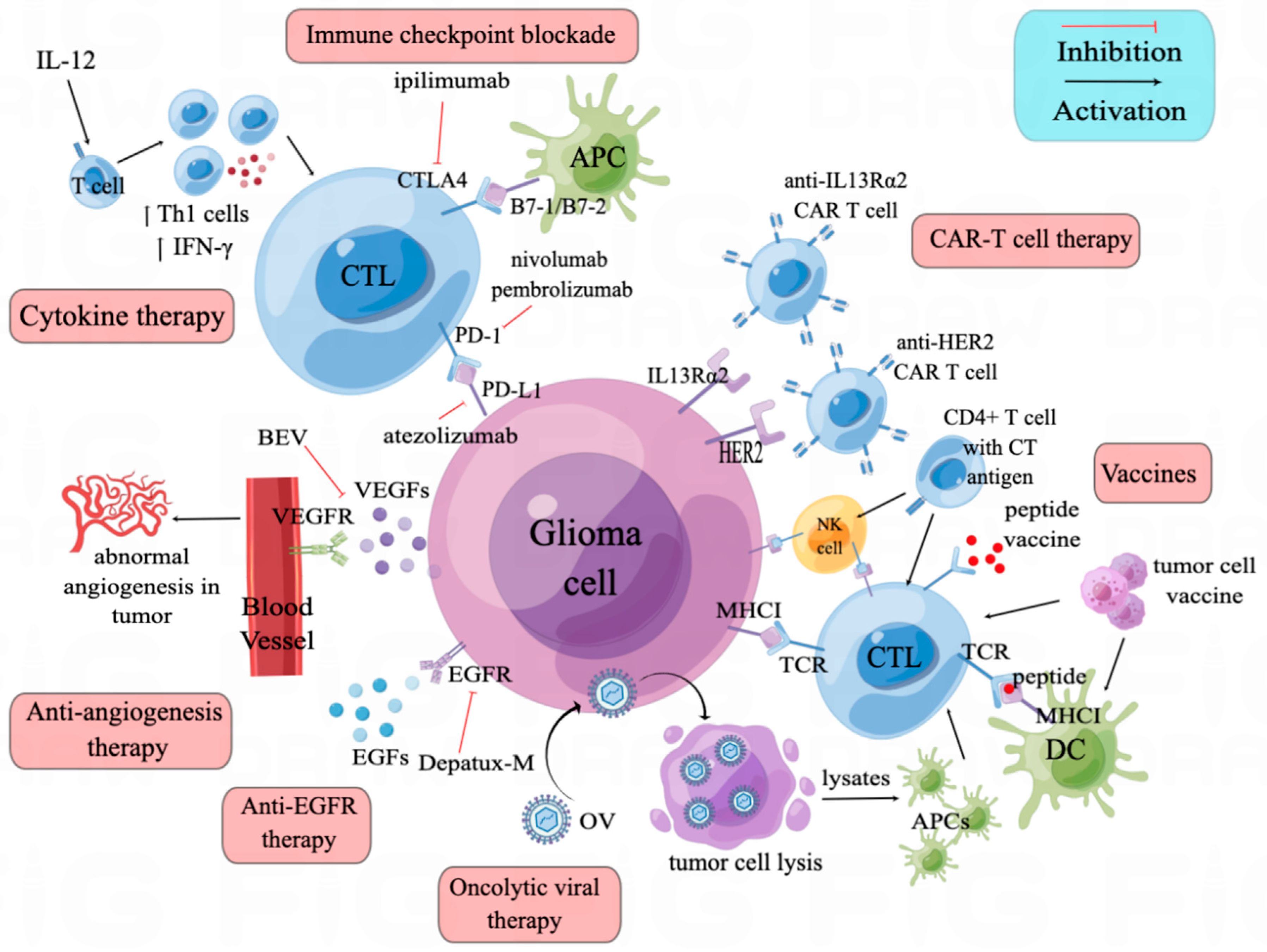
3.1. CTLA-4/B7 Axis
3.2. PD-1/PD-L1 Axis
4. Anti-Angiogenesis Therapy
5. Anti-EGFR Therapy
6. Cytokine Therapy
7. Oncolytic Viruses
7.1. Retrovirus
7.2. Adenovirus
7.3. HSV-1
7.4. Parvovirus
8. Vaccines and Cell-Based Immunotherapies
8.1. Immune Cell and Cancer Cell Vaccines/Cell-Based Immunotherapies
8.1.1. Dendritic Cell Vaccines
8.1.2. Glioma (Stem) Cell Vaccines
8.1.3. T-Cell-Based Immunotherapy
8.1.4. NK-Cell-Based Immunotherapy
8.2. Peptide Vaccines
9. CAR-T Therapy
10. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Marra, J.S.; Mendes, G.P.; Yoshinari, G.H.; Guimarães, F.D.S.; Mazin, S.C.; de Oliveira, H.F. Survival after radiation therapy for high-grade glioma. Rep. Pract. Oncol. Radiother. 2019, 24, 35–40. [Google Scholar] [CrossRef]
- Chen, W.; Wang, Y.; Zhao, B.; Liu, P.; Liu, L.; Wang, Y.; Ma, W. Optimal Therapies for Recurrent Glioblastoma: A Bayesian Network Meta-Analysis. Front. Oncol. 2021, 11, 641878. [Google Scholar] [CrossRef]
- TTan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Tang, L.; Li, X.; Fan, F.; Liu, Z. Immunotherapy for glioma: Current management and future application. Cancer Lett. 2020, 476, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Wang, W.; Li, H.; Jiao, Y.; Huo, R.; Yan, Z.; Wang, J.; Wang, S.; Wang, J.; Chen, D.; et al. Single-Cell Atlas Reveals Complexity of the Immunosuppressive Microenvironment of Initial and Recurrent Glioblastoma. Front. Immunol. 2020, 11, 835. [Google Scholar] [CrossRef]
- Lukas, R.V.; Rodon, J.; Becker, K.; Wong, E.T.; Shih, K.; Touat, M.; Fassò, M.; Osborne, S.; Molinero, L.; O’Hear, C.; et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J. Neuro Oncol. 2018, 140, 317–328. [Google Scholar] [CrossRef]
- Varn, F.S.; Johnson, K.C.; Martinek, J.; Huse, J.T.; Nasrallah, M.P.; Wesseling, P.; Cooper, L.A.; Malta, T.M.; Wade, T.E.; Sabedot, T.S.; et al. Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell 2022, 185, 2184–2199.e16. [Google Scholar] [CrossRef]
- Youssef, G.; Dietrich, J. Ipilimumab: An investigational immunotherapy for glioblastoma. Expert Opin. Investig. Drugs 2020, 29, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, C.S.; Hunn, M.K.; Ferguson, P.M.; Ruedl, C.; Ancelet, L.R.; Hermans, I.F. Blocking CTLA-4 while priming with a whole cell vaccine reshapes the oligoclonal T cell infiltrate and eradicates tumors in an orthotopic glioma model. Oncoimmunology 2017, 7, e1376154. [Google Scholar] [CrossRef] [PubMed]
- Genoud, V.; Espinoza, F.I.; Marinari, E.; Rochemont, V.; Dietrich, P.-Y.; McSheehy, P.; Bachmann, F.; Lane, H.A.; Walker, P.R. Treating ICB-resistant glioma with anti-CD40 and mitotic spindle checkpoint controller BAL101553 (lisavanbulin). JCI Insight 2021, 6, e142980. [Google Scholar] [CrossRef]
- Aslan, K.; Turco, V.; Blobner, J.; Sonner, J.K.; Liuzzi, A.R.; Núñez, N.G.; De Feo, D.; Kickingereder, P.; Fischer, M.; Green, E.; et al. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nat. Commun. 2020, 11, 931. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.F.; Ng, S.M.; Brooks, C.; Coutts, T.; Holmes, J.; Roberts, C.; Elhussein, L.; Hoskin, P.; Maughan, T.; Blagden, S.; et al. A phase II open label, randomised study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: The Ipi-Glio trial protocol. BMC Cancer 2020, 20, 198. [Google Scholar] [CrossRef] [Green Version]
- Litak, J.; Mazurek, M.; Grochowski, C.; Kamieniak, P.; Roliński, J. PD-L1/PD-1 Axis in Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 5347. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Dai, Z.; Wu, W.; Wang, Z.; Zhang, N.; Zhang, L.; Zeng, W.J.; Liu, Z.; Cheng, Q. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 184. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2019, 29, 3766. [Google Scholar] [CrossRef]
- Quandt, D.; Jasinski-Bergner, S.; Müller, U.; Schulze, B.; Seliger, B. Synergistic effects of IL-4 and TNFα on the induction of B7-H1 in renal cell carcinoma cells inhibiting allogeneic T cell proliferation. J. Transl. Med. 2014, 12, 151. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.Y.; Ma, T.T.; Wu, B.T.; Lin, Y.; Tu, Z.G. IL-12 regulates B7-H1 expression in ovarian cancer-associated macrophages by effects on NF-κB signalling. Asian Pac. J. Cancer Prev. 2014, 15, 5767–5772. [Google Scholar] [CrossRef] [Green Version]
- DiDomenico, J.; Lamano, J.B.; Oyon, D.; Li, Y.; Veliceasa, D.; Kaur, G.; Ampie, L.; Choy, W.; Lamano, J.B.; Bloch, O. The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology 2018, 7, e1448329. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.; Hasan, N.; Begum, G.; Kohanbash, G.; Carney, K.E.; Pigott, V.M.; Persson, A.I.; Castro, M.G.; Jia, W.; Sun, D. Blockade of Na/H exchanger stimulates glioma tumor immunogenicity and enhances combinatorial TMZ and anti-PD-1 therapy. Cell Death Dis. 2018, 9, 1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, B.; Qi, N.; Li, J.; Zhang, G. Temozolomide combined with PD-1 Antibody therapy for mouse orthotopic glioma model. Biochem. Biophys. Res. Commun. 2018, 501, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Riva, M.; Wouters, R.; Sterpin, E.; Giovannoni, R.; Boon, L.; Himmelreich, U.; Gsell, W.; Van Ranst, M.; Coosemans, A. Radiotherapy, Temozolomide, and Antiprogrammed Cell Death Protein 1 Treatments Modulate the Immune Microenvironment in Experimental High-Grade Glioma. Neurosurgery 2020, 88, E205–E215. [Google Scholar] [CrossRef] [PubMed]
- Kadiyala, P.; Carney, S.V.; Gauss, J.C.; Garcia-Fabiani, M.B.; Haase, S.; Alghamri, M.S.; Núñez, F.J.; Liu, Y.; Yu, M.; Taher, A.W.; et al. Inhibition of 2-hydroxyglutarate elicits metabolic reprogramming and mutant IDH1 glioma immunity in mice. J. Clin. Investig. 2021, 131, e139542. [Google Scholar] [CrossRef] [PubMed]
- Dang, W.; Xiao, J.; Ma, Q.; Miao, J.; Cao, M.; Chen, L.; Shi, Y.; Yao, X.; Yu, S.; Liu, X.; et al. Combination of p38 MAPK inhibitor with PD-L1 antibody effectively prolongs survivals of temozolomide-resistant glioma-bearing mice via reduction of infiltrating glioma-associated macrophages and PD-L1 expression on resident glioma-associated microglia. Brain Tumor Pathol. 2021, 38, 189–200. [Google Scholar] [CrossRef]
- Mantica, M.; Pritchard, A.; Lieberman, F.; Drappatz, J. Retrospective study of nivolumab for patients with recurrent high grade gliomas. J. Neuro-Oncol. 2018, 139, 625–631. [Google Scholar] [CrossRef]
- Reiss, S.N.; Yerram, P.; Modelevsky, L.; Grommes, C. Retrospective review of safety and efficacy of programmed cell death-1 inhibitors in refractory high grade gliomas. J. Immunother. Cancer 2017, 5, 99. [Google Scholar] [CrossRef]
- Aoki, T.; Kagawa, N.; Sugiyama, K.; Wakabayashi, T.; Arakawa, Y.; Yamaguchi, S.; Tanaka, S.; Ishikawa, E.; Muragaki, Y.; Nagane, M.; et al. Efficacy and safety of nivolumab in Japanese patients with first recurrence of glioblastoma: An open-label, non-comparative study. Int. J. Clin. Oncol. 2021, 26, 2205–2215. [Google Scholar] [CrossRef]
- Gorsi, H.S.; Malicki, D.M.; Barsan, V.; Tumblin, M.; Yeh-Nayre, L.; Milburn, M.; Elster, J.D.; Crawford, J.R. Nivolumab in the Treatment of Recurrent or Refractory Pediatric Brain Tumors: A Single Institutional Experience. J. Pediatr. Hematol. 2019, 41, e235–e241. [Google Scholar] [CrossRef] [PubMed]
- Kurz, S.C.; Cabrera, L.P.; Hastie, D.; Huang, R.; Unadkat, P.; Rinne, M.; Nayak, L.; Lee, E.Q.; Reardon, D.A.; Wen, P.Y. PD-1 inhibition has only limited clinical benefit in patients with recurrent high-grade glioma. Neurology 2018, 91, e1355–e1359. [Google Scholar] [CrossRef]
- Lombardi, G.; Barresi, V.; Indraccolo, S.; Simbolo, M.; Fassan, M.; Mandruzzato, S.; Simonelli, M.; Caccese, M.; Pizzi, M.; Fassina, A.; et al. Pembrolizumab Activity in Recurrent High-Grade Gliomas with Partial or Complete Loss of Mismatch Repair Protein Expression: A Monocentric, Observational and Prospective Pilot Study. Cancers 2020, 12, 2283. [Google Scholar] [CrossRef] [PubMed]
- Nayak, L.; Molinaro, A.M.; Peters, K.B.; Clarke, J.L.; Jordan, J.T.; de Groot, J.F.; Nghiemphu, P.L.; Kaley, T.J.; Colman, H.; McCluskey, C.; et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2021, 27, 1048–1057. [Google Scholar] [CrossRef]
- Reardon, D.A.; Kim, T.M.; Frenel, J.; Simonelli, M.; Lopez, J.; Subramaniam, D.S.; Siu, L.L.; Wang, H.; Krishnan, S.; Stein, K.; et al. Treatment with pembrolizumab in programmed death ligand 1–positive recurrent glioblastoma: Results from the multicohort phase 1 KEYNOTE-028 trial. Cancer 2021, 127, 1620–1629. [Google Scholar] [CrossRef]
- Park, J.; Kwon, M.; Kim, K.H.; Kim, T.-S.; Hong, S.-H.; Kim, C.G.; Kang, S.-G.; Moon, J.H.; Kim, E.H.; Park, S.-H.; et al. Immune Checkpoint Inhibitor-induced Reinvigoration of Tumor-infiltrating CD8+ T Cells is Determined by Their Differentiation Status in Glioblastoma. Clin. Cancer Res. 2019, 25, 2549–2559. [Google Scholar] [CrossRef] [Green Version]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef] [Green Version]
- Duerinck, J.; Schwarze, J.K.; Awada, G.; Tijtgat, J.; Vaeyens, F.; Bertels, C.; Geens, W.; Klein, S.; Seynaeve, L.; Cras, L.; et al. Intracerebral administration of CTLA-4 and PD-1 immune checkpoint blocking monoclonal antibodies in patients with recurrent glioblastoma: A phase I clinical trial. J. Immunother. Cancer 2021, 9, e002296. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Reardon, D.A.; Lassman, A.B.; Schiff, D.; Yunus, S.A.; Gerstner, E.R.; Cloughesy, T.F.; Lee, E.Q.; Bs, S.C.G.; Bs, J.B.; Ma, A.M.; et al. Phase 2 and biomarker study of trebananib, an angiopoietin-blocking peptibody, with and without bevacizumab for patients with recurrent glioblastoma. Cancer 2018, 124, 1438–1448. [Google Scholar] [CrossRef]
- Sahebjam, S.; Forsyth, P.A.; Tran, N.D.; Arrington, J.A.; Macaulay, R.; Etame, A.B.; Walko, C.M.; Boyle, T.; Peguero, E.N.; Jaglal, M.; et al. Hypofractionated stereotactic re-irradiation with pembrolizumab and bevacizumab in patients with recurrent high-grade gliomas: Results from a phase I study. Neuro-Oncology 2021, 23, 677–686. [Google Scholar] [CrossRef]
- Awada, G.; Ben Salama, L.; De Cremer, J.; Schwarze, J.K.; Fischbuch, L.; Seynaeve, L.; Du Four, S.; Vanbinst, A.-M.; Michotte, A.; Everaert, H.; et al. Axitinib plus avelumab in the treatment of recurrent glioblastoma: A stratified, open-label, single-center phase 2 clinical trial (GliAvAx). J. Immunother. Cancer 2020, 8, e001146. [Google Scholar] [CrossRef]
- Nayak, L.; Standifer, N.; Dietrich, J.; Clarke, J.L.; Dunn, G.P.; Lim, M.; Cloughesy, T.; Gan, H.K.; Flagg, E.; George, E.; et al. Circulating Immune Cell and Outcome Analysis from the Phase II Study of PD-L1 Blockade with Durvalumab for Newly Diagnosed and Recurrent Glioblastoma. Clin. Cancer Res. 2022, 28, 2567–2578. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Plate, K.H.; Scholz, A.; Dumont, D.J. Tumor angiogenesis and anti-angiogenic therapy in malignant gliomas revisited. Acta Neuropathol. 2012, 124, 763–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin(R)) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- Quillien, V.; Carpentier, A.F.; Gey, A.; Avril, T.; Tartour, E.; Sejalon, F.; Campillo-Gimenez, B.; Vauleon, E. Absolute numbers of regulatory T cells and neutrophils in corticosteroid-free patients are predictive for response to bevacizumab in recurrent glioblastoma patients. Cancer Immunol. Immunother. 2019, 68, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Badruddoja, M.A.; Pazzi, M.; Sanan, A.; Schroeder, K.; Kuzma, K.; Norton, T.; Scully, T.; Mahadevan, D.; Ahmadi, M.M. Phase II study of bi-weekly temozolomide plus bevacizumab for adult patients with recurrent glioblastoma. Cancer Chemother. Pharmacol. 2017, 80, 715–721. [Google Scholar] [CrossRef]
- Brandes, A.A.; Gil-Gil, M.; Saran, F.; Carpentier, A.F.; Nowak, A.K.; Mason, W.; Zagonel, V.; Dubois, F.; Finocchiaro, G.; Fountzilas, G.; et al. A Randomized Phase II Trial (TAMIGA) Evaluating the Efficacy and Safety of Continuous Bevacizumab Through Multiple Lines of Treatment for Recurrent Glioblastoma. Oncologist 2019, 24, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Bent, M.J.v.D.; Klein, M.; Smits, M.; Reijneveld, J.C.; French, P.J.; Clement, P.; de Vos, F.Y.F.; Wick, A.; Mulholland, P.J.; Taphoorn, M.J.B.; et al. Bevacizumab and temozolomide in patients with first recurrence of WHO grade II and III glioma, without 1p/19q co-deletion (TAVAREC): A randomised controlled phase 2 EORTC trial. Lancet Oncol. 2018, 19, 1170–1179. [Google Scholar] [CrossRef] [Green Version]
- Draaisma, K.; Tesileanu, C.M.S.; de Heer, I.; Klein, M.; Smits, M.; Reijneveld, J.C.; Clement, P.M.; de Vos, F.Y.F.; Wick, A.; Mulholland, P.J.; et al. Prognostic Significance of DNA Methylation Profiles at MRI Enhancing Tumor Recurrence: A Report from the EORTC 26091 TAVAREC Trial. Clin. Cancer Res. 2022, 28, 2440–2448. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Anderson, S.K.; Twohy, E.L.; Bs, X.W.C.; Dixon, J.G.; Tran, D.D.; Jeyapalan, S.A.; Anderson, D.M.; Kaufmann, T.J.; Bs, R.W.F.; et al. A phase 1 and randomized, placebo-controlled phase 2 trial of bevacizumab plus dasatinib in patients with recurrent glioblastoma: Alliance/North Central Cancer Treatment Group N0872. Cancer 2019, 125, 3790–3800. [Google Scholar] [CrossRef] [PubMed]
- Ghiaseddin, A.; Reardon, D.; Massey, W.; Mannerino, A.; Lipp, E.S.; Ii, J.E.H.; McSherry, F.; Desjardins, A.; Randazzo, D.; Friedman, H.S.; et al. Phase II Study of Bevacizumab and Vorinostat for Patients with Recurrent World Health Organization Grade 4 Malignant Glioma. Oncol. 2017, 23, 157-e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, K.B.; Lipp, E.S.; Miller, E.; Herndon, J.E.; McSherry, F.; Desjardins, A.; Reardon, D.A.; Friedman, H.S. Phase I/II trial of vorinostat, bevacizumab, and daily temozolomide for recurrent malignant gliomas. J. Neuro-Oncol. 2018, 137, 349–356. [Google Scholar] [CrossRef]
- Puduvalli, V.K.; Wu, J.; Yuan, Y.; Armstrong, T.S.; Vera, E.; Wu, J.; Xu, J.; Giglio, P.; Colman, H.; Walbert, T.; et al. A Bayesian adaptive randomized phase II multicenter trial of bevacizumab with or without vorinostat in adults with recurrent glioblastoma. Neuro-Oncology 2020, 22, 1505–1515. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Becker, K.P.; Mekhail, T.; Chowdhary, S.A.; Eakle, J.F.; Wright, D.; Langdon, R.M.; Yost, K.J.; Padula, G.D.A.; West-Osterfield, K.; et al. Phase I/II study of bevacizumab with BKM120, an oral PI3K inhibitor, in patients with refractory solid tumors (phase I) and relapsed/refractory glioblastoma (phase II). J. Neuro-Oncol. 2019, 144, 303–311. [Google Scholar] [CrossRef]
- Lee, E.Q.; Duda, D.G.; Muzikansky, A.; Gerstner, E.R.; Kuhn, J.G.; Reardon, D.A.; Nayak, L.; Norden, A.D.; Doherty, L.; LaFrankie, D.; et al. Phase I and Biomarker Study of Plerixafor and Bevacizumab in Recurrent High-Grade Glioma. Clin. Cancer Res. 2018, 24, 4643–4649. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Q.; Zhang, P.; Wen, P.Y.; Gerstner, E.R.; Reardon, D.A.; Aldape, K.D.; deGroot, J.F.; Pan, E.; Raizer, J.J.; Kim, L.J.; et al. NRG/RTOG 1122: A phase 2, double-blinded, placebo-controlled study of bevacizumab with and without trebananib in patients with recurrent glioblastoma or gliosarcoma. Cancer 2020, 126, 2821–2828. [Google Scholar] [CrossRef]
- Yoshimoto, K.; Dang, J.; Zhu, S.; Nathanson, D.; Huang, T.; Dumont, R.; Seligson, D.B.; Yong, W.H.; Xiong, Z.; Rao, N.; et al. Development of a real-time RT-PCR assay for detecting EGFRvIII in glioblastoma samples. Clin. Cancer Res. 2008, 14, 488–493. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current Therapeutic Advances Targeting EGFR and EGFRvIII in Glioblastoma. Front. Oncol. 2015, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Van den Bent, M.J.; Gao, Y.; Kerkhof, M.; Kros, J.M.; Gorlia, T.; Van Zwieten, K.; Prince, J.; Van Duinen, S.; Sillevis Smitt, P.A.; Taphoorn, M.; et al. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro-Oncology 2015, 17, 935–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidya, K.S.; Mitten, M.J.; Zelaya-Lazo, A.L.; Oleksijew, A.; Alvey, C.; Falls, H.D.; Mishra, S.; Palma, J.; Ansell, P.; Phillips, A.C.; et al. Synergistic therapeutic benefit by combining the antibody drug conjugate, depatux-m with temozolomide in pre-clinical models of glioblastoma with overexpression of EGFR. J. Neuro-Oncol. 2021, 152, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bent, M.; Gan, H.K.; Lassman, A.B.; Kumthekar, P.; Merrell, R.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L.; Papadopoulos, K.P.; et al. Efficacy of depatuxizumab mafodotin (ABT-414) monotherapy in patients with EGFR-amplified, recurrent glioblastoma: Results from a multi-center, international study. Cancer Chemother. Pharmacol. 2017, 80, 1209–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, H.K.; Reardon, D.A.; Lassman, A.B.; Merrell, R.; Van Den Bent, M.; Butowski, N.; Lwin, Z.; Wheeler, H.; Fichtel, L.; Scott, A.M.; et al. Safety, pharmacokinetics, and antitumor response of depatuxizumab mafodotin as monotherapy or in combination with temozolomide in patients with glioblastoma. Neuro-Oncology 2018, 20, 838–847. [Google Scholar] [CrossRef]
- Lassman, A.B.; van den Bent, M.J.; Gan, H.K.; Reardon, D.A.; Kumthekar, P.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L.B.; Papadopoulos, K.P.; et al. Safety and efficacy of depatuxizumab mafodotin + temozolomide in patients with EGFR-amplified, recurrent glioblastoma: Results from an international phase I multicenter trial. Neuro-Oncology 2019, 21, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Van Den Bent, M.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.S.; Franceschi, E.; Clement, P.M.; Chinot, O.; De Vos, F.; et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro-Oncology 2020, 22, 684–693. [Google Scholar] [CrossRef]
- Clement, P.M.J.; Dirven, L.; Eoli, M.; Sepulveda-Sanchez, J.M.; Walenkamp, A.M.E.; Frenel, J.S.; Franceschi, E.; Weller, M.; Chinot, O.; De Vos, F.Y.F.L.; et al. Impact of depatuxizumab mafodotin on health-related quality of life and neurological functioning in the phase II EORTC 1410/INTELLANCE 2 trial for EGFR-amplified recurrent glioblastoma. Eur. J. Cancer 2021, 147, 1–12. [Google Scholar] [CrossRef]
- Narita, Y.; Muragaki, Y.; Kagawa, N.; Asai, K.; Nagane, M.; Matsuda, M.; Ueki, K.; Kuroda, J.; Date, I.; Kobayashi, H.; et al. Safety and efficacy of depatuxizumab mafodotin in Japanese patients with malignant glioma: A nonrandomized, phase 1/2 trial. Cancer Sci. 2021, 112, 5020–5033. [Google Scholar] [CrossRef]
- Lasek, W.; Zagozdzon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef] [Green Version]
- Vom Berg, J.; Vrohlings, M.; Haller, S.; Haimovici, A.; Kulig, P.; Sledzinska, A.; Weller, M.; Becher, B. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. J. Exp. Med. 2013, 210, 2803–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiocca, E.A.; Yu, J.S.; Lukas, R.V.; Solomon, I.H.; Ligon, K.L.; Nakashima, H.; Triggs, D.A.; Reardon, D.A.; Wen, P.; Stopa, B.M.; et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci. Transl. Med. 2019, 11, eaaw568. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Gelb, A.B.; Chen, C.C.; Rao, G.; Reardon, D.A.; Wen, P.Y.; Bi, W.L.; Peruzzi, P.; Amidei, C.; Triggs, D.; et al. Combined immunotherapy with controlled interleukin-12 gene therapy and immune checkpoint blockade in recurrent glioblastoma: An open-label, multi-institutional phase I trial. Neuro-Oncology 2022, 24, 951–963. [Google Scholar] [CrossRef]
- Hogan, D.J.; Zhu, J.-J.; Diago, O.R.; Gammon, D.K.; Haghighi, A.; Lu, G.; Das, A.; Gruber, H.E.; Jolly, D.J.; Ostertag, D. Molecular Analyses Support the Safety and Activity of Retroviral Replicating Vector Toca 511 in Patients. Clin. Cancer Res. 2018, 24, 4680–4693. [Google Scholar] [CrossRef] [Green Version]
- Cloughesy, T.F.; Landolfi, J.; Vogelbaum, M.A.; Ostertag, D.; Elder, J.B.; Bloomfield, S.; Carter, B.; Chen, C.C.; Kalkanis, S.N.; Kesari, S.; et al. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro-Oncology 2018, 20, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Accomando, W.P.; Rao, A.R.; Hogan, D.J.; Newman, A.M.; Nakao, A.; Alizadeh, A.A.; Diehn, M.; Diago, O.R.; Gammon, D.K.; Haghighi, A.; et al. Molecular and Immunologic Signatures are Related to Clinical Benefit from Treatment with Vocimagene Amiretrorepvec (Toca 511) and 5-Fluorocytosine (Toca FC) in Patients with Glioma. Clin. Cancer Res. 2020, 26, 6176–6186. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Petrecca, K.; Walbert, T.; Butowski, N.; Salacz, M.; Perry, J.; Damek, D.; Bota, D.; Bettegowda, C.; Zhu, J.J.; et al. Effect of Vocimagene Amiretrorepvec in Combination With Flucytosine vs Standard of Care on Survival Following Tumor Resection in Patients With Recurrent High-Grade Glioma: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.J.; Peters, K.B.; Vredenburgh, J.; Bokstein, F.; Blumenthal, D.T.; Yust-Katz, S.; Peretz, I.; Oberman, B.; Freedman, L.S.; Ellingson, B.M.; et al. Safety and efficacy of VB-111, an anticancer gene therapy, in patients with recurrent glioblastoma: Results of a phase I/II study. Neuro-Oncology 2020, 22, 694–704. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Brenner, A.; de Groot, J.F.; Butowski, N.A.; Zach, L.; Campian, J.L.; Ellingson, B.M.; Freedman, L.S.; Cohen, Y.C.; Lowenton-Spier, N.; et al. A randomized controlled phase III study of VB-111 combined with bevacizumab vs bevacizumab monotherapy in patients with recurrent glioblastoma (GLOBE). Neuro-Oncology 2020, 22, 705–717. [Google Scholar] [CrossRef] [Green Version]
- Kieran, M.W.; Goumnerova, L.; Manley, P.; Chi, S.N.; Marcus, K.J.; Manzanera, A.G.; Polanco, M.L.S.; Guzik, B.W.; Aguilar-Cordova, E.; Diaz-Montero, C.M.; et al. Phase I study of gene-mediated cytotoxic immunotherapy with AdV-tk as adjuvant to surgery and radiation for pediatric malignant glioma and recurrent ependymoma. Neuro-Oncology 2019, 21, 537–546. [Google Scholar] [CrossRef]
- Trask, T.W.; Trask, R.P.; Aguilar-Cordova, E.; Shine, H.D.; Wyde, P.R.; Goodman, J.C.; Hamilton, W.J.; Rojas-Martinez, A.; Chen, S.H.; Woo, S.; et al. Phase I study of adenoviral delivery of the HSV-tk gene and ganciclovir administration in patients with current malignant brain tumors. Mol. Ther. 2000, 1, 195–203. [Google Scholar] [CrossRef]
- Todo, T.; Ino, Y.; Ohtsu, H.; Shibahara, J.; Tanaka, M. A phase I/II study of triple-mutated oncolytic herpes virus G47∆ in patients with progressive glioblastoma. Nat. Commun. 2022, 13, 4119. [Google Scholar] [CrossRef]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi–mutated herpes simplex virus–1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: Results of a phase I trial. Gene Ther. 2000, 7, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Todo, T.; Rabkin, S.; Sundaresan, P.; Wu, A.; Meehan, K.R.; Herscowitz, H.B.; Martuza, R.L. Systemic Antitumor Immunity in Experimental Brain Tumor Therapy Using a Multimutated, Replication-Competent Herpes Simplex Virus. Hum. Gene Ther. 1999, 10, 2741–2755. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.E.; Cassady, K.A.; Roth, J.C.; Clements, J.; Schieffer, K.M.; Leraas, K.; Miller, A.R.; Prasad, N.; Leavenworth, J.W.; Aban, I.B.; et al. Immune Activity and Response Differences of Oncolytic Viral Therapy in Recurrent Glioblastoma: Gene Expression Analyses of a Phase IB Study. Clin. Cancer Res. 2022, 28, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef] [Green Version]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.-O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef] [Green Version]
- Ardon, H.; Van Gool, S.W.; Verschuere, T.; Maes, W.; Fieuws, S.; Sciot, R.; Wilms, G.; Demaerel, P.; Goffin, J.; Van Calenbergh, F.; et al. Integration of autologous dendritic cell-based immunotherapy in the standard of care treatment for patients with newly diagnosed glioblastoma: Results of the HGG-2006 phase I/II trial. Cancer Immunol. Immunother. 2012, 61, 2033–2044. [Google Scholar] [CrossRef]
- Yao, Y.; Luo, F.; Tang, C.; Chen, D.; Qin, Z.; Hua, W.; Xu, M.; Zhong, P.; Yu, S.; Chen, D.; et al. Molecular subgroups and B7-H4 expression levels predict responses to dendritic cell vaccines in glioblastoma: An exploratory randomized phase II clinical trial. Cancer Immunol. Immunother. 2018, 67, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, Y.; Kikuchi, T.; Homma, S.; Koido, S.; Ohkusa, T.; Tasaki, T.; Hayashi, K.; Komita, H.; Watanabe, N.; Suzuki, Y.; et al. Phase I/II trial of combination of temozolomide chemotherapy and immunotherapy with fusions of dendritic and glioma cells in patients with glioblastoma. Cancer Immunol. Immunother. 2016, 65, 1499–1509. [Google Scholar] [CrossRef]
- Hu, J.L.; Omofoye, O.A.; Rudnick, J.D.; Kim, S.; Tighiouart, M.; Phuphanich, S.; Wang, H.; Mazer, M.; Ganaway, T.; Chu, R.M.; et al. A Phase I Study of Autologous Dendritic Cell Vaccine Pulsed with Allogeneic Stem-like Cell Line Lysate in Patients with Newly Diagnosed or Recurrent Glioblastoma. Clin. Cancer Res. 2022, 28, 689–696. [Google Scholar] [CrossRef]
- Rudnick, J.D.; Sarmiento, J.M.; Uy, B.; Nuno, M.; Wheeler, C.J.; Mazer, M.J.; Wang, H.; Hu, J.L.; Chu, R.M.; Phuphanich, S.; et al. A phase I trial of surgical resection with Gliadel Wafer placement followed by vaccination with dendritic cells pulsed with tumor lysate for patients with malignant glioma. J. Clin. Neurosci. 2020, 74, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, M.; VAN Gool, S.W.; Dionysiou, D.; Graf, N.; Stamatakos, G. Immune Phenotype Correlates With Survival in Patients With GBM Treated With Standard Temozolomide-based Therapy and Immunotherapy. Anticancer Res. 2019, 39, 2043–2051. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef]
- Lucas, K.G.; Bao, L.; Bruggeman, R.; Dunham, K.; Specht, C. The detection of CMV pp65 and IE1 in glioblastoma multiforme. J. Neuro-Oncol. 2010, 103, 231–238. [Google Scholar] [CrossRef]
- Dziurzynski, K.; Chang, S.M.; Heimberger, A.B.; Kalejta, R.F.; Dallas, S.R.M.; Smit, M.; Soroceanu, L.; Cobbs, C.S.; the HCMV and Gliomas Symposium. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro-Oncology 2012, 14, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E., 2nd; Sampson, J.H. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin. Cancer Res. 2020, 26, 5297–5303. [Google Scholar] [CrossRef]
- Stathopoulos, A.; Samuelson, C.; Milbouw, G.; Hermanne, J.P.; Schijns, V.E.J.; Chen, T.C. Therapeutic vaccination against malignant gliomas based on allorecognition and syngeneic tumor antigens: Proof of principle in two strains of rat. Vaccine 2008, 26, 1764–1772. [Google Scholar] [CrossRef]
- Bota, D.A.; Chung, J.; Dandekar, M.; Carrillo, J.A.; Kong, X.-T.; Fu, B.D.; Hsu, F.P.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C.; et al. Phase II study of ERC1671 plus bevacizumab versus bevacizumab plus placebo in recurrent glioblastoma: Interim results and correlations with CD4+T-lymphocyte counts. CNS Oncol. 2018, 7, CNS22. [Google Scholar] [CrossRef] [Green Version]
- Ogino, H.; Taylor, J.W.; Nejo, T.; Gibson, D.; Watchmaker, P.B.; Okada, K.; Saijo, A.; Tedesco, M.R.; Shai, A.; Wong, C.M.; et al. Randomized trial of neoadjuvant vaccination with tumor-cell lysate induces T cell response in low-grade gliomas. J. Clin. Investig. 2022, 132, e151239. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, A.F.; Dzhandzhugazyan, K.N.; Guldberg, P.; Fang, J.J.; Andersen, R.S.; Dahl, C.; Mortensen, J.; Lundby, T.; Wagner, A.; Law, I.; et al. Adoptive cancer immunotherapy using DNA-demethylated T helper cells as antigen-presenting cells. Nat. Commun. 2018, 9, 785. [Google Scholar] [CrossRef] [Green Version]
- Kennis, B.A.; Michel, K.A.; Brugmann, W.B.; Laureano, A.; Tao, R.-H.; Somanchi, S.S.; Einstein, S.A.; Bravo-Alegria, J.B.; Maegawa, S.; Wahba, A.; et al. Monitoring of intracerebellarly-administered natural killer cells with fluorine-19 MRI. J. Neuro-Oncol. 2019, 142, 395–407. [Google Scholar] [CrossRef]
- Ishikawa, E.; Tsuboi, K.; Saijo, K.; Harada, H.; Takano, S.; Nose, T.; Ohno, T. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer Res. 2004, 24, 1861–1871. [Google Scholar]
- Khatua, S.; Cooper, L.J.N.; Sandberg, D.I.; Ketonen, L.; Johnson, J.M.; Rytting, M.E.; Liu, D.D.; Meador, H.; Trikha, P.; Nakkula, R.J.; et al. Phase I study of intraventricular infusions of autologous ex vivo expanded NK cells in children with recurrent medulloblastoma and ependymoma. Neuro-Oncology 2020, 22, 1214–1225. [Google Scholar] [CrossRef]
- Fenstermaker, R.A.; Ciesielski, M.J.; Qiu, J.; Yang, N.; Frank, C.L.; Lee, K.P.; Mechtler, L.R.; Belal, A.; Ahluwalia, M.S.; Hutson, A.D. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol. Immunother. 2016, 65, 1339–1352. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, A.; Hashimoto, N.; Fujiki, F.; Morimoto, S.; Kagawa, N.; Nakajima, H.; Hosen, N.; Nishida, S.; Nakata, J.; Morita, S.; et al. A phase I clinical study of a cocktail vaccine of Wilms’ tumor 1 (WT1) HLA class I and II peptides for recurrent malignant glioma. Cancer Immunol. Immunother. 2018, 68, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Narita, Y.; Arakawa, Y.; Yamasaki, F.; Nishikawa, R.; Aoki, T.; Kanamori, M.; Nagane, M.; Kumabe, T.; Hirose, Y.; Ichikawa, T.; et al. A randomized, double-blind, phase III trial of personalized peptide vaccination for recurrent glioblastoma. Neuro-Oncology 2019, 21, 348–359. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Bunse, L.; Rupp, A.-K.; Poschke, I.; Bunse, T.; Lindner, K.; Wick, A.; Blobner, J.; Misch, M.; Tabatabai, G.; Glas, M.; et al. AMPLIFY-NEOVAC: A randomized, 3-arm multicenter phase I trial to assess safety, tolerability and immunogenicity of IDH1-vac combined with an immune checkpoint inhibitor targeting programmed death-ligand 1 in isocitrate dehydrogenase 1 mutant gliomas. Neurol. Res. Pract. 2022, 4, 20. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.-C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor–transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Johnson, A.J.; Wilson, A.L.; Brown, C.; Yokoyama, J.K.; Künkele, A.; Chang, C.A.; Rawlings-Rhea, S.; Huang, W.; Seidel, K.; et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: An interim analysis. Nat. Med. 2021, 27, 1544–1552. [Google Scholar] [CrossRef]
- Brown, C.E.; Rodriguez, A.; Palmer, J.; Ostberg, J.R.; Naranjo, A.; Wagner, J.R.; Aguilar, B.; Starr, R.; Weng, L.; Synold, T.W.; et al. Off-the-shelf, steroid-resistant, IL13Rα2-specific CAR T cells for treatment of glioblastoma. Neuro-Oncology 2022, 24, 1318–1330. [Google Scholar] [CrossRef]
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. Checkpoint Blockade Reverses Anergy in IL-13Rα2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol. Ther. Oncolytics 2018, 11, 20–38. [Google Scholar] [CrossRef] [Green Version]
- Okonogi, N.; Shirai, K.; Oike, T.; Murata, K.; Noda, S.-E.; Suzuki, Y.; Nakano, T. Topics in chemotherapy, molecular-targeted therapy, and immunotherapy for newly-diagnosed glioblastoma multiforme. Anticancer Res. 2015, 35, 1229–1236. [Google Scholar]
- Di Giacomo, A.M.; Mair, M.J.; Ceccarelli, M.; Anichini, A.; Ibrahim, R.; Weller, M.; Lahn, M.; Eggermont, A.M.; Fox, B.; Maio, M. Immunotherapy for brain metastases and primary brain tumors. Eur. J. Cancer 2023, 179, 113–120. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Study Number | Intervention | Disease | Phase | Status |
|---|---|---|---|---|
| NCT04145115 | Ipilimumab + nivolumab | Recurrent Astrocytoma, IDH-Mutant, Grade 4/Recurrent Glioblastoma, IDH Wildtype/Secondary Glioblastoma | 2 | Recruiting |
| NCT04323046 | Ipilimumab + nivolumab | Recurrent Glioblastoma/Recurrent Malignant Glioma/Recurrent Grade III Glioma | 1 | Active, not recruiting |
| NCT03890952 | Nivolumab + bevacizumab (BEV) | Recurrent Adult Brain Tumor | 2 | Active, not recruiting |
| NCT05540275 | Sintilimab plus Bevacizumab | Recurrent Glioblastoma | 2 | Not yet recruiting |
| NCT02658279 | Pembrolizumab | Glioma/Recurrent Malignant Glioma | Not applicable | Active, not recruiting |
| NCT02359565 | Pembrolizumab | Recurrent (Refractory) Brain Neoplasm/Recurrent Childhood Ependymoma/Recurrent (Refractory) Diffuse Intrinsic Pontine Glioma/Recurrent (Refractory) Medulloblastoma/Refractory Ependymoma | 1 | Recruiting |
| NCT03722342 | Pembrolizumab + TTAC-0001 | Recurrent Glioblastoma | 1 | Active, not recruiting |
| NCT02337686 | Pembrolizumab | Recurrent Glioblastoma/Recurrent Gliosarcoma | 2 | Active, not recruiting |
| NCT04201873 | Pembrolizumab + dendritic cell tumor cell lysate vaccine | Recurrent Glioblastoma | 1 | Recruiting |
| NCT04013672 | Pembrolizumab + SurVaxM + Sargramostim + Montanide ISA 51 | Recurrent Glioblastoma | 2 | Active, not recruiting |
| NCT05465954 | Pembrolizumab + efineptakin alfa | Recurrent Glioblastoma, IDH-Wildtype/Recurrent Gliosarcoma | 2 | Recruiting |
| NCT04479241 | Pembrolizumab + lerapolturev | Recurrent Glioblastoma | 2 | Active, not recruiting |
| NCT05053880 | Pembrolizumab + ACT001 | Recurrent Glioblastoma Multiforme (GBM) | 1/2 | Recruiting |
| NCT05463848 | Pembrolizumab + Olaparib + Temozolomide | Recurrent Glioblastoma | 2 | Recruiting |
| Study Number | Intervention | Disease | Phase | Status |
|---|---|---|---|---|
| NCT03152318 | rQNestin 34.5 (HSV-1) + Cyclophosphamide | Malignant Glioma of Brain/Astrocytoma/Malignant Astrocytoma/Oligodendroglioma/Anaplastic Oligodendroglioma of Brain (Diagnosis)/Mixed Oligoastrocytoma/Ependymoma/Ganglioglioma/Pylocytic/Pylomyxoid Astrocytoma | 1 | Recruiting |
| NCT04482933 | G207 (HSV-1) | High Grade Glioma/Glioblastoma Multiforme/Malignant Glioma of Brain/Anaplastic Astrocytoma of Brain/High-Grade Glioma/Anaplastic Glioma/Giant Cell Glioblastoma | 2 | Not yet recruiting |
| NCT02062827 | M032 (NSC 733972, HSV-1) | Recurrent Glioblastoma Multiforme/Progressive Glioblastoma Multiforme/Anaplastic Astrocytoma or Gliosarcoma | 1 | Active, not recruiting |
| NCT03657576 | C134 (HSV-1) | Glioblastoma Multiforme of Brain/Anaplastic Astrocytoma of Brain/Gliosarcoma of Brain | 1 | Active, not recruiting |
| NCT03896568 | Ad5-DNX-2401 (Adenovirus) | IDH1 wt Allele/Recurrent Anaplastic Astrocytoma/Recurrent Glioblastoma/Recurrent Gliosarcoma/Recurrent Malignant Glioma | 1 | Recruiting |
| NCT02457845 | G207 | Supratentorial Neoplasms, Malignant/Malignant Glioma/Glioblastoma/Anaplastic Astrocytoma/PNET/Cerebral Primitive/Neuroectodermal Tumor/Embryonal Tumor | 1 | Active, not recruiting |
| NCT03043391 | Polio/Rhinovirus Recombinant (PVSRIPO) | Malignant Glioma/Anaplastic Astrocytoma/Anaplastic Oligoastrocytoma/Anaplastic Oligodendroglioma/Glioblastoma/Gliosarcoma/Atypical Teratoid/Rhabdoid Tumor of Brain/Medulloblastoma/Ependymoma/Pleomorphic Xanthoastrocytoma of Brain/Embryonal Tumor of Brain | 1 | Active, not recruiting |
| NCT03911388 | G207 | Neoplasms, Brain/Glioblastoma Multiforme/Glioblastoma of Cerebellum/Neoplasms/Astrocytoma/Astrocytoma, Cerebellar/Neuroectodermal Tumors/Neuroectodermal Tumors, Primitive/Cerebellar PNET, Childhood/Cerebellar Neoplasms | 1 | Recruiting |
| Study Number | Intervention | Disease | Phase | Status |
|---|---|---|---|---|
| NCT04943718 | Personalized vaccine | Malignant Glioma/Recurrent Glioma | 1 | Recruiting |
| NCT05609994 | PEPIDH1M vaccine + vorasidenib | Recurrent IDH1 Mutant Low-Grade Glioma | 1 | Not yet recruiting |
| NCT03299309 | PEP-CMV | Recurrent Medulloblastoma/Recurrent Brain Tumor, Childhood | 1 | Recruiting |
| NCT05096481 | PEP-CMV + temozolomide + tetanus diphtheria vaccine | High Grade Glioma/Diffuse Intrinsic Pontine Glioma/Recurrent Medulloblastoma | 2 | Not yet recruiting |
| NCT01130077 | HLA-A2 restricted glioma antigen peptides vaccine + poly-ICLC | Newly Diagnosed and Recurrent Pediatric Glioma | 1 | Active, not recruiting |
| NCT05457959 | Dendritic cell tumor peptide vaccine + ipilimumab + nivolumab | Diffuse Hemispheric Glioma, H3 G34-Mutant | 1 | Not yet recruiting |
| NCT04888611 | GSC-DCV + camrelizumab | Recurrent Glioblastoma | 2 | Recruiting |
| NCT03360708 | Malignant glioma tumor lysate pulsed autologous dendritic cell vaccine | Giant Cell Glioblastoma/Recurrent Glioblastoma/Recurrent Gliosarcoma | 1 | Active, not recruiting |
| NCT04201873 | Dendritic cell tumor cell lysate vaccine + pembrolizumab | Recurrent Glioblastoma | 1 | Recruiting |
| NCT01814813 | HSPPC-96 + bevacizumab | Recurrent Glioblastoma/Recurrent Adult Brain Tumor/Gliosarcoma | 2 | Active, not recruiting |
| NCT05540873 | YYB-103 | Recurrent Malignant Glioma | 1 | Recruiting |
| NCT04003649 | IL13Rα2-specific Hingeoptimized 4-1BB-co-stimulatory CAR/Truncated CD19-expressing Autologous TN/MEM Cells + ipilimumab + nivolumab | Recurrent Glioblastoma/Refractory Glioblastoma | 1 | Recruiting |
| NCT04214392 | Chlorotoxin (EQ)-CD28 CD3zeta-CD19texpressing CAR T lymphocytes | Recurrent Glioblastoma/Recurrent Malignant Glioma/Recurrent WHO Grade II/III Glioma | 1 | Recruiting |
| NCT04185038 | SCRICARB7H3(s); B7H3-specific chimeric antigen receptor (CAR) T cell | Diffuse Intrinsic Pontine Glioma/Diffuse Midline Glioma/Recurrent or Refractory Pediatric CNS Tumors | 1 | Recruiting |
| NCT03389230 | HER2(EQ)BB#/CD19t+ T cells | Glioblastoma/Malignant Glioma/Recurrent or Refractory Glioma/WHO Grade III Glioma | 1 | Recruiting |
| NCT03638167 | EGFR806-specific chimeric antigen receptor (CAR) T cell | Recurrent or Refractory Pediatric CNS tumors | 1 | Active, not recruiting |
| NCT03500991 | HER2-specific chimeric antigen receptor (CAR) T cell | Recurrent or Refractory Pediatric CNS tumors | 1 | Recruiting |
| NCT05577091 | Inverse correlated dual target, truncated IL7Ra modified CAR-expressing autologous T lymphocytes | Recurrent Glioblastoma | 1 | Not yet recruiting |
| NCT04717999 | NKG2D CAR-T | Recurrent Glioblastoma | Not applicable | Not yet recruiting |
| NCT04385173 | B7-H3 CAR T + Temozolomide | Recurrent/Refractory Glioblastoma | 1 | Recruiting |
| NCT04077866 | B7-H3 CAR T + Temozolomide | Recurrent/Refractory Glioblastoma | 1/2 | Recruiting |
| NCT02208362 | IL13Rα2-specific Hingeoptimized 4-1BB-co-stimulatory CAR/Truncated CD19-expressing Autologous TN/MEM cells/T lymphocytes | Recurrent (Refractory) Glioblastoma/Recurrent (Refractory) Malignant Glioma/Recurrent (Refractory) WHO II/III Glioma | 1 | Active, not recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pu, Y.; Zhou, G.; Zhao, K.; Chen, Y.; Shen, S. Immunotherapy for Recurrent Glioma—From Bench to Bedside. Cancers 2023, 15, 3421. https://doi.org/10.3390/cancers15133421
Pu Y, Zhou G, Zhao K, Chen Y, Shen S. Immunotherapy for Recurrent Glioma—From Bench to Bedside. Cancers. 2023; 15(13):3421. https://doi.org/10.3390/cancers15133421
Chicago/Turabian StylePu, Yi, Guanyu Zhou, Kejia Zhao, Yaohui Chen, and Shensi Shen. 2023. "Immunotherapy for Recurrent Glioma—From Bench to Bedside" Cancers 15, no. 13: 3421. https://doi.org/10.3390/cancers15133421
APA StylePu, Y., Zhou, G., Zhao, K., Chen, Y., & Shen, S. (2023). Immunotherapy for Recurrent Glioma—From Bench to Bedside. Cancers, 15(13), 3421. https://doi.org/10.3390/cancers15133421