
Thrombin Cleavage of Osteopontin and the Host Anti-Tumor Immune Response


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
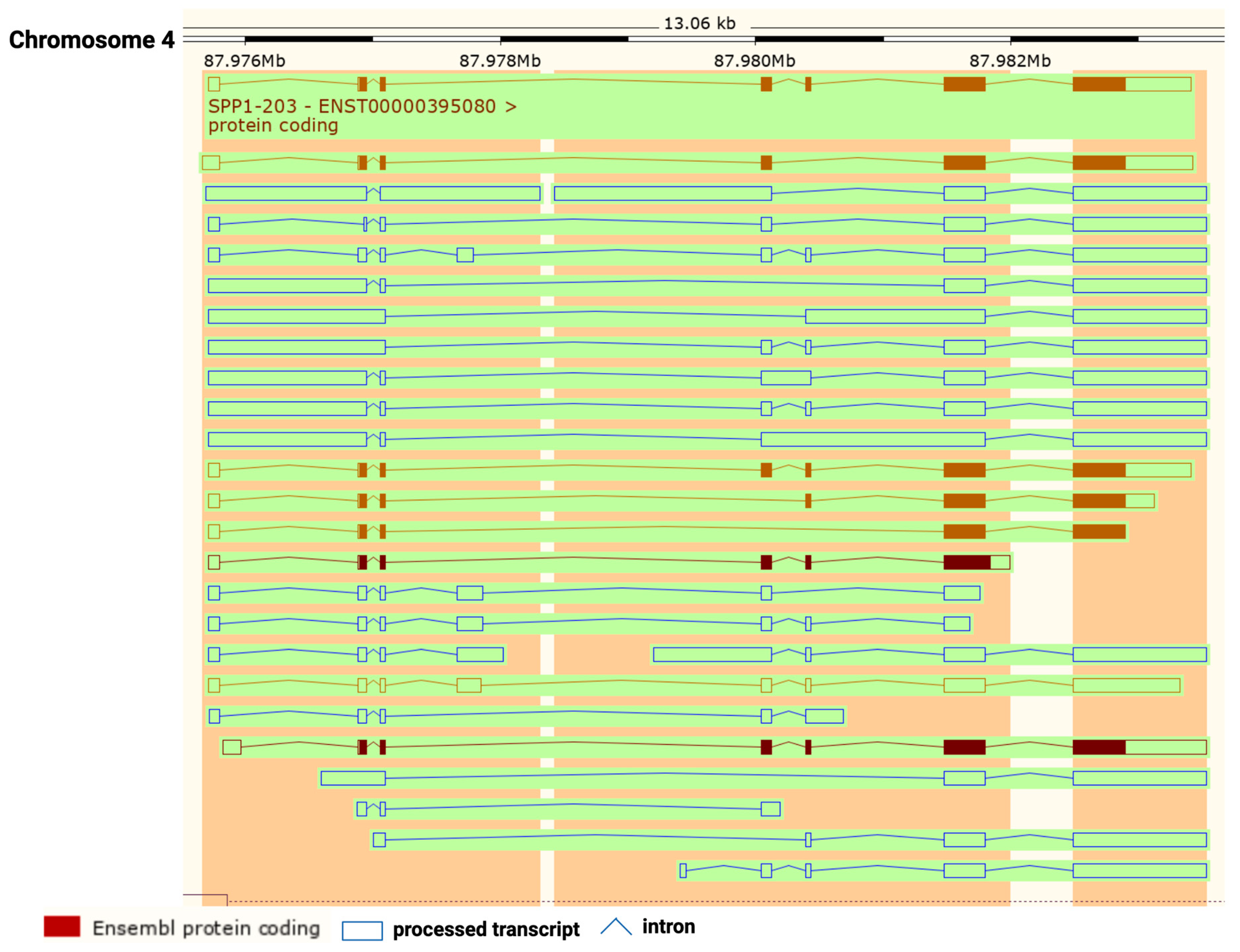
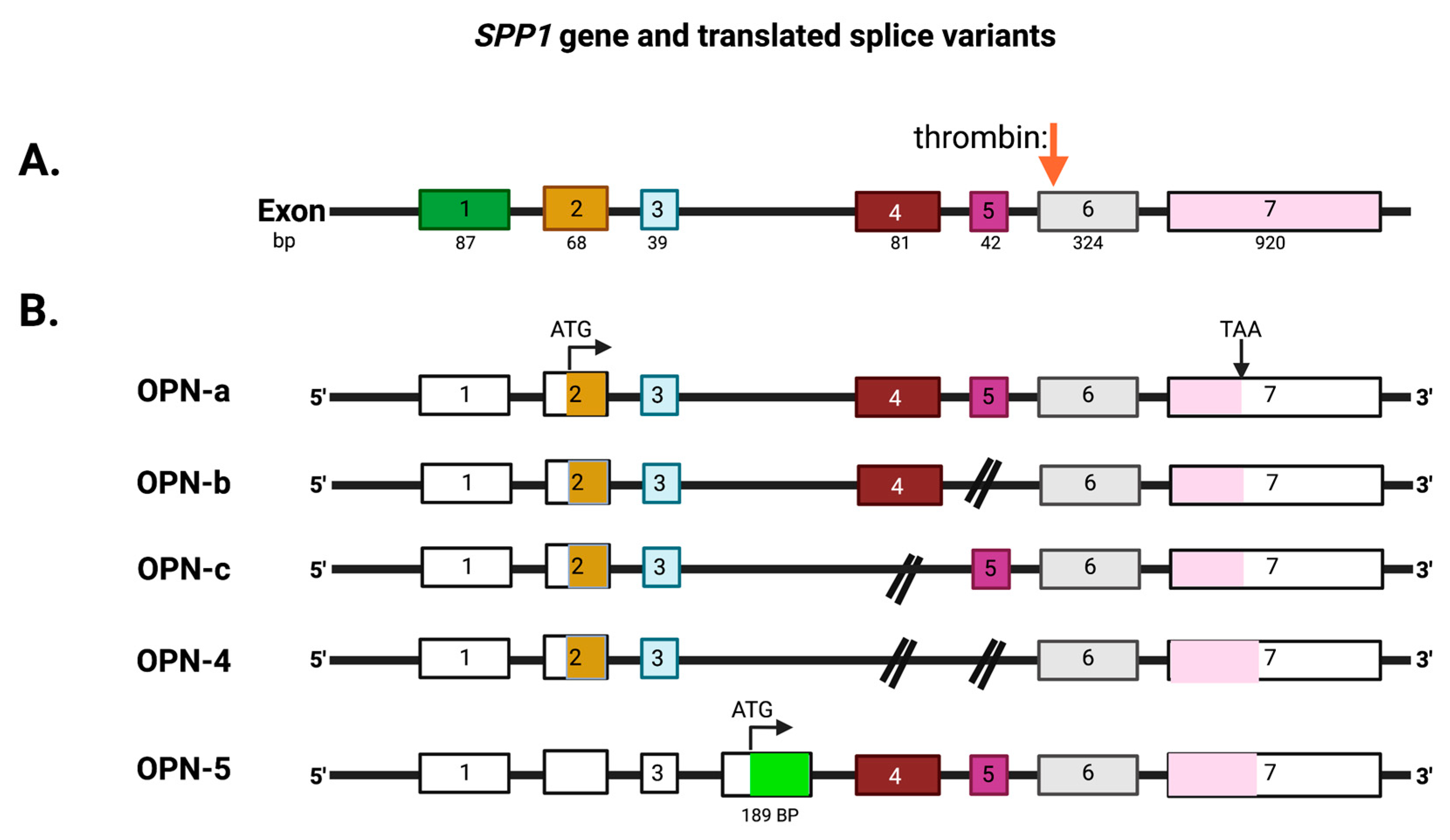
2. OPN Protein Structure
3. OPN Post-Translational Modifications
4. OPN Binding to Extracellular Matrix Components
5. OPN Modulates the Innate and Adaptive Arms of the Immune System
6. The Role of OPN in Cancer
7. Increased Expression of OPN and Cancer
8. OPN mRNA Expression in Tumor Cells and Tumor-Associated Cells
9. OPN in Tumor Cell Culture Models: Its Expression and Its Effects
10. Characterization of the Functions of OPN in Animal Models of Cancer
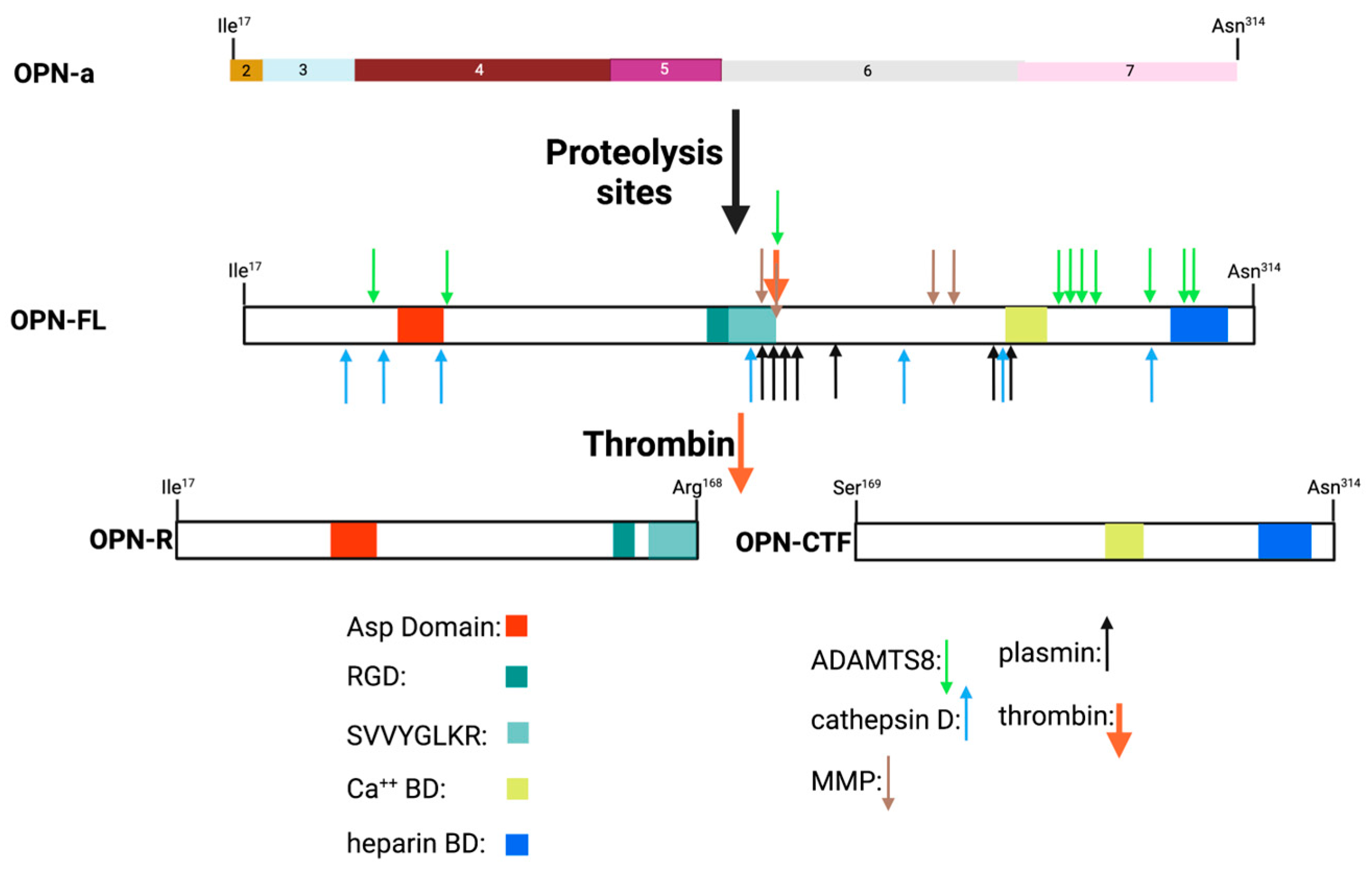
11. Protease Cleavages of OPN
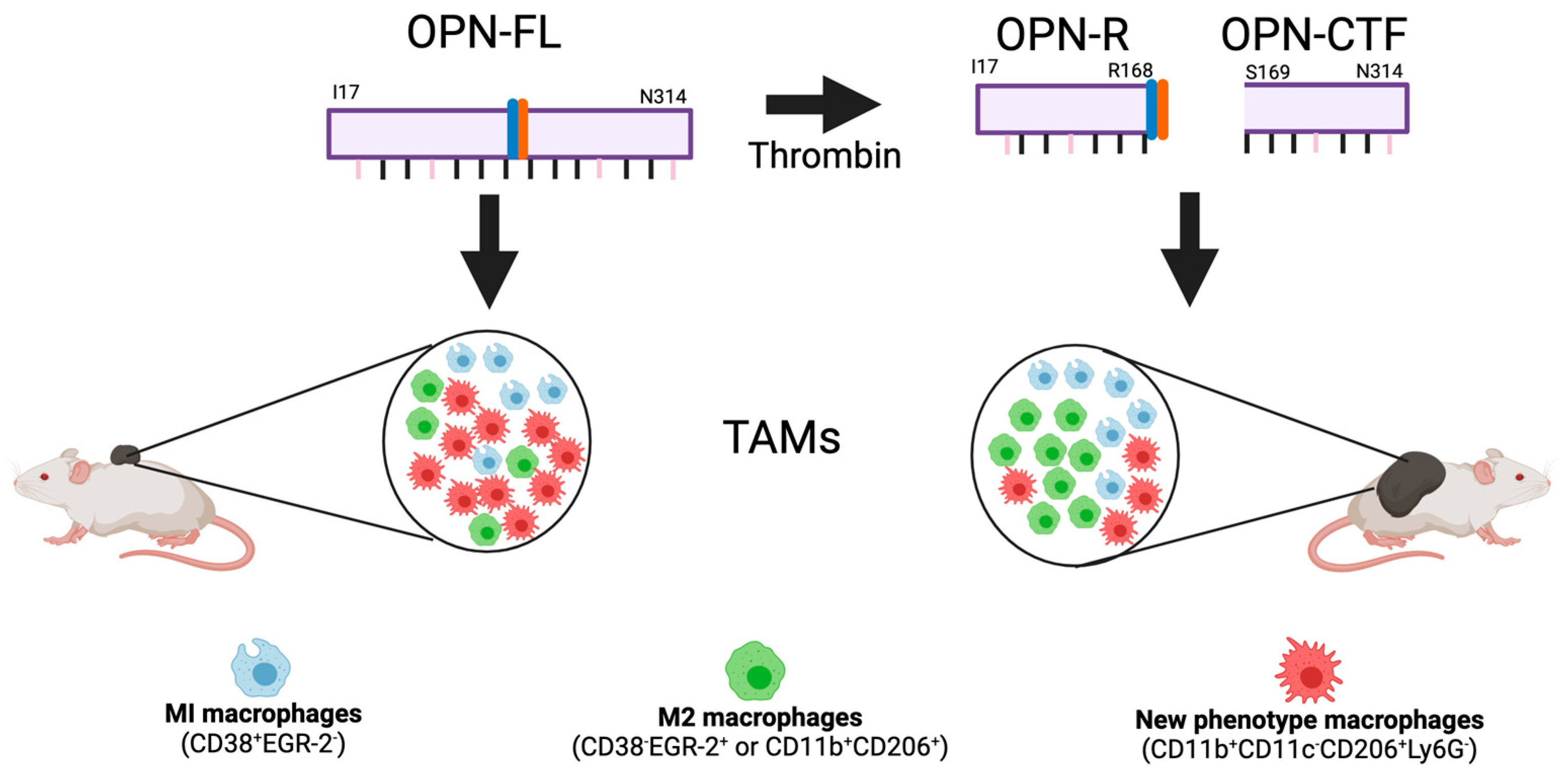
12. Properties of the Thrombin Cleavage Fragments of OPN
13. Thrombin-Resistant OPNR153A Knock-in (KI) Mouse
14. Therapies Targeting OPN in Cancer
15. Conclusions
16. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patarca, R.; Freeman, G.J.; Singh, R.P.; Wei, F.Y.; Durfee, T.; Blattner, F.; Regnier, D.C.; Kozak, C.A.; Mock, B.A.; Morse, H.C., 3rd; et al. Structural and functional studies of the early T lymphocyte activation 1 (Eta-1) gene. Definition of a novel T cell-dependent response associated with genetic resistance to bacterial infection. J. Exp. Med. 1989, 170, 145–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzen, A.; Heinegard, D. Isolation and characterization of two sialoproteins present only in bone calcified matrix. Biochem. J. 1985, 232, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Zohar, R.; Lee, W.; Arora, P.; Cheifetz, S.; McCulloch, C.; Sodek, J. Single cell analysis of intracellular osteopontin in osteogenic cultures of fetal rat calvarial cells. J. Cell. Physiol. 1997, 170, 88–100. [Google Scholar] [CrossRef]
- Oldberg, A.; Franzen, A.; Heinegard, D. Cloning and sequence analysis of rat bone sialoprotein (osteopontin) cDNA reveals an Arg-Gly-Asp cell-binding sequence. Proc. Natl. Acad. Sci. USA 1986, 83, 8819–8823. [Google Scholar] [CrossRef]
- Uede, T. Osteopontin, intrinsic tissue regulator of intractable inflammatory diseases. Pathol. Int. 2011, 61, 265–280. [Google Scholar] [CrossRef]
- Jiang, R.; Lonnerdal, B. Osteopontin in human milk and infant formula affects infant plasma osteopontin concentrations. Pediatr. Res. 2019, 85, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Liaw, L.; Birk, D.E.; Ballas, C.B.; Whitsitt, J.S.; Davidson, J.M.; Hogan, B.L. Altered wound healing in mice lacking a functional osteopontin gene (spp1). J. Clin. Investig. 1998, 101, 1468–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Lonnerdal, B. Evaluation of Bioactivities of Bovine Milk Osteopontin Using a Knockout Mouse Model. J. Pediatr. Gastroenterol. Nutr. 2020, 71, 125–131. [Google Scholar] [CrossRef]
- Jiang, R.; Lo, J.; Prell, C.; Lonnerdal, B. Milk osteopontin promotes intestinal development by up-regulating the expression of integrin alphavbeta3 and CD44. FASEB J. 2023, 37, e22988. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Tran, M.; Lonnerdal, B. Recombinant Bovine and Human Osteopontin Generated by Chlamydomonas reinhardtii Exhibit Bioactivities Similar to Bovine Milk Osteopontin When Assessed in Mouse Pups Fed Osteopontin-Deficient Milk. Mol. Nutr. Food Res. 2021, 65, e2000644. [Google Scholar] [CrossRef]
- Moorman, H.R.; Poschel, D.; Klement, J.D.; Lu, C.; Redd, P.S.; Liu, K. Osteopontin: A Key Regulator of Tumor Progression and Immunomodulation. Cancers 2020, 12, 3379. [Google Scholar] [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic. Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef]
- Liu, Y.N.; Kang, B.B.; Chen, J.H. Transcriptional regulation of human osteopontin promoter by C/EBPalpha and AML-1 in metastatic cancer cells. Oncogene 2004, 23, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Vietor, I.; Kurzbauer, R.; Brosch, G.; Huber, L.A. TIS7 regulation of the beta-catenin/Tcf-4 target gene osteopontin (OPN) is histone deacetylase-dependent. J. Biol. Chem. 2005, 280, 39795–39801. [Google Scholar] [CrossRef] [Green Version]
- Wai, P.Y.; Mi, Z.; Gao, C.; Guo, H.; Marroquin, C.; Kuo, P.C. Ets-1 and runx2 regulate transcription of a metastatic gene, osteopontin, in murine colorectal cancer cells. J. Biol. Chem. 2006, 281, 18973–18982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabiati, M.; Salvadori, C.; Sapio, A.; Burchielli, S.; Carlucci, L.; Moscato, S.; Sabatino, L.; Caselli, C.; Mattii, L.; Del Ry, S. Aging and biomarkers: Transcriptional levels evaluation of Osteopontin/miRNA-181a axis in hepatic tissue of rats in different age ranges. Exp. Gerontol. 2020, 133, 110879. [Google Scholar] [CrossRef]
- Marisetty, A.; Wei, J.; Kong, L.Y.; Ott, M.; Fang, D.; Sabbagh, A.; Heimberger, A.B. MiR-181 Family Modulates Osteopontin in Glioblastoma Multiforme. Cancers 2020, 12, 3813. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, H.; Mi, Z.; Gao, C.; Bhattacharya, S.; Li, J.; Kuo, P.C. EF1A1-actin interactions alter mRNA stability to determine differential osteopontin expression in HepG2 and Hep3B cells. Exp. Cell Res. 2009, 315, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Arjomandi, M.; Galanter, J.M.; Choudhry, S.; Eng, C.; Hu, D.; Beckman, K.; Chapela, R.; Rodriguez-Santana, J.R.; Rodriguez-Cintron, W.; Ford, J.; et al. Polymorphism in Osteopontin Gene (SPP1) Is Associated with Asthma and Related Phenotypes in a Puerto Rican Population. Pediatr. Allergy Immunol. Pulmonol. 2011, 24, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Gazal, S.; Sacre, K.; Allanore, Y.; Teruel, M.; Goodall, A.H.; Tohma, S.; Alfredsson, L.; Okada, Y.; Xie, G.; Constantin, A.; et al. Identification of secreted phosphoprotein 1 gene as a new rheumatoid arthritis susceptibility gene. Ann. Rheum. Dis. 2015, 74, e19. [Google Scholar] [CrossRef] [PubMed]
- Glas, J.; Seiderer, J.; Bayrle, C.; Wetzke, M.; Fries, C.; Tillack, C.; Olszak, T.; Beigel, F.; Steib, C.; Friedrich, M.; et al. The role of osteopontin (OPN/SPP1) haplotypes in the susceptibility to Crohn’s disease. PLoS ONE 2011, 6, e29309. [Google Scholar] [CrossRef] [Green Version]
- Amar, A.; Afzal, A.; Hameed, A.; Ahmad, M.; Khan, A.R.; Najma, H.; Abid, A.; Khaliq, S. Osteopontin promoter polymorphisms and risk of urolithiasis: A candidate gene association and meta-analysis study. BMC Med. Genet. 2020, 21, 172. [Google Scholar] [CrossRef]
- Konya, E.; Umekawa, T.; Iguchi, M.; Kurita, T. The role of osteopontin on calcium oxalate crystal formation. Eur. Urol. 2003, 43, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Bastos, A.; Gomes, A.V.P.; Silva, G.R.; Emerenciano, M.; Ferreira, L.B.; Gimba, E.R.P. The Intracellular and Secreted Sides of Osteopontin and Their Putative Physiopathological Roles. Int. J. Mol. Sci. 2023, 24, 2942. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.L.; Kim, H.J.; Kim, J.H.; Garcia, V.A.; Cantor, H. Alternative translation of osteopontin generates intracellular and secreted isoforms that mediate distinct biological activities in dendritic cells. Proc. Natl. Acad. Sci. USA 2008, 105, 7235–7239. [Google Scholar] [CrossRef] [PubMed]
- Leavenworth, J.W.; Verbinnen, B.; Yin, J.; Huang, H.; Cantor, H. A p85alpha-osteopontin axis couples the receptor ICOS to sustained Bcl-6 expression by follicular helper and regulatory T cells. Nat. Immunol. 2015, 16, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Zduniak, K.; Ziolkowski, P.; Ahlin, C.; Agrawal, A.; Agrawal, S.; Blomqvist, C.; Fjallskog, M.L.; Weber, G.F. Nuclear osteopontin-c is a prognostic breast cancer marker. Br. J. Cancer 2015, 112, 729–738. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Zhang, M.; Zhang, L.; Wang, P.; Song, G.; Liu, B.; Wu, H.; Yin, Z.; Gao, C. Intracellular osteopontin stabilizes TRAF3 to positively regulate innate antiviral response. Sci. Rep. 2016, 6, 23771. [Google Scholar] [CrossRef] [Green Version]
- Dong, Q.Z.; Zhang, X.F.; Zhao, Y.; Jia, H.L.; Zhou, H.J.; Dai, C.; Sun, H.J.; Qin, Y.; Zhang, W.D.; Ren, N.; et al. Osteopontin promoter polymorphisms at locus -443 significantly affect the metastasis and prognosis of human hepatocellular carcinoma. Hepatology 2013, 57, 1024–1034. [Google Scholar] [CrossRef]
- Lee, T.Y.; Lin, J.T.; Wu, C.C.; Yu, C.C.; Wu, M.S.; Lee, T.C.; Chen, H.P.; Wu, C.Y. Osteopontin promoter polymorphisms are associated with susceptibility to gastric cancer. J. Clin. Gastroenterol. 2013, 47, e55–e59. [Google Scholar] [CrossRef]
- Zhao, F.; Chen, X.; Meng, T.; Hao, B.; Zhang, Z.; Zhang, G. Genetic polymorphisms in the osteopontin promoter increases the risk of distance metastasis and death in Chinese patients with gastric cancer. BMC Cancer 2012, 12, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.L.; Nong, L.G.; Tang, Y.J.; Wei, Y.S.; Yang, F.L.; Wang, C.F. Correlation between OPN gene polymorphisms and the risk of nasopharyngeal carcinoma. Med. Oncol. 2014, 31, 20. [Google Scholar] [CrossRef]
- Chen, J.; Wu, Q.; Lu, Y.; Xu, T.; Huang, Y.; Ribas, J.; Ni, X.; Hu, G.; Huang, F.; Zhou, L.; et al. SPP1 promoter polymorphisms and glioma risk in a Chinese Han population. J. Hum. Genet. 2010, 55, 456–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.G.; Godbey, W.T. The potential of the human osteopontin promoter and single-nucleotide polymorphisms for targeted cancer gene therapy. Curr. Gene Ther. 2015, 15, 82–92. [Google Scholar] [CrossRef]
- Giacopelli, F.; Marciano, R.; Pistorio, A.; Catarsi, P.; Canini, S.; Karsenty, G.; Ravazzolo, R. Polymorphisms in the osteopontin promoter affect its transcriptional activity. Physiol. Genom. 2004, 20, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briones-Orta, M.A.; Avendano-Vazquez, S.E.; Aparicio-Bautista, D.I.; Coombes, J.D.; Weber, G.F.; Syn, W.K. Osteopontin splice variants and polymorphisms in cancer progression and prognosis. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Briones-Orta, M.A.; Avendano-Vazquez, S.E.; Ivette Aparicio-Bautista, D.; Coombes, J.D.; Weber, G.F.; Syn, W.K. Prediction of transcription factor bindings sites affected by SNPs located at the osteopontin promoter. Data Brief 2017, 14, 538–542. [Google Scholar] [CrossRef]
- Beninati, S.; Senger, D.R.; Cordella-Miele, E.; Mukherjee, A.B.; Chackalaparampil, I.; Shanmugam, V.; Singh, K.; Mukherjee, B.B. Osteopontin: Its transglutaminase-catalyzed posttranslational modifications and cross-linking to fibronectin. J. Biochem. 1994, 115, 675–682. [Google Scholar] [CrossRef]
- Prince, C.W.; Dickie, D.; Krumdieck, C.L. Osteopontin, a substrate for transglutaminase and factor XIII activity. Biochem. Biophys. Res. Commun. 1991, 177, 1205–1210. [Google Scholar] [CrossRef]
- Gimba, E.R.; Tilli, T.M. Human osteopontin splicing isoforms: Known roles, potential clinical applications and activated signaling pathways. Cancer Lett. 2013, 331, 11–17. [Google Scholar] [CrossRef]
- Gimba, E.R.P.; Brum, M.C.M.; Nestal De Moraes, G. Full-length osteopontin and its splice variants as modulators of chemoresistance and radioresistance (Review). Int. J. Oncol. 2019, 54, 420–430. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Fnu, G.; Xie, C.; Weber, G.F. Meta-analysis of Osteopontin splice variants in cancer. BMC Cancer 2023, 23, 373. [Google Scholar] [CrossRef]
- Weber, G.F. The Phylogeny of Osteopontin-Analysis of the Protein Sequence. Int. J. Mol. Sci. 2018, 19, 2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schedlbauer, A.; Ozdowy, P.; Kontaxis, G.; Hartl, M.; Bister, K.; Konrat, R. Backbone assignment of osteopontin, a cytokine and cell attachment protein implicated in tumorigenesis. Biomol. NMR Assign. 2008, 2, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Platzer, G.; Zerko, S.; Saxena, S.; Kozminski, W.; Konrat, R. (1)H, (15)N, (13)C resonance assignment of human osteopontin. Biomol. NMR Assign. 2015, 9, 289–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, Y.; Hanashima, S.; Yagi, H.; Takahashi, Y.; Sasakawa, H.; Kurimoto, E.; Iguchi, T.; Kon, S.; Uede, T.; Kato, K. NMR characterization of intramolecular interaction of osteopontin, an intrinsically disordered protein with cryptic integrin-binding motifs. Biochem. Biophys. Res. Commun. 2010, 393, 487–491. [Google Scholar] [CrossRef]
- Iline-Vul, T.; Nanda, R.; Mateos, B.; Hazan, S.; Matlahov, I.; Perelshtein, I.; Keinan-Adamsky, K.; Althoff-Ospelt, G.; Konrat, R.; Goobes, G. Osteopontin regulates biomimetic calcium phosphate crystallization from disordered mineral layers covering apatite crystallites. Sci. Rep. 2020, 10, 15722. [Google Scholar] [CrossRef]
- Holzinger, J.; Kotisch, H.; Richter, K.W.; Konrat, R. Binding Mode Characterization of Osteopontin on Hydroxyapatite by Solution NMR Spectroscopy. Chembiochem 2021, 22, 2300–2305. [Google Scholar] [CrossRef]
- Mateos, B.; Holzinger, J.; Conrad-Billroth, C.; Platzer, G.; Zerko, S.; Sealey-Cardona, M.; Anrather, D.; Kozminski, W.; Konrat, R. Hyperphosphorylation of Human Osteopontin and Its Impact on Structural Dynamics and Molecular Recognition. Biochemistry 2021, 60, 1347–1355. [Google Scholar] [CrossRef]
- Du, J.; Hou, S.; Zhong, C.; Lai, Z.; Yang, H.; Dai, J.; Zhang, D.; Wang, H.; Guo, Y.; Ding, J. Molecular basis of recognition of human osteopontin by 23C3, a potential therapeutic antibody for treatment of rheumatoid arthritis. J. Mol. Biol. 2008, 382, 835–842. [Google Scholar] [CrossRef] [Green Version]
- Hotta, H.; Kon, S.; Katagiri, Y.U.; Tosa, N.; Tsukamoto, T.; Chambers, A.F.; Uede, T. Detection of various epitopes of murine osteopontin by monoclonal antibodies. Biochem. Biophys. Res. Commun. 1999, 257, 6–11. [Google Scholar] [CrossRef]
- Gorski, J.; Hankenson, K. Secreted non-collagenous proteins of bone. In Principles of Bone Biology, 4th ed.; Bilezikian, J.P., Martin, T.J., Clemens, T.L., CJ, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 359–378. [Google Scholar]
- Fisher, L.W.; Torchia, D.A.; Fohr, B.; Young, M.F.; Fedarko, N.S. Flexible structures of SIBLING proteins, bone sialoprotein, and osteopontin. Biochem. Biophys. Res. Commun. 2001, 280, 460–465. [Google Scholar] [CrossRef] [Green Version]
- Kariya, Y.; Kanno, M.; Matsumoto-Morita, K.; Konno, M.; Yamaguchi, Y.; Hashimoto, Y. Osteopontin O-glycosylation contributes to its phosphorylation and cell-adhesion properties. Biochem. J. 2014, 463, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Shen, H.; Yan, G.; Zhang, Y.; Liu, M.; Fang, P.; Yu, H.; Yang, P. Site-specific structural characterization of O-glycosylation and identification of phosphorylation sites of recombinant osteopontin. Biochim. Biophys. Acta 2015, 1854, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Nagata, T.; Todescan, R.; Goldberg, H.A.; Zhang, Q.; Sodek, J. Sulphation of secreted phosphoprotein I (SPPI, osteopontin) is associated with mineralized tissue formation. Biochem. Biophys. Res. Commun. 1989, 165, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Keykhosravani, M.; Doherty-Kirby, A.; Zhang, C.; Brewer, D.; Goldberg, H.A.; Hunter, G.K.; Lajoie, G. Comprehensive identification of post-translational modifications of rat bone osteopontin by mass spectrometry. Biochemistry 2005, 44, 6990–7003. [Google Scholar] [CrossRef] [PubMed]
- Oyama, M.; Kariya, Y.; Kariya, Y.; Matsumoto, K.; Kanno, M.; Yamaguchi, Y.; Hashimoto, Y. Biological role of site-specific O-glycosylation in cell adhesion activity and phosphorylation of osteopontin. Biochem. J. 2018, 475, 1583–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, B.; Nielsen, M.S.; Haselmann, K.F.; Petersen, T.E.; Sorensen, E.S. Post-translationally modified residues of native human osteopontin are located in clusters: Identification of 36 phosphorylation and five O-glycosylation sites and their biological implications. Biochem. J. 2005, 390, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froehlich, J.W.; Chu, C.S.; Tang, N.; Waddell, K.; Grimm, R.; Lebrilla, C.B. Label-free liquid chromatography-tandem mass spectrometry analysis with automated phosphopeptide enrichment reveals dynamic human milk protein phosphorylation during lactation. Anal. Biochem. 2011, 408, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Grimm, G.; Vila, G.; Bieglmayer, C.; Riedl, M.; Luger, A.; Clodi, M. Changes in osteopontin and in biomarkers of bone turnover during human endotoxemia. Bone 2010, 47, 388–391. [Google Scholar] [CrossRef]
- Kazanecki, C.C.; Uzwiak, D.J.; Denhardt, D.T. Control of osteopontin signaling and function by post-translational phosphorylation and protein folding. J. Cell. Biochem. 2007, 102, 912–924. [Google Scholar] [CrossRef] [PubMed]
- Schytte, G.N.; Christensen, B.; Bregenov, I.; Kjoge, K.; Scavenius, C.; Petersen, S.V.; Enghild, J.J.; Sorensen, E.S. FAM20C phosphorylation of the RGDSVVYGLR motif in osteopontin inhibits interaction with the alphavbeta3 integrin. J. Cell. Biochem. 2020, 121, 4809–4818. [Google Scholar] [CrossRef]
- Schytte, G.N.; Christensen, B.; Bregenov, I.; Sorensen, E.S. Ras-transformation reduce FAM20C expression and osteopontin phosphorylation. Biosci. Rep. 2020, 40, BSR20194378. [Google Scholar] [CrossRef]
- Raineri, D.; Dianzani, C.; Cappellano, G.; Maione, F.; Baldanzi, G.; Iacobucci, I.; Clemente, N.; Baldone, G.; Boggio, E.; Gigliotti, C.L.; et al. Osteopontin binds ICOSL promoting tumor metastasis. Commun. Biol. 2020, 3, 615. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.X.; Denhardt, D.T. Osteopontin: Role in immune regulation and stress responses. Cytokine Growth Factor Rev. 2008, 19, 333–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, S.A.; Du, X.; Myles, T.; Song, J.J.; Price, E.; Lee, D.M.; Goodman, S.B.; Nagashima, M.; Morser, J.; Robinson, W.H.; et al. Thrombin-activatable carboxypeptidase B cleavage of osteopontin regulates neutrophil survival and synoviocyte binding in rheumatoid arthritis. Arthritis Rheum. 2009, 60, 2902–2912. [Google Scholar] [CrossRef] [Green Version]
- Grassinger, J.; Haylock, D.N.; Storan, M.J.; Haines, G.O.; Williams, B.; Whitty, G.A.; Vinson, A.R.; Be, C.L.; Li, S.; Sorensen, E.S.; et al. Thrombin-cleaved osteopontin regulates hemopoietic stem and progenitor cell functions through interactions with alpha9beta1 and alpha4beta1 integrins. Blood 2009, 114, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, T.; Sorensen, E.S.; Rittling, S.R. Osteopontin binding to the alpha 4 integrin requires highest affinity integrin conformation, but is independent of post-translational modifications of osteopontin. Matrix Biol. J. Int. Soc. Matrix Biol. 2015, 41, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Yokosaki, Y.; Matsuura, N.; Sasaki, T.; Murakami, I.; Schneider, H.; Higashiyama, S.; Saitoh, Y.; Yamakido, M.; Taooka, Y.; Sheppard, D. The integrin alpha(9)beta(1) binds to a novel recognition sequence (SVVYGLR) in the thrombin-cleaved amino-terminal fragment of osteopontin. J. Biol. Chem. 1999, 274, 36328–36334. [Google Scholar] [CrossRef] [Green Version]
- Myles, T.; Nishimura, T.; Yun, T.H.; Nagashima, M.; Morser, J.; Patterson, A.J.; Pearl, R.G.; Leung, L.L. Thrombin activatable fibrinolysis inhibitor, a potential regulator of vascular inflammation. J. Biol. Chem. 2003, 278, 51059–51067. [Google Scholar] [CrossRef] [Green Version]
- Rangaswami, H.; Bulbule, A.; Kundu, G.C. Osteopontin: Role in cell signaling and cancer progression. Trends Cell Biol. 2006, 16, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuck, A.B.; Hota, C.; Wilson, S.M.; Chambers, A.F. Osteopontin-induced migration of human mammary epithelial cells involves activation of EGF receptor and multiple signal transduction pathways. Oncogene 2003, 22, 1198–1205. [Google Scholar] [CrossRef] [Green Version]
- Moro, L.; Dolce, L.; Cabodi, S.; Bergatto, E.; Boeri Erba, E.; Smeriglio, M.; Turco, E.; Retta, S.F.; Giuffrida, M.G.; Venturino, M.; et al. Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J. Biol. Chem. 2002, 277, 9405–9414. [Google Scholar] [CrossRef] [Green Version]
- Das, R.; Mahabeleshwar, G.H.; Kundu, G.C. Osteopontin induces AP-1-mediated secretion of urokinase-type plasminogen activator through c-Src-dependent epidermal growth factor receptor transactivation in breast cancer cells. J. Biol. Chem. 2004, 279, 11051–11064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribe, M.L.; Marrocco, I.; Yarden, Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers 2021, 13, 2748. [Google Scholar] [CrossRef]
- Das, R.; Mahabeleshwar, G.H.; Kundu, G.C. Osteopontin stimulates cell motility and nuclear factor kappaB-mediated secretion of urokinase type plasminogen activator through phosphatidylinositol 3-kinase/Akt signaling pathways in breast cancer cells. J. Biol. Chem. 2003, 278, 28593–28606. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.H.; Yang-Yen, H.F. The osteopontin-CD44 survival signal involves activation of the phosphatidylinositol 3-kinase/Akt signaling pathway. J. Biol. Chem. 2001, 276, 46024–46030. [Google Scholar] [CrossRef] [Green Version]
- Sugatani, T.; Alvarez, U.; Hruska, K.A. PTEN regulates RANKL- and osteopontin-stimulated signal transduction during osteoclast differentiation and cell motility. J. Biol. Chem. 2003, 278, 5001–5008. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-kappaB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Rangaswami, H.; Bulbule, A.; Kundu, G.C. Nuclear factor-inducing kinase plays a crucial role in osteopontin-induced MAPK/IkappaBalpha kinase-dependent nuclear factor kappaB-mediated promatrix metalloproteinase-9 activation. J. Biol. Chem. 2004, 279, 38921–38935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemente, N.; Raineri, D.; Cappellano, G.; Boggio, E.; Favero, F.; Soluri, M.F.; Dianzani, C.; Comi, C.; Dianzani, U.; Chiocchetti, A. Osteopontin Bridging Innate and Adaptive Immunity in Autoimmune Diseases. J. Immunol. Res. 2016, 2016, 7675437. [Google Scholar] [CrossRef] [Green Version]
- Murugaiyan, G.; Mittal, A.; Weiner, H.L. Increased osteopontin expression in dendritic cells amplifies IL-17 production by CD4+ T cells in experimental autoimmune encephalomyelitis and in multiple sclerosis. J. Immunol. 2008, 181, 7480–7488. [Google Scholar] [CrossRef] [Green Version]
- Guan, H.; Nagarkatti, P.S.; Nagarkatti, M. Role of CD44 in the differentiation of Th1 and Th2 cells: CD44-deficiency enhances the development of Th2 effectors in response to sheep RBC and chicken ovalbumin. J. Immunol. 2009, 183, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Rittling, S.R. Osteopontin in macrophage function. Expert. Rev. Mol. Med. 2011, 13, e15. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, J.; Sato, K.; Nakayama, Y.; Kimura, C.; Kajino, K.; Matsui, Y.; Miyazaki, T.; Uede, T. Osteopontin modulates the generation of memory CD8+ T cells during influenza virus infection. J. Immunol. 2011, 187, 5671–5683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuka, M.; De Giovanni, M.; Iannacone, M. The role of type I interferons in CD4(+) T cell differentiation. Immunol. Lett. 2019, 215, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Klement, J.D.; Paschall, A.V.; Redd, P.S.; Ibrahim, M.L.; Lu, C.; Yang, D.; Celis, E.; Abrams, S.I.; Ozato, K.; Liu, K. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J. Clin. Investig. 2018, 128, 5549–5560. [Google Scholar] [CrossRef] [Green Version]
- Shurin, M.R. Osteopontin controls immunosuppression in the tumor microenvironment. J. Clin. Investig. 2018, 128, 5209–5212. [Google Scholar] [CrossRef]
- Scutera, S.; Salvi, V.; Lorenzi, L.; Piersigilli, G.; Lonardi, S.; Alotto, D.; Casarin, S.; Castagnoli, C.; Dander, E.; D’Amico, G.; et al. Adaptive Regulation of Osteopontin Production by Dendritic Cells Through the Bidirectional Interaction With Mesenchymal Stromal Cells. Front. Immunol. 2018, 9, 1207. [Google Scholar] [CrossRef]
- Kawamura, K.; Iyonaga, K.; Ichiyasu, H.; Nagano, J.; Suga, M.; Sasaki, Y. Differentiation, maturation, and survival of dendritic cells by osteopontin regulation. Clin. Diagn. Lab. Immunol. 2005, 12, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Samant, R.S.; Shevde, L.A. Hedgehog signaling induced by breast cancer cells promotes osteoclastogenesis and osteolysis. J. Biol. Chem. 2011, 286, 9612–9622. [Google Scholar] [CrossRef] [Green Version]
- Zunich, S.M.; Douglas, T.; Valdovinos, M.; Chang, T.; Bushman, W.; Walterhouse, D.; Iannaccone, P.; Lamm, M.L. Paracrine sonic hedgehog signalling by prostate cancer cells induces osteoblast differentiation. Mol. Cancer 2009, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.; Wong, J.P.C.; Kwok, H.F. Osteopontin—A promising biomarker for cancer therapy. J. Cancer 2017, 8, 2173–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiodoni, C.; Colombo, M.P.; Sangaletti, S. Matricellular proteins: From homeostasis to inflammation, cancer, and metastasis. Cancer Metastasis Rev. 2010, 29, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Lamort, A.S.; Giopanou, I.; Psallidas, I.; Stathopoulos, G.T. Osteopontin as a Link between Inflammation and Cancer: The Thorax in the Spotlight. Cells 2019, 8, 815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shevde, L.A.; Samant, R.S. Role of osteopontin in the pathophysiology of cancer. Matrix Biol. J. Int. Soc. Matrix Biol. 2014, 37, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [Green Version]
- Atai, N.A.; Bansal, M.; Lo, C.; Bosman, J.; Tigchelaar, W.; Bosch, K.S.; Jonker, A.; De Witt Hamer, P.C.; Troost, D.; McCulloch, C.A.; et al. Osteopontin is up-regulated and associated with neutrophil and macrophage infiltration in glioblastoma. Immunology 2011, 132, 39–48. [Google Scholar] [CrossRef]
- Rohde, F.; Rimkus, C.; Friederichs, J.; Rosenberg, R.; Marthen, C.; Doll, D.; Holzmann, B.; Siewert, J.R.; Janssen, K.P. Expression of osteopontin, a target gene of de-regulated Wnt signaling, predicts survival in colon cancer. Int. J. Cancer 2007, 121, 1717–1723. [Google Scholar] [CrossRef]
- Klement, J.D.; Poschel, D.B.; Lu, C.; Merting, A.D.; Yang, D.; Redd, P.S.; Liu, K. Osteopontin Blockade Immunotherapy Increases Cytotoxic T Lymphocyte Lytic Activity and Suppresses Colon Tumor Progression. Cancers 2021, 13, 1006. [Google Scholar] [CrossRef]
- Sun, J.; Chen, X.; Wang, Y. Comparison of the diagnostic value of CEA combined with OPN or DKK1 in non-small cell lung cancer. Oncol. Lett. 2020, 20, 3046–3052. [Google Scholar] [CrossRef]
- Xu, C.; Yuan, Q.; Wang, W.; Chi, C.; Zhang, Q.; Li, L.; Yang, R.; Wang, Y. Prognostic significance of serum osteopontin levels in small cell lung cancer. BMC Pulm. Med. 2020, 20, 235. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Liu, Y.; Mei, F.; Li, X.; Zhang, M.; Yao, B.; Wu, R.; You, J.; Pei, F. SPP1 overexpression is associated with poor outcomes in ALK fusion lung cancer patients without receiving targeted therapy. Sci. Rep. 2021, 11, 14031. [Google Scholar] [CrossRef] [PubMed]
- Creaney, J.; Yeoman, D.; Musk, A.W.; de Klerk, N.; Skates, S.J.; Robinson, B.W. Plasma versus serum levels of osteopontin and mesothelin in patients with malignant mesothelioma—Which is best? Lung Cancer 2011, 74, 55–60. [Google Scholar] [CrossRef]
- Kerenidi, T.; Kazakou, A.P.; Lada, M.; Tsilioni, I.; Daniil, Z.; Gourgoulianis, K.I. Clinical Significance of Circulating Osteopontin Levels in Patients With Lung Cancer and Correlation With VEGF and MMP-9. Cancer Investig. 2016, 34, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Rong, W.; Zhang, Y.; Yang, L.; Feng, L.; Wei, B.; Wu, F.; Wang, L.; Gao, Y.; Cheng, S.; Wu, J.; et al. Post-surgical resection prognostic value of combined OPN, MMP7, and PSG9 plasma biomarkers in hepatocellular carcinoma. Front. Med. 2019, 13, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Li, P.; Sun, D.; Bu, Q.; Li, G. Prognostic value of osteopontin in patients with hepatocellular carcinoma: A systematic review and meta-analysis. Medicine 2018, 97, e12954. [Google Scholar] [CrossRef]
- Cabiati, M.; Gaggini, M.; De Simone, P.; Del Ry, S. Data mining of key genes expression in hepatocellular carcinoma: Novel potential biomarkers of diagnosis prognosis or progression. Clin. Exp. Metastasis 2022, 39, 589–602. [Google Scholar] [CrossRef]
- Cabiati, M.; Gaggini, M.; Cesare, M.M.; Caselli, C.; De Simone, P.; Filipponi, F.; Basta, G.; Gastaldelli, A.; Del Ry, S. Osteopontin in hepatocellular carcinoma: A possible biomarker for diagnosis and follow-up. Cytokine 2017, 99, 59–65. [Google Scholar] [CrossRef]
- Cabiati, M.; Di Giorgi, N.; Salvadori, C.; Finamore, F.; Del Turco, S.; Cecchettini, A.; Rocchiccioli, S.; Del Ry, S. Transcriptional level evaluation of osteopontin/miRNA-181a axis in hepatocellular carcinoma cell line-secreted extracellular vesicles. Pathol. Res. Pract. 2022, 238, 154088. [Google Scholar] [CrossRef]
- Anborgh, P.H.; Lee, D.J.; Stam, P.F.; Tuck, A.B.; Chambers, A.F. Role of osteopontin as a predictive biomarker for anti-EGFR therapy in triple-negative breast cancer. Expert Opin. Ther. Targets 2018, 22, 727–734. [Google Scholar] [CrossRef]
- Bellahcene, A.; Castronovo, V. Increased expression of osteonectin and osteopontin, two bone matrix proteins, in human breast cancer. Am. J. Pathol. 1995, 146, 95–100. [Google Scholar]
- Singhal, H.; Bautista, D.S.; Tonkin, K.S.; O’Malley, F.P.; Tuck, A.B.; Chambers, A.F.; Harris, J.F. Elevated plasma osteopontin in metastatic breast cancer associated with increased tumor burden and decreased survival. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1997, 3, 605–611. [Google Scholar]
- Tuck, A.B.; O’Malley, F.P.; Singhal, H.; Tonkin, K.S.; Harris, J.F.; Bautista, D.; Chambers, A.F. Osteopontin and p53 expression are associated with tumor progression in a case of synchronous, bilateral, invasive mammary carcinomas. Arch. Pathol. Lab. Med. 1997, 121, 578–584. [Google Scholar]
- Lindahl, G.; Rzepecka, A.; Dabrosin, C. Increased Extracellular Osteopontin Levels in Normal Human Breast Tissue at High Risk of Developing Cancer and Its Association With Inflammatory Biomarkers in situ. Front. Oncol. 2019, 9, 746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gothlin Eremo, A.; Lagergren, K.; Othman, L.; Montgomery, S.; Andersson, G.; Tina, E. Evaluation of SPP1/osteopontin expression as predictor of recurrence in tamoxifen treated breast cancer. Sci. Rep. 2020, 10, 1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.D.; Chen, H.; Liu, H.Q.; Hao, M. Correlation between ovarian neoplasm and serum levels of osteopontin: A meta-analysis. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 11799–11808. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Liu, D.; Sun, H.; Shao, Z.; Shi, P.; Li, T.; Yin, S.; Zhu, T. SPP1 is a prognostic related biomarker and correlated with tumor-infiltrating immune cells in ovarian cancer. BMC Cancer 2022, 22, 1367. [Google Scholar] [CrossRef]
- Rani, S.; Sehgal, A.; Kaur, J.; Pandher, D.K.; Punia, R.S. Osteopontin as a Tumor Marker in Ovarian Cancer. J. Midlife Health 2022, 13, 200–205. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, C. The role of osteopontin in the development and metastasis of melanoma. Melanoma Res. 2021, 31, 283–289. [Google Scholar] [CrossRef]
- Kiss, T.; Ecsedi, S.; Vizkeleti, L.; Koroknai, V.; Emri, G.; Kovacs, N.; Adany, R.; Balazs, M. The role of osteopontin expression in melanoma progression. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 7841–7847. [Google Scholar] [CrossRef] [Green Version]
- Abildgaard, S.K.; Vorum, H. Proteomics of uveal melanoma: A minireview. J. Oncol. 2013, 2013, 820953. [Google Scholar] [CrossRef] [Green Version]
- Guarneri, C.; Bevelacqua, V.; Polesel, J.; Falzone, L.; Cannavo, P.S.; Spandidos, D.A.; Malaponte, G.; Libra, M. NF-kappaB inhibition is associated with OPN/MMP-9 downregulation in cutaneous melanoma. Oncol. Rep. 2017, 37, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Szasz, I.; Koroknai, V.; Kiss, T.; Vizkeleti, L.; Adany, R.; Balazs, M. Molecular alterations associated with acquired resistance to BRAFV600E targeted therapy in melanoma cells. Melanoma Res. 2019, 29, 390–400. [Google Scholar] [CrossRef]
- Ouyang, X.; Huang, Y.; Jin, X.; Zhao, W.; Hu, T.; Wu, F.; Huang, J. Osteopontin promotes cancer cell drug resistance, invasion, and lactate production and is associated with poor outcome of patients with advanced non-small-cell lung cancer. Onco Targets Ther. 2018, 11, 5933–5941. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.; Szasz, I.; Koroknai, V.; Kiss, T.; Balazs, M. Molecular Alterations Associated with Acquired Drug Resistance during Combined Treatment with Encorafenib and Binimetinib in Melanoma Cell Lines. Cancers 2021, 13, 6058. [Google Scholar] [CrossRef]
- Chen, J.; Hou, C.; Zheng, Z.; Lin, H.; Lv, G.; Zhou, D. Identification of Secreted Phosphoprotein 1 (SPP1) as a Prognostic Factor in Lower-Grade Gliomas. World Neurosurg. 2019, 130, e775–e785. [Google Scholar] [CrossRef]
- Sreekanthreddy, P.; Srinivasan, H.; Kumar, D.M.; Nijaguna, M.B.; Sridevi, S.; Vrinda, M.; Arivazhagan, A.; Balasubramaniam, A.; Hegde, A.S.; Chandramouli, B.A.; et al. Identification of potential serum biomarkers of glioblastoma: Serum osteopontin levels correlate with poor prognosis. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1409–1422. [Google Scholar] [CrossRef] [Green Version]
- Kohata, T.; Ito, S.; Masuda, T.; Furuta, T.; Nakada, M.; Ohtsuki, S. Laminin Subunit Alpha-4 and Osteopontin Are Glioblastoma-Selective Secreted Proteins That Are Increased in the Cerebrospinal Fluid of Glioblastoma Patients. J. Proteome Res. 2020, 19, 3542–3553. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Shao, Z.; Sharif, S.; Du, X.Y.; Myles, T.; Merchant, M.; Harsh, G.; Glantz, M.; Recht, L.; Morser, J.; et al. Thrombin-cleaved fragments of osteopontin are overexpressed in malignant glial tumors and provide a molecular niche with survival advantage. J. Biol. Chem. 2013, 288, 3097–3111. [Google Scholar] [CrossRef] [Green Version]
- Colin, C.; Baeza, N.; Bartoli, C.; Fina, F.; Eudes, N.; Nanni, I.; Martin, P.M.; Ouafik, L.; Figarella-Branger, D. Identification of genes differentially expressed in glioblastoma versus pilocytic astrocytoma using Suppression Subtractive Hybridization. Oncogene 2006, 25, 2818–2826. [Google Scholar] [CrossRef] [Green Version]
- Noda, M.; Yoon, K.; Prince, C.W.; Butler, W.T.; Rodan, G.A. Transcriptional regulation of osteopontin production in rat osteosarcoma cells by type beta transforming growth factor. J. Biol. Chem. 1988, 263, 13916–13921. [Google Scholar] [CrossRef]
- Brown, L.F.; Papadopoulos-Sergiou, A.; Berse, B.; Manseau, E.J.; Tognazzi, K.; Perruzzi, C.A.; Dvorak, H.F.; Senger, D.R. Osteopontin expression and distribution in human carcinomas. Am. J. Pathol. 1994, 145, 610–623. [Google Scholar]
- Craig, A.M.; Smith, J.H.; Denhardt, D.T. Osteopontin, a transformation-associated cell adhesion phosphoprotein, is induced by 12-O-tetradecanoylphorbol 13-acetate in mouse epidermis. J. Biol. Chem. 1989, 264, 9682–9689. [Google Scholar] [CrossRef]
- Rittling, S.R.; Novick, K.E. Osteopontin expression in mammary gland development and tumorigenesis. Cell Growth Differ. 1997, 8, 1061–1069. [Google Scholar]
- Ue, T.; Yokozaki, H.; Kitadai, Y.; Yamamoto, S.; Yasui, W.; Ishikawa, T.; Tahara, E. Co-expression of osteopontin and CD44v9 in gastric cancer. Int. J. Cancer 1998, 79, 127–132. [Google Scholar] [CrossRef]
- Das, S.; Harris, L.G.; Metge, B.J.; Liu, S.; Riker, A.I.; Samant, R.S.; Shevde, L.A. The hedgehog pathway transcription factor GLI1 promotes malignant behavior of cancer cells by up-regulating osteopontin. J. Biol. Chem. 2009, 284, 22888–22897. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Yang, H.; Wang, Z.; Zhang, Z.; Lu, X.; Yang, W.; Xu, X.; Jiang, Y.; Li, Y.; Fan, X.; et al. A Noncanonical Hedgehog Signaling Exerts a Tumor-Promoting Effect on Pancreatic Cancer Cells Via Induction of Osteopontin Expression. Cancer Biother. Radiopharm. 2021. [Google Scholar] [CrossRef]
- Manda, K.R.; Tripathi, P.; Hsi, A.C.; Ning, J.; Ruzinova, M.B.; Liapis, H.; Bailey, M.; Zhang, H.; Maher, C.A.; Humphrey, P.A.; et al. NFATc1 promotes prostate tumorigenesis and overcomes PTEN loss-induced senescence. Oncogene 2016, 35, 3282–3292. [Google Scholar] [CrossRef] [Green Version]
- Sathe, A.; Mason, K.; Grimes, S.M.; Zhou, Z.; Lau, B.T.; Bai, X.; Su, A.; Tan, X.; Lee, H.; Suarez, C.J.; et al. Colorectal Cancer Metastases in the Liver Establish Immunosuppressive Spatial Networking between Tumor-Associated SPP1+ Macrophages and Fibroblasts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2023, 29, 244–260. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, Q.; Liu, X.; Wang, F.; Ma, Y.; Zhang, S.; Shao, Z.; Yang, Y.; Tian, X. Single Cell RNA-Seq Identifies Immune-Related Prognostic Model and Key Signature-SPP1 in Pancreatic Ductal Adenocarcinoma. Genes 2022, 13, 1760. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Q.; Chen, G.; Luo, D. Multi-Omics Analysis Showed the Clinical Value of Gene Signatures of C1QC(+) and SPP1(+) TAMs in Cervical Cancer. Front. Immunol. 2021, 12, 694801. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Z.; Sun, Y.; Pang, S.; Yao, Q.; Lin, P.; Cheng, J.; Li, J.; Ding, G.; Hui, L.; et al. Integrative analysis reveals novel driver genes and molecular subclasses of hepatocellular carcinoma. Aging 2020, 12, 23849–23871. [Google Scholar] [CrossRef]
- Xie, L.; Ning, Z.; Hua, Y.; Wang, P.; Meng, Z. Single-cell transcriptome analysis revealed the immune profile of PD-1 blockade in gallbladder carcinoma liver metastasis. Hepatol. Commun. 2023, 7, e0131. [Google Scholar] [CrossRef] [PubMed]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1, e85841. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. A J. Virtual Libr. 2008, 13, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, H.; Wei, T.; Lin, A.; Sun, Y.; Luo, P.; Zhang, J. Single-Cell RNA Sequencing Reveals the Heterogeneity of Tumor-Associated Macrophage in Non-Small Cell Lung Cancer and Differences Between Sexes. Front. Immunol. 2021, 12, 756722. [Google Scholar] [CrossRef]
- Araujo, J.M.; Prado, A.; Cardenas, N.K.; Zaharia, M.; Dyer, R.; Doimi, F.; Bravo, L.; Pinillos, L.; Morante, Z.; Aguilar, A.; et al. Repeated observation of immune gene sets enrichment in women with non-small cell lung cancer. Oncotarget 2016, 7, 20282–20292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leader, A.M.; Grout, J.A.; Maier, B.B.; Nabet, B.Y.; Park, M.D.; Tabachnikova, A.; Chang, C.; Walker, L.; Lansky, A.; Le Berichel, J.; et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 2021, 39, 1594–1609.e12. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zayas, G.; Almeida, F.A.; Yarmus, L.; Steinfort, D.; Lazarus, D.R.; Simoff, M.J.; Saettele, T.; Murgu, S.; Dammad, T.; Duong, D.K.; et al. Predicting Lymph Node Metastasis in Non-small Cell Lung Cancer: Prospective External and Temporal Validation of the HAL and HOMER Models. Chest 2021, 160, 1108–1120. [Google Scholar] [CrossRef]
- Dong, B.; Wu, C.; Huang, L.; Qi, Y. Macrophage-Related SPP1 as a Potential Biomarker for Early Lymph Node Metastasis in Lung Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9, 739358. [Google Scholar] [CrossRef]
- Jain, S.; Rick, J.W.; Joshi, R.S.; Beniwal, A.; Spatz, J.; Gill, S.; Chang, A.C.; Choudhary, N.; Nguyen, A.T.; Sudhir, S.; et al. Single-cell RNA sequencing and spatial transcriptomics reveal cancer-associated fibroblasts in glioblastoma with protumoral effects. J. Clin. Investig. 2023, 133, e147087. [Google Scholar] [CrossRef] [PubMed]
- Ozato, Y.; Kojima, Y.; Kobayashi, Y.; Hisamatsu, Y.; Toshima, T.; Yonemura, Y.; Masuda, T.; Kagawa, K.; Goto, Y.; Utou, M.; et al. Spatial and single-cell transcriptomics decipher the cellular environment containing HLA-G+ cancer cells and SPP1+ macrophages in colorectal cancer. Cell Rep. 2023, 42, 111929. [Google Scholar] [CrossRef] [PubMed]
- Butti, R.; Kumar, T.V.S.; Nimma, R.; Banerjee, P.; Kundu, I.G.; Kundu, G.C. Osteopontin Signaling in Shaping Tumor Microenvironment Conducive to Malignant Progression. Adv. Exp. Med. Biol. 2021, 1329, 419–441. [Google Scholar] [CrossRef]
- Robertson, B.W.; Bonsal, L.; Chellaiah, M.A. Regulation of Erk1/2 activation by osteopontin in PC3 human prostate cancer cells. Mol. Cancer 2010, 9, 260. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Wei, Y.Y.; Chen, H.T.; Fong, Y.C.; Hsu, C.J.; Tsai, C.H.; Hsu, H.C.; Liu, S.H.; Tang, C.H. Osteopontin increases migration and MMP-9 up-regulation via alphavbeta3 integrin, FAK, ERK, and NF-kappaB-dependent pathway in human chondrosarcoma cells. J. Cell. Physiol. 2009, 221, 98–108. [Google Scholar] [CrossRef]
- Zhang, H.; Guo, M.; Chen, J.H.; Wang, Z.; Du, X.F.; Liu, P.X.; Li, W.H. Osteopontin knockdown inhibits alphav, beta3 integrin-induced cell migration and invasion and promotes apoptosis of breast cancer cells by inducing autophagy and inactivating the PI3K/Akt/mTOR pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2014, 33, 991–1002. [Google Scholar] [CrossRef]
- Robertson, B.W.; Chellaiah, M.A. Osteopontin induces beta-catenin signaling through activation of Akt in prostate cancer cells. Exp. Cell Res. 2010, 316, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Zhang, Y.; Lei, Z.; Liu, T.; Cai, T.; Wang, A.; Du, W.; Zeng, Y.; Zhu, J.; Liu, Z.; et al. Abnormally activated OPN/integrin alphaVbeta3/FAK signalling is responsible for EGFR-TKI resistance in EGFR mutant non-small-cell lung cancer. J. Hematol. Oncol. 2020, 13, 169. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, F.; Yang, X.; Xue, M.; Li, X.; Gao, Y.; Liu, L. Secreted Phosphoprotein 1 (SPP1) Contributes to Second-Generation EGFR Tyrosine Kinase Inhibitor Resistance in Non-Small Cell Lung Cancer. Oncol. Res. 2019, 27, 871–877. [Google Scholar] [CrossRef]
- Wang, Y.J.; Wang, Q.W.; Yu, D.H.; Song, C.K.; Guo, Z.X.; Liu, X.P.; Chen, C.; Yao, J.; Wang, A.F.; Hu, W.D. Osteopontin improves sensitivity to tyrosine kinase inhibitor in lung adenocarcinoma in vitro by promoting epidermal growth factor receptor phosphorylation. J. Cancer Res. Clin. Oncol. 2021, 147, 3245–3254. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Wisniewski, P.; Kijewska, M.; Gajdanowicz, P.; Pszczolkowska, D.; Przanowski, P.; Dabrowski, M.; Maleszewska, M.; Kaminska, B. Tumour-processed osteopontin and lactadherin drive the protumorigenic reprogramming of microglia and glioma progression. Oncogene 2016, 35, 6366–6377. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Stewart, J., Jr.; Prince, C.W.; Chang, P.L.; Trikha, M.; Han, X.; Grammer, J.R.; Gladson, C.L. Promotion of malignant astrocytoma cell migration by osteopontin expressed in the normal brain: Differences in integrin signaling during cell adhesion to osteopontin versus vitronectin. Cancer Res. 2002, 62, 5336–5343. [Google Scholar] [CrossRef] [PubMed]
- Tuck, A.B.; Arsenault, D.M.; O’Malley, F.P.; Hota, C.; Ling, M.C.; Wilson, S.M.; Chambers, A.F. Osteopontin induces increased invasiveness and plasminogen activator expression of human mammary epithelial cells. Oncogene 1999, 18, 4237–4246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irby, R.B.; McCarthy, S.M.; Yeatman, T.J. Osteopontin regulates multiple functions contributing to human colon cancer development and progression. Clin. Exp. Metastasis 2004, 21, 515–523. [Google Scholar] [CrossRef]
- Chae, S.; Jun, H.O.; Lee, E.G.; Yang, S.J.; Lee, D.C.; Jung, J.K.; Park, K.C.; Yeom, Y.I.; Kim, K.W. Osteopontin splice variants differentially modulate the migratory activity of hepatocellular carcinoma cell lines. Int. J. Oncol. 2009, 35, 1409–1416. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Cai, C.Q.; Schroeder, R.A.; Kuo, P.C. Osteopontin is a negative feedback regulator of nitric oxide synthesis in murine macrophages. J. Immunol. 2001, 166, 1079–1086. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Marroquin, C.E.; Wai, P.Y.; Kuo, P.C. Nitric oxide-dependent osteopontin expression induces metastatic behavior in HepG2 cells. Dig. Dis. Sci. 2005, 50, 1288–1298. [Google Scholar] [CrossRef]
- Ikeguchi, M.; Ueta, T.; Yamane, Y.; Hirooka, Y.; Kaibara, N. Inducible nitric oxide synthase and survivin messenger RNA expression in hepatocellular carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 3131–3136. [Google Scholar]
- Ortiz-Martinez, F.; Sanmartin, E.; Pomares-Navarro, E.; Perez-Balaguer, A.; Andres, L.; Sanchez-Paya, J.; Aranda, F.I.; Lerma, E.; Peiro, G. Osteopontin Regulates VEGFA and ICAM-1 mRNA Expression in Breast Carcinoma. Am. J. Clin. Pathol. 2015, 143, 812–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.L.; Lin, K.J.; Bai, A.P.; Wang, W.X.; Meng, X.K.; Su, X.L.; Hou, M.X.; Dong, P.D.; Zhang, J.J.; Wang, Z.Y.; et al. Osteopontin knockdown suppresses the growth and angiogenesis of colon cancer cells. World J. Gastroenterol. 2014, 20, 10440–10448. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Zhou, C.Q.; Chellaiah, M.A. Osteopontin and MMP9: Associations with VEGF Expression/Secretion and Angiogenesis in PC3 Prostate Cancer Cells. Cancers 2013, 5, 617–638. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Peng, L.; Fan, K.; Wang, H.; Wei, R.; Ji, G.; Cai, J.; Lu, B.; Li, B.; Zhang, D.; et al. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene 2009, 28, 3412–3422. [Google Scholar] [CrossRef] [Green Version]
- Ruzafa, N.; Pereiro, X.; Aspichueta, P.; Araiz, J.; Vecino, E. The Retina of Osteopontin deficient Mice in Aging. Mol. Neurobiol. 2018, 55, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Crawford, H.C.; Matrisian, L.M.; Liaw, L. Distinct roles of osteopontin in host defense activity and tumor survival during squamous cell carcinoma progression in vivo. Cancer Res. 1998, 58, 5206–5215. [Google Scholar]
- Feng, B.; Rollo, E.E.; Denhardt, D.T. Osteopontin (OPN) may facilitate metastasis by protecting cells from macrophage NO-mediated cytotoxicity: Evidence from cell lines down-regulated for OPN expression by a targeted ribozyme. Clin. Exp. Metastasis 1995, 13, 453–462. [Google Scholar] [CrossRef]
- Hsieh, Y.H.; Juliana, M.M.; Hicks, P.H.; Feng, G.; Elmets, C.; Liaw, L.; Chang, P.L. Papilloma development is delayed in osteopontin-null mice: Implicating an antiapoptosis role for osteopontin. Cancer Res. 2006, 66, 7119–7127. [Google Scholar] [CrossRef] [Green Version]
- Kale, S.; Raja, R.; Thorat, D.; Soundararajan, G.; Patil, T.V.; Kundu, G.C. Osteopontin signaling upregulates cyclooxygenase-2 expression in tumor-associated macrophages leading to enhanced angiogenesis and melanoma growth via alpha9beta1 integrin. Oncogene 2014, 33, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Kale, S.; Raja, R.; Thorat, D.; Soundararajan, G.; Patil, T.V.; Kundu, G.C. Osteopontin signaling upregulates cyclooxygenase-2 expression in tumor-associated macrophages leading to enhanced angiogenesis and melanoma growth via alpha9beta1 integrin. Oncogene 2015, 34, 5408–5410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danzaki, K.; Kanayama, M.; Alcazar, O.; Shinohara, M.L. Osteopontin has a protective role in prostate tumor development in mice. Eur. J. Immunol. 2016, 46, 2669–2678. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; LeSavage, B.L.; Hubka, K.M.; Ma, C.; Natarajan, S.; Eggold, J.T.; Xiao, Y.; Fuh, K.C.; Krishnan, V.; Enejder, A.; et al. Cancer-associated mesothelial cells promote ovarian cancer chemoresistance through paracrine osteopontin signaling. J. Clin. Investig. 2021, 131, e146186. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Perruzzi, C.A.; Papadopoulos-Sergiou, A.; Van de Water, L. Adhesive properties of osteopontin: Regulation by a naturally occurring thrombin-cleavage in close proximity to the GRGDS cell-binding domain. Mol. Biol. Cell 1994, 5, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Myles, T.; Leung, L.L. Thrombin hydrolysis of human osteopontin is dependent on thrombin anion-binding exosites. J. Biol. Chem. 2008, 283, 17789–17796. [Google Scholar] [CrossRef] [Green Version]
- Agnihotri, R.; Crawford, H.C.; Haro, H.; Matrisian, L.M.; Havrda, M.C.; Liaw, L. Osteopontin, a novel substrate for matrix metalloproteinase-3 (stromelysin-1) and matrix metalloproteinase-7 (matrilysin). J. Biol. Chem. 2001, 276, 28261–28267. [Google Scholar] [CrossRef] [Green Version]
- Christensen, B.; Sorensen, E.S. Osteopontin is highly susceptible to cleavage in bovine milk and the proteolytic fragments bind the alphaVbeta(3)-integrin receptor. J. Dairy Sci. 2014, 97, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Santamaria, S.; Martin, D.R.; Dong, X.; Yamamoto, K.; Apte, S.S.; Ahnstrom, J. Post-translational regulation and proteolytic activity of the metalloproteinase ADAMTS8. J. Biol. Chem. 2021, 297, 101323. [Google Scholar] [CrossRef]
- Takafuji, V.; Forgues, M.; Unsworth, E.; Goldsmith, P.; Wang, X.W. An osteopontin fragment is essential for tumor cell invasion in hepatocellular carcinoma. Oncogene 2007, 26, 6361–6371. [Google Scholar] [CrossRef] [Green Version]
- Christensen, B.; Schack, L.; Klaning, E.; Sorensen, E.S. Osteopontin is cleaved at multiple sites close to its integrin-binding motifs in milk and is a novel substrate for plasmin and cathepsin D. J. Biol. Chem. 2010, 285, 7929–7937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, Z.; Oliver, T.; Guo, H.; Gao, C.; Kuo, P.C. Thrombin-cleaved COOH(-) terminal osteopontin peptide binds with cyclophilin C to CD147 in murine breast cancer cells. Cancer Res. 2007, 67, 4088–4097. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.; Morser, J.; Leung, L.L.K. Thrombin cleavage of osteopontin disrupts a pro-chemotactic sequence for dendritic cells, which is compensated by the release of its pro-chemotactic C-terminal fragment. J. Biol. Chem. 2014, 289, 27146–27158. [Google Scholar] [CrossRef] [PubMed]
- Hamias, R.; Rudich, A.; Greenberg, G.; Szendro, G.; Wolak, T. Angiotensin 1-7, but not the thrombin-cleaved osteopontin C-terminal fragment, attenuates osteopontin-mediated macrophage-induced endothelial-cell inflammation. Inflamm. Res. 2018, 67, 265–275. [Google Scholar] [CrossRef]
- Christensen, B.; Karlsen, N.J.; Jorgensen, S.D.S.; Jacobsen, L.N.; Ostenfeld, M.S.; Petersen, S.V.; Mullertz, A.; Sorensen, E.S. Milk osteopontin retains integrin-binding activity after in vitro gastrointestinal transit. J. Dairy Sci. 2020, 103, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Christensen, B.; Nielsen, N.R.; Sorensen, M.R.; Jacobsen, L.N.; Ostenfeld, M.S.; Sorensen, E.S. Naturally Occurring N-Terminal Fragments of Bovine Milk Osteopontin Are Transported across Models of the Intestinal Barrier. Biomedicines 2023, 11, 893. [Google Scholar] [CrossRef]
- Kubota, T.; Zhang, Q.; Wrana, J.L.; Ber, R.; Aubin, J.E.; Butler, W.T.; Sodek, J. Multiple forms of SppI (secreted phosphoprotein, osteopontin) synthesized by normal and transformed rat bone cell populations: Regulation by TGF-beta. Biochem. Biophys. Res. Commun. 1989, 162, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Perruzzi, C.A.; Papadopoulos, A.; Tenen, D.G. Purification of a human milk protein closely similar to tumor-secreted phosphoproteins and osteopontin. Biochim. Biophys. Acta 1989, 996, 43–48. [Google Scholar] [CrossRef]
- Zhang, Q.; Domenicucci, C.; Goldberg, H.A.; Wrana, J.L.; Sodek, J. Characterization of fetal porcine bone sialoproteins, secreted phosphoprotein I (SPPI, osteopontin), bone sialoprotein, and a 23-kDa glycoprotein. Demonstration that the 23-kDa glycoprotein is derived from the carboxyl terminus of SPPI. J. Biol. Chem. 1990, 265, 7583–7589. [Google Scholar] [CrossRef]
- Ullrich, O.; Mann, K.; Haase, W.; Koch-Brandt, C. Biosynthesis and secretion of an osteopontin-related 20-kDa polypeptide in the Madin-Darby canine kidney cell line. J. Biol. Chem. 1991, 266, 3518–3525. [Google Scholar] [CrossRef]
- Bayless, K.J.; Meininger, G.A.; Scholtz, J.M.; Davis, G.E. Osteopontin is a ligand for the alpha4beta1 integrin. J. Cell Sci. 1998, 111 Pt 9, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.L.; Cheung, H.K.; Ling, L.E.; Chen, J.; Sheppard, D.; Pytela, R.; Giachelli, C.M. Osteopontin N-terminal domain contains a cryptic adhesive sequence recognized by alpha9beta1 integrin. J. Biol. Chem. 1996, 271, 28485–28491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokasaki, Y.; Sheppard, D. Mapping of the cryptic integrin-binding site in osteopontin suggests a new mechanism by which thrombin can regulate inflammation and tissue repair. Trends. Cardiovasc. Med. 2000, 10, 155–159. [Google Scholar] [CrossRef]
- Bajzar, L.; Manuel, R.; Nesheim, M.E. Purification and characterization of TAFI, a thrombin-activable fibrinolysis inhibitor. J. Biol. Chem. 1995, 270, 14477–14484. [Google Scholar] [CrossRef] [Green Version]
- Morser, J. Thrombomodulin links coagulation to inflammation and immunity. Curr. Drug Targets 2012, 13, 421–431. [Google Scholar] [CrossRef]
- Boggio, E.; Dianzani, C.; Gigliotti, C.L.; Soluri, M.F.; Clemente, N.; Cappellano, G.; Toth, E.; Raineri, D.; Ferrara, B.; Comi, C.; et al. Thrombin Cleavage of Osteopontin Modulates Its Activities in Human Cells In Vitro and Mouse Experimental Autoimmune Encephalomyelitis In Vivo. J. Immunol. Res. 2016, 2016, 9345495. [Google Scholar] [CrossRef] [Green Version]
- Cui, G.; Chen, J.; Wu, Z.; Huang, H.; Wang, L.; Liang, Y.; Zeng, P.; Yang, J.; Uede, T.; Diao, H. Thrombin cleavage of osteopontin controls activation of hepatic stellate cells and is essential for liver fibrogenesis. J. Cell. Physiol. 2019, 234, 8988–8997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, B.B.; Sharma, S.; Vasishta, R.K.; Sen, R.K.; Sharma, A.; Luthra-Guptasarma, M. Blocking osteopontin-fibronectin interactions reduce extracellular fibronectin deployment and arthritic immunopathology. Int. Immunopharmacol. 2018, 55, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Storan, M.J.; Heazlewood, S.Y.; Heazlewood, C.K.; Haylock, D.N.; Alexander, W.S.; Neaves, R.J.; Oteiza, A.; Nilsson, S.K. Brief Report: Factors Released by Megakaryocytes Thrombin Cleave Osteopontin to Negatively Regulate Hematopoietic Stem Cells. Stem Cells 2015, 33, 2351–2357. [Google Scholar] [CrossRef]
- Nilsson, S.K.; Johnston, H.M.; Whitty, G.A.; Williams, B.; Webb, R.J.; Denhardt, D.T.; Bertoncello, I.; Bendall, L.J.; Simmons, P.J.; Haylock, D.N. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 2005, 106, 1232–1239. [Google Scholar] [CrossRef]
- Stier, S.; Ko, Y.; Forkert, R.; Lutz, C.; Neuhaus, T.; Grunewald, E.; Cheng, T.; Dombkowski, D.; Calvi, L.M.; Rittling, S.R.; et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J. Exp. Med. 2005, 201, 1781–1791. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2019, 129, 137–149. [Google Scholar] [CrossRef]
- Hur, E.M.; Youssef, S.; Haws, M.E.; Zhang, S.Y.; Sobel, R.A.; Steinman, L. Osteopontin-induced relapse and progression of autoimmune brain disease through enhanced survival of activated T cells. Nat. Immunol. 2007, 8, 74–83. [Google Scholar] [CrossRef]
- Peraramelli, S.; Zhou, Q.; Zhou, Q.; Wanko, B.; Zhao, L.; Nishimura, T.; Leung, T.H.; Mizuno, S.; Ito, M.; Myles, T.; et al. Thrombin cleavage of osteopontin initiates osteopontin’s tumor-promoting activity. J. Thromb. Haemost. 2022, 20, 1256–1270. [Google Scholar] [CrossRef]
- Nemoto, H.; Rittling, S.R.; Yoshitake, H.; Furuya, K.; Amagasa, T.; Tsuji, K.; Nifuji, A.; Denhardt, D.T.; Noda, M. Osteopontin deficiency reduces experimental tumor cell metastasis to bone and soft tissues. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2001, 16, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Giavazzi, R.; Decio, A. Syngeneic murine metastasis models: B16 melanoma. Methods Mol. Biol. 2014, 1070, 131–140. [Google Scholar] [CrossRef]
- Li, Z.; Xu, X.; Feng, X.; Murphy, P.M. The Macrophage-depleting Agent Clodronate Promotes Durable Hematopoietic Chimerism and Donor-specific Skin Allograft Tolerance in Mice. Sci. Rep. 2016, 6, 22143. [Google Scholar] [CrossRef] [Green Version]
- van Rooijen, N.; Hendrikx, E. Liposomes for specific depletion of macrophages from organs and tissues. Methods Mol. Biol. 2010, 605, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K.; et al. NOD/SCID/gamma(c)(null) mouse: An excellent recipient mouse model for engraftment of human cells. Blood 2002, 100, 3175–3182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debacker, J.M.; Gondry, O.; Lahoutte, T.; Keyaerts, M.; Huvenne, W. The Prognostic Value of CD206 in Solid Malignancies: A Systematic Review and Meta-Analysis. Cancers 2021, 13, 3422. [Google Scholar] [CrossRef]
- Diaz, A.H.; Rodgers, G.M.; Gilreath, J.A. Enoxaparin once daily vs. twice daily dosing for the treatment of venous thromboembolism in cancer patients: A literature summary. J. Oncol. Pharm. Pract. 2012, 18, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Schulze, E.B.; Hedley, B.D.; Goodale, D.; Postenka, C.O.; Al-Katib, W.; Tuck, A.B.; Chambers, A.F.; Allan, A.L. The thrombin inhibitor Argatroban reduces breast cancer malignancy and metastasis via osteopontin-dependent and osteopontin-independent mechanisms. Breast Cancer Res. Treat. 2008, 112, 243–254. [Google Scholar] [CrossRef]
- Niers, T.M.; Bruggemann, L.W.; GL, V.A.N.S.; Liu, R.D.; Versteeg, H.H.; Buller, H.R.; CJ, V.A.N.N.; Reitsma, P.H.; Spek, C.A.; Richel, D.J. Long-term thrombin inhibition promotes cancer cell extravasation in a mouse model of experimental metastasis. J. Thromb. Haemost. 2009, 7, 1595–1597. [Google Scholar] [CrossRef]
- Kahale, L.A.; Matar, C.F.; Tsolakian, I.; Hakoum, M.B.; Barba, M.; Yosuico, V.E.; Terrenato, I.; Sperati, F.; Schunemann, H.; Akl, E.A. Oral anticoagulation in people with cancer who have no therapeutic or prophylactic indication for anticoagulation. Cochrane Database Syst. Rev. 2021, 10, CD006466. [Google Scholar] [CrossRef]
- Chiasakul, T.; Zwicker, J.I. The impact of warfarin on overall survival in cancer patients. Thromb. Res. 2022, 213, S113–S119. [Google Scholar] [CrossRef] [PubMed]
- Prandoni, P.; Lensing, A.W.; Piccioli, A.; Bernardi, E.; Simioni, P.; Girolami, B.; Marchiori, A.; Sabbion, P.; Prins, M.H.; Noventa, F.; et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002, 100, 3484–3488. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- Viallard, C.; Larrivee, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Farge, D.; Debourdeau, P.; Beckers, M.; Baglin, C.; Bauersachs, R.M.; Brenner, B.; Brilhante, D.; Falanga, A.; Gerotzafias, G.T.; Haim, N.; et al. International clinical practice guidelines for the treatment and prophylaxis of venous thromboembolism in patients with cancer. J. Thromb. Haemost. 2013, 11, 56–70. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.J.; Green, D.L.; Shah, G.L. Therapeutic Anticoagulation in Patients with Primary Brain Tumors or Secondary Brain Metastasis. Oncologist 2018, 23, 468–473. [Google Scholar] [CrossRef] [Green Version]
- Maeda, N.; Ohashi, T.; Chagan-Yasutan, H.; Hattori, T.; Takahashi, Y.; Harigae, H.; Hasegawa, H.; Yamada, Y.; Fujii, M.; Maenaka, K.; et al. Osteopontin-integrin interaction as a novel molecular target for antibody-mediated immunotherapy in adult T-cell leukemia. Retrovirology 2015, 12, 99. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Li, B.; Shi, J.; Peng, L.; Zhang, D.; Qian, W.; Hou, S.; Zhao, L.; Gao, J.; Cao, Z.; et al. A humanized anti-osteopontin antibody inhibits breast cancer growth and metastasis in vivo. Cancer Immunol. Immunother. 2010, 59, 355–366. [Google Scholar] [CrossRef]
- Shojaei, F.; Scott, N.; Kang, X.; Lappin, P.B.; Fitzgerald, A.A.; Karlicek, S.; Simmons, B.H.; Wu, A.; Lee, J.H.; Bergqvist, S.; et al. Osteopontin induces growth of metastatic tumors in a preclinical model of non-small lung cancer. J. Exp. Clin. Cancer Res. 2012, 31, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kon, S.; Yokosaki, Y.; Maeda, M.; Segawa, T.; Horikoshi, Y.; Tsukagoshi, H.; Rashid, M.M.; Morimoto, J.; Inobe, M.; Shijubo, N.; et al. Mapping of functional epitopes of osteopontin by monoclonal antibodies raised against defined internal sequences. J. Cell. Biochem. 2002, 84, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Nakashima, T.; Torikai, M.; Naruse, T.; Morimoto, J.; Kon, S.; Sakai, F.; Uede, T. Successful treatment of collagen-induced arthritis in non-human primates by chimeric anti-osteopontin antibody. Int. Immunopharmacol. 2007, 7, 1460–1470. [Google Scholar] [CrossRef]
- Boumans, M.J.; Houbiers, J.G.; Verschueren, P.; Ishikura, H.; Westhovens, R.; Brouwer, E.; Rojkovich, B.; Kelly, S.; den Adel, M.; Isaacs, J.; et al. Safety, tolerability, pharmacokinetics, pharmacodynamics and efficacy of the monoclonal antibody ASK8007 blocking osteopontin in patients with rheumatoid arthritis: A randomised, placebo controlled, proof-of-concept study. Ann. Rheum. Dis. 2012, 71, 180–185. [Google Scholar] [CrossRef]
- Farrokhi, V.; Chabot, J.R.; Neubert, H.; Yang, Z. Assessing the Feasibility of Neutralizing Osteopontin with Various Therapeutic Antibody Modalities. Sci. Rep. 2018, 8, 7781. [Google Scholar] [CrossRef]
- Wu, J.; Pungaliya, P.; Kraynov, E.; Bates, B. Identification and quantification of osteopontin splice variants in the plasma of lung cancer patients using immunoaffinity capture and targeted mass spectrometry. Biomark. Biochem. Indic. Expo. Response Susceptibility Chem. 2012, 17, 125–133. [Google Scholar] [CrossRef]
- Graf, C.; Wilgenbus, P.; Pagel, S.; Pott, J.; Marini, F.; Reyda, S.; Kitano, M.; Macher-Goppinger, S.; Weiler, H.; Ruf, W. Myeloid cell-synthesized coagulation factor X dampens antitumor immunity. Sci. Immunol. 2019, 4, eaaw8405. [Google Scholar] [CrossRef]
- Hisada, Y.; Mackman, N. Cancer cell-derived tissue factor-positive extracellular vesicles: Biomarkers of thrombosis and survival. Curr. Opin. Hematol. 2019, 26, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Ruf, W.; Rothmeier, A.S.; Graf, C. Targeting clotting proteins in cancer therapy—Progress and challenges. Thromb. Res. 2016, 140 (Suppl. S1), S1–S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Z.; Chaikof, E.L. Interface between hemostasis and adaptive immunity. Curr. Opin. Immunol. 2010, 22, 634–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niers, T.M.; Bruggemann, L.W.; Klerk, C.P.; Muller, F.J.; Buckle, T.; Reitsma, P.H.; Richel, D.J.; Spek, C.A.; Van Tellingen, O.; Van Noorden, C.J. Differential effects of anticoagulants on tumor development of mouse cancer cell lines B16, K1735 and CT26 in lung. Clin. Exp. Metastasis 2009, 26, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Akl, E.A.; Rohilla, S.; Barba, M.; Sperati, F.; Terrenato, I.; Muti, P.; Schunemann, H.J. Anticoagulation for the initial treatment of venous thromboembolism in patients with cancer. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef]
- O’Rorke, M.A.; Murray, L.J.; Hughes, C.M.; Cantwell, M.M.; Cardwell, C.R. The effect of warfarin therapy on breast, colorectal, lung, and prostate cancer survival: A population-based cohort study using the Clinical Practice Research Datalink. Cancer Causes Control CCC 2015, 26, 355–366. [Google Scholar] [CrossRef]
- Chai-Adisaksopha, C.; Hillis, C.; Isayama, T.; Lim, W.; Iorio, A.; Crowther, M. Mortality outcomes in patients receiving direct oral anticoagulants: A systematic review and meta-analysis of randomized controlled trials. J. Thromb. Haemost. 2015, 13, 2012–2020. [Google Scholar] [CrossRef] [Green Version]
- Honda, M.; Kimura, C.; Uede, T.; Kon, S. Neutralizing antibody against osteopontin attenuates non-alcoholic steatohepatitis in mice. J. Cell Commun. Signal. 2020, 14, 223–232. [Google Scholar] [CrossRef]
- Herum, K.M.; Romaine, A.; Wang, A.; Melleby, A.O.; Strand, M.E.; Pacheco, J.; Braathen, B.; Duner, P.; Tonnessen, T.; Lunde, I.G.; et al. Syndecan-4 Protects the Heart From the Profibrotic Effects of Thrombin-Cleaved Osteopontin. J. Am. Heart Assoc. 2020, 9, e013518. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leung, L.L.; Myles, T.; Morser, J. Thrombin Cleavage of Osteopontin and the Host Anti-Tumor Immune Response. Cancers 2023, 15, 3480. https://doi.org/10.3390/cancers15133480
Leung LL, Myles T, Morser J. Thrombin Cleavage of Osteopontin and the Host Anti-Tumor Immune Response. Cancers. 2023; 15(13):3480. https://doi.org/10.3390/cancers15133480
Chicago/Turabian StyleLeung, Lawrence L., Timothy Myles, and John Morser. 2023. "Thrombin Cleavage of Osteopontin and the Host Anti-Tumor Immune Response" Cancers 15, no. 13: 3480. https://doi.org/10.3390/cancers15133480
APA StyleLeung, L. L., Myles, T., & Morser, J. (2023). Thrombin Cleavage of Osteopontin and the Host Anti-Tumor Immune Response. Cancers, 15(13), 3480. https://doi.org/10.3390/cancers15133480