


Understanding WT1 Alterations and Expression Profiles in Hematological Malignancies
 , ,
, ,  , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Preparation of Genomic Material
2.2. Patient Samples
2.3. Complementary DNA Synthesis and RT-PCR
2.4. Intracellular Staining of WT1 Protein and Flow Cytometry
2.5. Fragment Length Analysis
2.6. Generating WT1 Expression Constructs
2.7. Production of Lentiviral Particles
2.8. Next-Generation Sequencing
2.9. Sanger Sequencing
3. Results
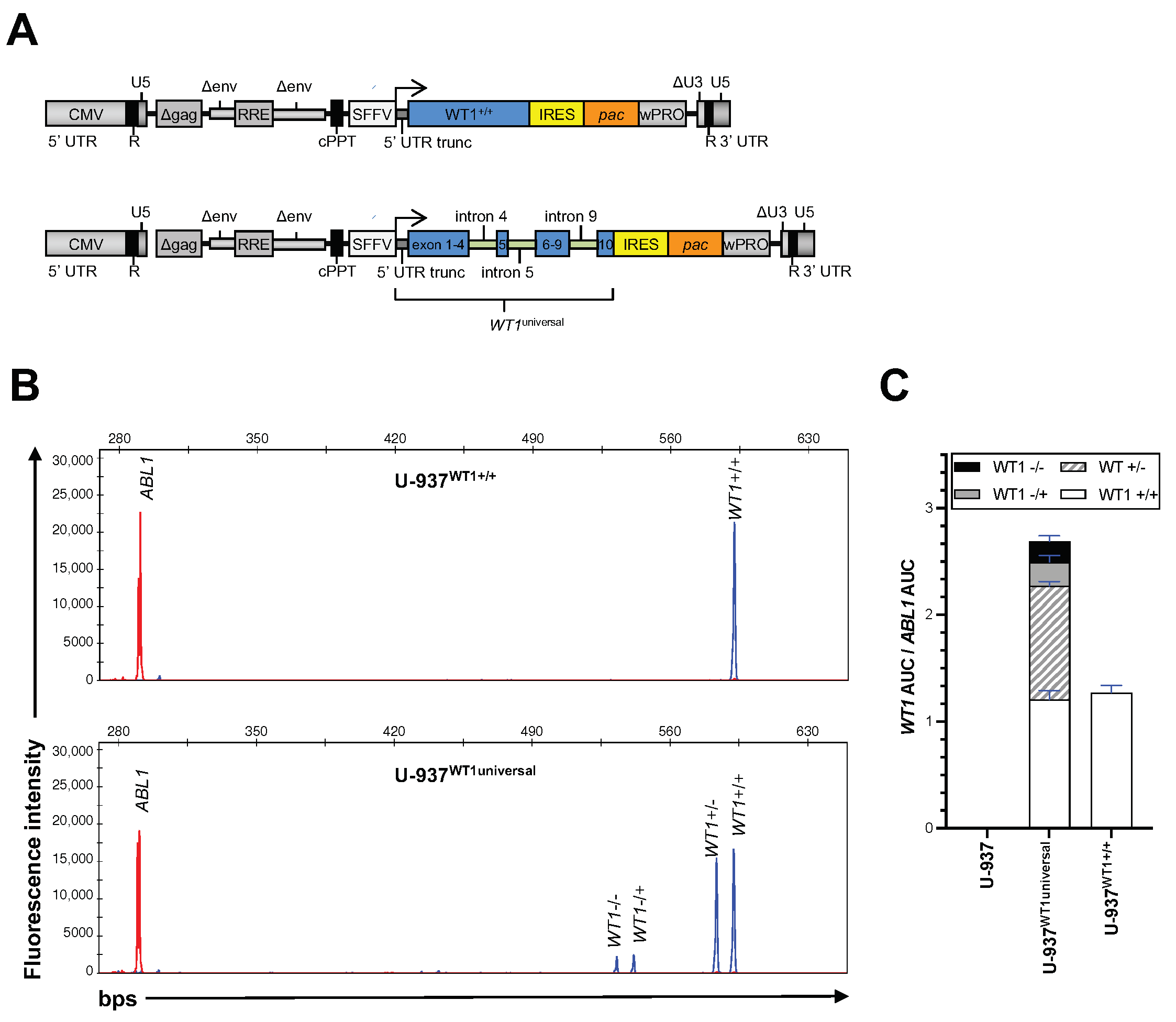
3.1. The Expression Profile of WT1 in Leukemia and Lymphoma Cells
3.2. WT1 Isoform Expression Pattern in Leukemic Cell Lines and Primary AML Blasts
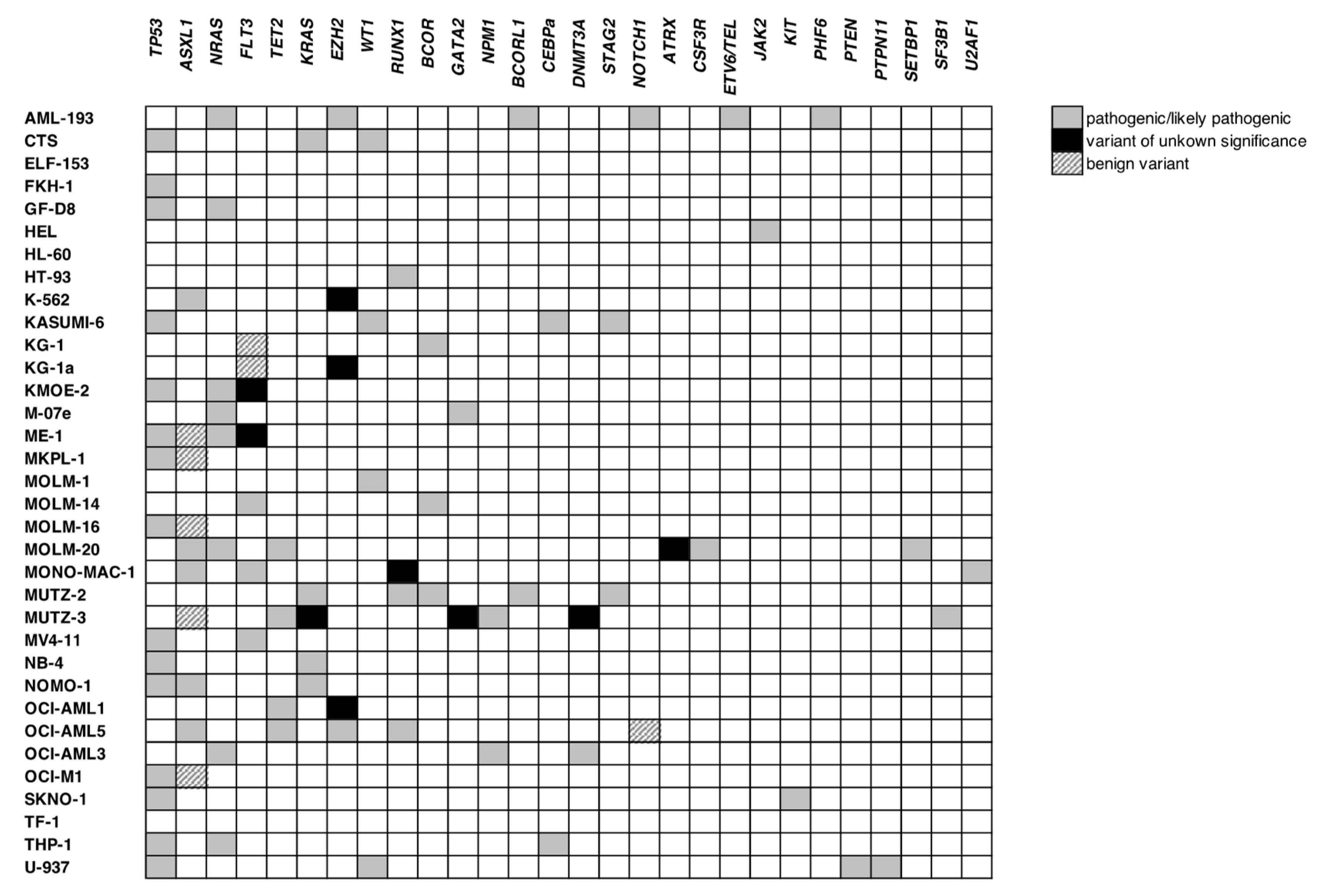
3.3. Screening for WT1 Mutations
3.4. Expression of WT1 Mutated Alleles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huff, V. Wilms’ tumours: About tumour suppressor genes, an oncogene and a chameleon gene. Nat. Rev. Cancer 2011, 11, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Kreidberg, J.A.; Sariola, H.; Loring, J.M.; Maeda, M.; Pelletier, J.; Housman, D.; Jaenisch, R. WT-1 is required for early kidney development. Cell 1993, 74, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Chau, Y.Y.; Brownstein, D.; Mjoseng, H.; Lee, W.C.; Buza-Vidas, N.; Nerlov, C.; Jacobsen, S.E.; Perry, P.; Berry, R.; Thornburn, A.; et al. Acute multiple organ failure in adult mice deleted for the developmental regulator Wt1. PLoS Genet. 2011, 7, e1002404. [Google Scholar] [CrossRef] [Green Version]
- Niksic, M.; Slight, J.; Sanford, J.R.; Caceres, J.F.; Hastie, N.D. The Wilms’ tumour protein (WT1) shuttles between nucleus and cytoplasm and is present in functional polysomes. Hum. Mol. Genet. 2004, 13, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haber, D.A.; Sohn, R.L.; Buckler, A.J.; Pelletier, J.; Call, K.M.; Housman, D.E. Alternative splicing and genomic structure of the Wilms tumor gene WT1. Proc. Natl. Acad. Sci. USA 1991, 88, 9618–9622. [Google Scholar] [CrossRef] [PubMed]
- Dutton, J.R.; Lahiri, D.; Ward, A. Different isoforms of the Wilms’ tumour protein WT1 have distinct patterns of distribution and trafficking within the nucleus. Cell Prolif. 2006, 39, 519–535. [Google Scholar] [CrossRef]
- Bor, Y.C.; Swartz, J.; Morrison, A.; Rekosh, D.; Ladomery, M.; Hammarskjöld, M.L. The Wilms’ tumor 1 (WT1) gene (+KTS isoform) functions with a CTE to enhance translation from an unspliced RNA with a retained intron. Genes Dev. 2006, 20, 1597–1608. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Romaniuk, P.J. The ratio of +/−KTS splice variants of the Wilms’ tumour suppressor protein WT1 mRNA is determined by an intronic enhancer. Biochem. Cell Biol. 2008, 86, 312–321. [Google Scholar] [CrossRef]
- Ullmark, T.; Montano, G.; Gullberg, U. DNA and RNA binding by the Wilms’ tumour gene 1 (WT1) protein +KTS and -KTS isoforms-From initial observations to recent global genomic analyses. Eur. J. Haematol. 2018, 100, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Han, Y.; Suarez Saiz, F.; Minden, M.D. A tumor suppressor and oncogene: The WT1 story. Leukemia 2007, 21, 868–876. [Google Scholar] [CrossRef] [Green Version]
- Rampal, R.; Figueroa, M.E. Wilms tumor 1 mutations in the pathogenesis of acute myeloid leukemia. Haematologica 2016, 101, 672–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagami, T.; Sugiyama, H.; Inoue, K.; Ogawa, H.; Tatekawa, T.; Hirata, M.; Kudoh, T.; Akiyama, T.; Murakami, A.; Maekawa, T. Growth inhibition of human leukemic cells by WT1 (Wilms tumor gene) antisense oligodeoxynucleotides: Implications for the involvement of WT1 in leukemogenesis. Blood 1996, 87, 2878–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svedberg, H.; Richter, J.; Gullberg, U. Forced expression of the Wilms tumor 1 (WT1) gene inhibits proliferation of human hematopoietic CD34(+) progenitor cells. Leukemia 2001, 15, 1914–1922. [Google Scholar] [CrossRef] [Green Version]
- Nishida, S.; Hosen, N.; Shirakata, T.; Kanato, K.; Yanagihara, M.; Nakatsuka, S.; Hoshida, Y.; Nakazawa, T.; Harada, Y.; Tatsumi, N.; et al. AML1-ETO rapidly induces acute myeloblastic leukemia in cooperation with the Wilms tumor gene, WT1. Blood 2006, 107, 3303–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosen, N.; Shirakata, T.; Nishida, S.; Yanagihara, M.; Tsuboi, A.; Kawakami, M.; Oji, Y.; Oka, Y.; Okabe, M.; Tan, B.; et al. The Wilms’ tumor gene WT1-GFP knock-in mouse reveals the dynamic regulation of WT1 expression in normal and leukemic hematopoiesis. Leukemia 2007, 21, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Lopotová, T.; Polák, J.; Schwarz, J.; Klamová, H.; Moravcová, J. Expression of four major WT1 splicing variants in acute and chronic myeloid leukemia patients analyzed by newly developed four real-time RT PCRs. Blood Cells Mol. Dis. 2012, 49, 41–47. [Google Scholar] [CrossRef]
- Kramarzova, K.; Stuchly, J.; Willasch, A.; Gruhn, B.; Schwarz, J.; Cermak, J.; Machova-Polakova, K.; Fuchs, O.; Stary, J.; Trka, J.; et al. Real-time PCR quantification of major Wilms’ tumor gene 1 (WT1) isoforms in acute myeloid leukemia, their characteristic expression patterns and possible functional consequences. Leukemia 2012, 26, 2086–2095. [Google Scholar] [CrossRef] [Green Version]
- Luna, I.; Such, E.; Cervera, J.; Barragán, E.; Ibañez, M.; Gómez-Seguí, I.; López-Pavía, M.; Llop, M.; Fuster, O.; Dolz, S.; et al. WT1 isoform expression pattern in acute myeloid leukemia. Leuk. Res. 2013, 37, 1744–1749. [Google Scholar] [CrossRef]
- Hou, H.A.; Huang, T.C.; Lin, L.I.; Liu, C.Y.; Chen, C.Y.; Chou, W.C.; Tang, J.L.; Tseng, M.H.; Huang, C.F.; Chiang, Y.C.; et al. WT1 mutation in 470 adult patients with acute myeloid leukemia: Stability during disease evolution and implication of its incorporation into a survival scoring system. Blood 2010, 115, 5222–5231. [Google Scholar] [CrossRef] [Green Version]
- Gaidzik, V.I.; Schlenk, R.F.; Moschny, S.; Becker, A.; Bullinger, L.; Corbacioglu, A.; Krauter, J.; Schlegelberger, B.; Ganser, A.; Döhner, H.; et al. Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: A study of the German-Austrian AML Study Group. Blood 2009, 113, 4505–4511. [Google Scholar] [CrossRef] [Green Version]
- Niktoreh, N.; Walter, C.; Zimmermann, M.; von Neuhoff, C.; von Neuhoff, N.; Rasche, M.; Waack, K.; Creutzig, U.; Hanenberg, H.; Reinhardt, D. Mutated WT1, FLT3-ITD, and NUP98-NSD1 Fusion in Various Combinations Define a Poor Prognostic Group in Pediatric Acute Myeloid Leukemia. J. Oncol. 2019, 2019, 1609128. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.A.; Zeng, R.; Alonzo, T.A.; Gerbing, R.B.; Miller, K.L.; Pollard, J.A.; Stirewalt, D.L.; Heerema, N.A.; Raimondi, S.C.; Hirsch, B.; et al. Prevalence and prognostic implications of WT1 mutations in pediatric acute myeloid leukemia (AML): A report from the Children’s Oncology Group. Blood 2010, 116, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidovic, K.; Ullmark, T.; Rosberg, B.; Lennartsson, A.; Olofsson, T.; Nilsson, B.; Gullberg, U. Leukemia associated mutant Wilms’ tumor gene 1 protein promotes expansion of human hematopoietic progenitor cells. Leuk. Res. 2013, 37, 1341–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, J.L.; Korch, C.T. Authentication of Human and Mouse Cell Lines by Short Tandem Repeat (STR) DNA Genotype Analysis. In Assay Guidance Manual [Internet]; Markossian, S., Grossman, A.K.B., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2023; pp. 2004–2057. [Google Scholar]
- Creutzig, U.; Zimmermann, M.; Bourquin, J.P.; Dworzak, M.N.; Fleischhack, G.; Graf, N.; Klingebiel, T.; Kremens, B.; Lehrnbecher, T.; von Neuhoff, C.; et al. Randomized trial comparing liposomal daunorubicin with idarubicin as induction for pediatric acute myeloid leukemia: Results from Study AML-BFM 2004. Blood 2013, 122, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweitzer, J.; Zimmermann, M.; Rasche, M.; von Neuhoff, C.; Creutzig, U.; Dworzak, M.; Reinhardt, D.; Klusmann, J.H. Improved outcome of pediatric patients with acute megakaryoblastic leukemia in the AML-BFM 04 trial. Ann. Hematol. 2015, 94, 1327–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaal, H. HEXplorer—Institute of Virology—Heinrich Heine University Düsseldorf. Available online: https://www2.hhu.de/rna/html/hexplorer_score.php (accessed on 1 February 2023).
- Erkelenz, S.; Theiss, S.; Kaisers, W.; Ptok, J.; Walotka, L.; Müller, L.; Hillebrand, F.; Brillen, A.L.; Sladek, M.; Schaal, H. Ranking noncanonical 5′ splice site usage by genome-wide RNA-seq analysis and splicing reporter assays. Genome Res. 2018, 28, 1826–1840. [Google Scholar] [CrossRef]
- Haist, C.; Schulte, E.; Bartels, N.; Bister, A.; Poschinski, Z.; Ibach, T.C.; Geipel, K.; Wiek, C.; Wagenmann, M.; Monzel, C.; et al. CD44v6-targeted CAR T-cells specifically eliminate CD44 isoform 6 expressing head/neck squamous cell carcinoma cells. Oral Oncol. 2021, 116, 105259. [Google Scholar] [CrossRef]
- Walter, C.; Pozzorini, C.; Reinhardt, K.; Geffers, R.; Xu, Z.; Reinhardt, D.; von Neuhoff, N.; Hanenberg, H. Single-cell whole exome and targeted sequencing in NPM1/FLT3 positive pediatric acute myeloid leukemia. Pediatr. Blood Cancer 2018, 65, e26848. [Google Scholar] [CrossRef]
- Luthra, R.; Patel, K.P.; Reddy, N.G.; Haghshenas, V.; Routbort, M.J.; Harmon, M.A.; Barkoh, B.A.; Kanagal-Shamanna, R.; Ravandi, F.; Cortes, J.E.; et al. Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: Applicability for diagnostics and disease monitoring. Haematologica 2014, 99, 465–473. [Google Scholar] [CrossRef]
- Wagner, K.D.; Wagner, N.; Schedl, A. The complex life of WT1. J. Cell Sci. 2003, 116, 1653–1658. [Google Scholar] [CrossRef] [Green Version]
- Schneider-Poetsch, T.; Ju, J.; Eyler, D.E.; Dang, Y.; Bhat, S.; Merrick, W.C.; Green, R.; Shen, B.; Liu, J.O. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 2010, 6, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Grimwade, D.; Vyas, P.; Freeman, S. Assessment of minimal residual disease in acute myeloid leukemia. Curr. Opin. Oncol. 2010, 22, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Candoni, A.; De Marchi, F.; Zannier, M.E.; Lazzarotto, D.; Filì, C.; Dubbini, M.V.; Rabassi, N.; Toffoletti, E.; Lau, B.W.; Fanin, R. High prognostic value of pre-allogeneic stem cell transplantation minimal residual disease detection by WT1 gene expression in AML transplanted in cytologic complete remission. Leuk. Res. 2017, 63, 22–27. [Google Scholar] [CrossRef]
- Deng, D.X.; Wen, J.J.; Cheng, Y.F.; Zhang, X.H.; Xu, L.P.; Wang, Y.; Yan, C.H.; Chen, Y.H.; Chen, H.; Han, W.; et al. Wilms’ tumor gene 1 is an independent prognostic factor for pediatric acute myeloid leukemia following allogeneic hematopoietic stem cell transplantation. BMC Cancer 2021, 21, 292. [Google Scholar] [CrossRef] [PubMed]
- Virappane, P.; Gale, R.; Hills, R.; Kakkas, I.; Summers, K.; Stevens, J.; Allen, C.; Green, C.; Quentmeier, H.; Drexler, H.; et al. Mutation of the Wilms’ tumor 1 gene is a poor prognostic factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: The United Kingdom Medical Research Council Adult Leukaemia Working Party. J. Clin. Oncol. 2008, 26, 5429–5435. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Marcucci, G.; Ruppert, A.S.; Whitman, S.P.; Mrózek, K.; Maharry, K.; Langer, C.; Baldus, C.D.; Zhao, W.; Powell, B.L.; et al. Wilms’ tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: A cancer and leukemia group B study. J. Clin. Oncol. 2008, 26, 4595–4602. [Google Scholar] [CrossRef]
- Hollink, I.H.; van den Heuvel-Eibrink, M.M.; Zimmermann, M.; Balgobind, B.V.; Arentsen-Peters, S.T.; Alders, M.; Willasch, A.; Kaspers, G.J.; Trka, J.; Baruchel, A.; et al. Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood 2009, 113, 5951–5960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, N.; Chen, W.M.; Li, L.D.; Long, L.Y.; Wang, X.; Jiang, Q.; Jiang, H.; Huang, X.J.; Qin, Y.Z. High WT1 expression predicted induction chemotherapy failure in acute myeloid leukemia patients with non-favorable cytogenetic risk. Clin. Exp. Med. 2023, 13, 1–10. [Google Scholar] [CrossRef]
- Chen, S.; Nagel, S.; Schneider, B.; Dai, H.; Geffers, R.; Kaufmann, M.; Meyer, C.; Pommerenke, C.; Thress, K.S.; Li, J.; et al. A new ETV6-NTRK3 cell line model reveals MALAT1 as a novel therapeutic target—A short report. Cell Oncol. 2018, 41, 93–101. [Google Scholar] [CrossRef]
- Qi, X.W.; Zhang, F.; Wu, H.; Liu, J.L.; Zong, B.G.; Xu, C.; Jiang, J. Wilms’ tumor 1 (WT1) expression and prognosis in solid cancer patients: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 8924. [Google Scholar] [CrossRef] [Green Version]
- Shebl, F.M.; Pinto, L.A.; García-Piñeres, A.; Lempicki, R.; Williams, M.; Harro, C.; Hildesheim, A. Comparison of mRNA and protein measures of cytokines following vaccination with human papillomavirus-16 L1 virus-like particles. Cancer Epidemiol. Biomark. Prev. 2010, 19, 978–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankov, D.; Sjöström, L.; Kalidindi, T.; Lee, S.G.; Sjöström, K.; Gardner, R.; McDevitt, M.R.; O’Reilly, R.; Thorek, D.L.J.; Larson, S.M.; et al. In vivo immuno-targeting of an extracellular epitope of membrane bound preferentially expressed antigen in melanoma (PRAME). Oncotarget 2017, 8, 65917–65931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehra, A.; Lee, K.H.; Hatzimanikatis, V. Insights into the relation between mRNA and protein expression patterns: I. Theoretical considerations. Biotechnol. Bioeng. 2003, 84, 822–833. [Google Scholar] [CrossRef]
- Kozak, M. Some thoughts about translational regulation: Forward and backward glances. J. Cell. Biochem. 2007, 102, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Stepaniants, S.B.; Mao, M.; Weng, L.; Feetham, M.C.; Doyle, M.J.; Yi, E.C.; Dai, H.; Thorsson, V.; Eng, J.; et al. Integrated genomic and proteomic analyses of gene expression in Mammalian cells. Mol. Cell. Proteom. 2004, 3, 960–969. [Google Scholar] [CrossRef] [Green Version]
- Barbaux, S.; Niaudet, P.; Gubler, M.C.; Grünfeld, J.P.; Jaubert, F.; Kuttenn, F.; Fékété, C.N.; Souleyreau-Therville, N.; Thibaud, E.; Fellous, M.; et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat. Genet. 1997, 17, 467–470. [Google Scholar] [CrossRef]
- Shao, Q.; Xie, X.; Geng, J.; Yang, X.; Li, W.; Zhang, Y. Frasier Syndrome: A 15-Year-Old Phenotypically Female Adolescent Presenting with Delayed Puberty and Nephropathy. Children 2023, 10, 577. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Erkelenz, S.; Poschmann, G.; Ptok, J.; Müller, L.; Schaal, H. Profiling of cis- and trans-acting factors supporting noncanonical splice site activation. RNA Biol. 2021, 18, 118–130. [Google Scholar] [CrossRef]
- Terenziani, M.; Sardella, M.; Gamba, B.; Testi, M.A.; Spreafico, F.; Ardissino, G.; Fedeli, F.; Fossati-Bellani, F.; Radice, P.; Perotti, D. A novel WT1 mutation in a 46,XY boy with congenital bilateral cryptorchidism, nystagmus and Wilms tumor. Pediatr. Nephrol. 2009, 24, 1413–1417. [Google Scholar] [CrossRef]
- Royer-Pokora, B.; Beier, M.; Henzler, M.; Alam, R.; Schumacher, V.; Weirich, A.; Huff, V. Twenty-four new cases of WT1 germline mutations and review of the literature: Genotype/phenotype correlations for Wilms tumor development. Am. J. Med. Genet. A 2004, 127, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Englert, C.; Vidal, M.; Maheswaran, S.; Ge, Y.; Ezzell, R.M.; Isselbacher, K.J.; Haber, D.A. Truncated WT1 mutants alter the subnuclear localization of the wild-type protein. Proc. Natl. Acad. Sci. USA 1995, 92, 11960–11964. [Google Scholar] [CrossRef]
- Reddy, J.C.; Morris, J.C.; Wang, J.; English, M.A.; Haber, D.A.; Shi, Y.; Licht, J.D. WT1-mediated transcriptional activation is inhibited by dominant negative mutant proteins. J. Biol. Chem. 1995, 270, 10878–10884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajinda, K.; Carroll, J.; Roberts, C.T., Jr. Regulation of insulin-like growth factor I receptor promoter activity by wild-type and mutant versions of the WT1 tumor suppressor. Endocrinology 1999, 140, 4713–4724. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.; Eriksson, H.; Gekas, C.; Olofsson, T.; Richter, J.; Gullberg, U. DNA-binding dependent and independent functions of WT1 protein during human hematopoiesis. Exp. Cell Res. 2005, 308, 211–221. [Google Scholar] [CrossRef]
- Rampal, R.; Alkalin, A.; Madzo, J.; Vasanthakumar, A.; Pronier, E.; Patel, J.; Li, Y.; Ahn, J.; Abdel-Wahab, O.; Shih, A.; et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014, 9, 1841–1855. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, M.; Chen, X.; Chen, L.; Xu, Y.; Lv, L.; Wang, P.; Yang, H.; Ma, S.; Lin, H.; et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol. Cell 2015, 57, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Pronier, E.; Bowman, R.L.; Ahn, J.; Glass, J.; Kandoth, C.; Merlinsky, T.R.; Whitfield, J.T.; Durham, B.H.; Gruet, A.; Hanasoge Somasundara, A.V.; et al. Genetic and epigenetic evolution as a contributor to WT1-mutant leukemogenesis. Blood 2018, 132, 1265–1278. [Google Scholar] [CrossRef]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, S.; Erpelinck-Verschueren, C.A.; Goudswaard, C.S.; Löwenberg, B.; Valk, P.J. Mutant Wilms’ tumor 1 (WT1) mRNA with premature termination codons in acute myeloid leukemia (AML) is sensitive to nonsense-mediated RNA decay (NMD). Leukemia 2010, 24, 660–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willasch, A.M.; Gruhn, B.; Coliva, T.; Kalinova, M.; Schneider, G.; Kreyenberg, H.; Steinbach, D.; Weber, G.; Hollink, I.H.; Zwaan, C.M.; et al. Standardization of WT1 mRNA quantitation for minimal residual disease monitoring in childhood AML and implications of WT1 gene mutations: A European multicenter study. Leukemia 2009, 23, 1472–1479. [Google Scholar] [CrossRef] [PubMed]
- Rein, L.A.; Chao, N.J. WT1 vaccination in acute myeloid leukemia: New methods of implementing adoptive immunotherapy. Expert Opin. Investig. Drugs 2014, 23, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Oka, Y.; Tsuboi, A.; Taguchi, T.; Osaki, T.; Kyo, T.; Nakajima, H.; Elisseeva, O.A.; Oji, Y.; Kawakami, M.; Ikegame, K.; et al. Induction of WT1 (Wilms’ tumor gene)-specific cytotoxic T lymphocytes by WT1 peptide vaccine and the resultant cancer regression. Proc. Natl. Acad. Sci. USA 2004, 101, 13885–13890. [Google Scholar] [CrossRef]
- Rezvani, K.; Yong, A.S.; Mielke, S.; Savani, B.N.; Musse, L.; Superata, J.; Jafarpour, B.; Boss, C.; Barrett, A.J. Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood 2008, 111, 236–242. [Google Scholar] [CrossRef] [Green Version]
- Keilholz, U.; Letsch, A.; Busse, A.; Asemissen, A.M.; Bauer, S.; Blau, I.W.; Hofmann, W.K.; Uharek, L.; Thiel, E.; Scheibenbogen, C. A clinical and immunologic phase 2 trial of Wilms tumor gene product 1 (WT1) peptide vaccination in patients with AML and MDS. Blood 2009, 113, 6541–6548. [Google Scholar] [CrossRef] [Green Version]
- Maslak, P.G.; Dao, T.; Krug, L.M.; Chanel, S.; Korontsvit, T.; Zakhaleva, V.; Zhang, R.; Wolchok, J.D.; Yuan, J.; Pinilla-Ibarz, J.; et al. Vaccination with synthetic analog peptides derived from WT1 oncoprotein induces T-cell responses in patients with complete remission from acute myeloid leukemia. Blood 2010, 116, 171–179. [Google Scholar] [CrossRef]
- Tsuboi, A.; Oka, Y.; Kyo, T.; Katayama, Y.; Elisseeva, O.A.; Kawakami, M.; Nishida, S.; Morimoto, S.; Murao, A.; Nakajima, H.; et al. Long-term WT1 peptide vaccination for patients with acute myeloid leukemia with minimal residual disease. Leukemia 2012, 26, 1410–1413. [Google Scholar] [CrossRef] [Green Version]
- Maslak, P.G.; Dao, T.; Bernal, Y.; Chanel, S.M.; Zhang, R.; Frattini, M.; Rosenblat, T.; Jurcic, J.G.; Brentjens, R.J.; Arcila, M.E.; et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv. 2018, 2, 224–234. [Google Scholar] [CrossRef]
- Dao, T.; Yan, S.; Veomett, N.; Pankov, D.; Zhou, L.; Korontsvit, T.; Scott, A.; Whitten, J.; Maslak, P.; Casey, E.; et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci. Transl. Med. 2013, 5, 176ra133. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Ahmed, M.; Tassev, D.V.; Hasan, A.; Kuo, T.Y.; Guo, H.F.; O’Reilly, R.J.; Cheung, N.K. Affinity maturation of T-cell receptor-like antibodies for Wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia 2015, 29, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Oren, R.; Hod-Marco, M.; Haus-Cohen, M.; Thomas, S.; Blat, D.; Duvshani, N.; Denkberg, G.; Elbaz, Y.; Benchetrit, F.; Eshhar, Z.; et al. Functional comparison of engineered T cells carrying a native TCR versus TCR-like antibody-based chimeric antigen receptors indicates affinity/avidity thresholds. J. Immunol. 2014, 193, 5733–5743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dao, T.; Pankov, D.; Scott, A.; Korontsvit, T.; Zakhaleva, V.; Xu, Y.; Xiang, J.; Yan, S.; de Morais Guerreiro, M.D.; Veomett, N.; et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat. Biotechnol. 2015, 33, 1079–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiq, S.; Purdon, T.J.; Daniyan, A.F.; Koneru, M.; Dao, T.; Liu, C.; Scheinberg, D.A.; Brentjens, R.J. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia 2017, 31, 1788–1797. [Google Scholar] [CrossRef] [Green Version]
- Augsberger, C.; Hänel, G.; Xu, W.; Pulko, V.; Hanisch, L.J.; Augustin, A.; Challier, J.; Hunt, K.; Vick, B.; Rovatti, P.E.; et al. Targeting intracellular WT1 in AML with a novel RMF-peptide-MHC-specific T-cell bispecific antibody. Blood 2021, 138, 2655–2669. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P# | FAB | Variant 1 | Variant 2 | Loc. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| cDNA 1 | AA 2 | VF% | Exon | cDNA | AA | VF% | Exon | |||
| 1 | M1 | c.1057_1058insTA | p.Arg353LeufsTer6 | 45.8 | 7 | c.1123dupA | p.Met375AsnfsTer9 | 44.5 | 7 | biallelic |
| 2 | M4 | c.1121delT | p.Phe374SerfsTer58 | 91.84 | 7 | |||||
| 3 | M1 | c.1133dupA | p.Tyr378fsTer1 | 46.56 | 7 | |||||
| 4 | M4 | c.1059dupT | p.Val354CysfsTer14 | 46.94 | 7 | c.1082_1091dup TTGTACGGTC | p.Ala365CysfsTer6 | 40.65 | 7 | biallelic |
| 5 | M1 | c.1054_1055insT | p.Arg352LeufsTer16 | 67.2 | 7 | |||||
| 6 | M0 | c.1058_1059insGGTGCCGCTCG | p.Gly356LeufsTer6 | 48.49 | 7 | c.1082_1091dup TTGTACGGTC | p.Ala365CysfsTer6 | 41.83 | 7 | biallelic |
| 7 | M1 | c.1078_1079insGCCGA | p.Thr360SerfsTer74 | 38.73 | 7 | c.1084_1085insGC | p.Val362Glyfs* 71 | 52.92 | 7 | biallelic |
| 8 | M1/M2 | c.1087delCinsGGG | p.Arg363GlyfsTer70 | 52.11 | 7 | c.1057delCinsGG | p.Arg353GlyfsTer15 | 42.78 | 7 | biallelic |
| 9 | M1 | c.1322_1332dupGAAAGTTCTCC | p.Arg445GlufsTer9 | 40.5 | 9 | c.1323_1338dup AAAGTTCTCCCGGTCC | p.Asp447LysfsTer18 | 40.1 | 9 | biallelic |
| 10 | M5 | c.1068_1076del9ins GACGGTCGTTATTA | p.Val357ThrfsTer77 | 42.14 | 7 | |||||
| 11 | M2/M4 | c.1325dupA | p.Phe443ValfsTer17 | 13.87 | 9 | |||||
| 12 | M4 | c.1048-4_1056dup GCAGGATGTGCGA | p.Arg353AlafsTer19 | 30.25 | 7 | |||||
| 13 | M2 | c.1090_1091dupTC | p.Ala365ArgfsTer68 | 44.25 | 7 | |||||
| 14 | M5 | c.1079_1089dupCTCTTGTACGG | p.Ser364LeufsTer72 | 39.58 | 7 | |||||
| 15 | M4 | c.1080_1087dupTCTTGTAC | p.Arg363LeufsTer72 | 34.57 | 7 | |||||
| 16 | M0 | c.1077_1078ins TGTTTCTTCCGCCCAG | p.Thr360CysfsTer13 | 36.95 | 7 | |||||
| 17 | M1 | c.1086_1087insGAACTCTTGTA | p.Arg363GlufsTer73 | 30.15 | 7 | |||||
| 18 | M4 | c.1090_1091insAGGT | p.Ser364fsTer1 | 42.97 | 7 | |||||
| 19 | M4eo | c.1056_1058delACGinsC | p.Arg353CysfsTer14 | 26.44 | 7 | c.1089dupG | p.Ser364ValfsTer4 | 9.59 | 7 | biallelic |
| 20 | M2 | c.1079_1090del CTCTTGTACGGTinsTGGG | p.Thr360MetfsTer5 | 55.23 | 7 | |||||
| 21 | M4 | c.1074_1077dupCCCG | p.Thr360ProfsTer9 | 9.9 | 7 | |||||
| 22 | M1/M2 | c.1090_1093dupTCGG | p.Ala365ValfsTer4 | 6.94 | 7 | c.1091dupC | p.Ala365GlyfsTer3 | 97.42 | 7 | biallelic |
| 23 | M0 | c.1087delCinsGG | p.Arg363GlyfsTer5 | 41.88 | 7 | |||||
| 24 | M1/M2 | c.1058delGinsCC | p.Arg353ProfsTer15 | 44.78 | 7 | |||||
| Location | Variant (DNA) 1 | Variant (AA) 2 | Zygosity | Cell Line | Previous Reported Variant? | Variant Status in Analyzed Cell Lines | ||
|---|---|---|---|---|---|---|---|---|
| Variant Ref. | Relevance | |||||||
| Exonic Variants | exon1 | c.330C > T | p.Pro110Pro | Heterozygous | CMK, FKH-1, HL-60, HAT-93, KASUMI-6, KG-1, ME-1, ML-2, MOLM-1, NOMO-1, SKNO-1, TF-1, THP-1, YNH-1 | LOVD 3 (WT1_000137) | Benign | Current data set |
| c.198G > T | p.Pro66Pro | Heterozygous | HL-60, HT-93, K-562, MOLM-1, M-07e, NB-4, SKNO-1, UOC-M1, U-937 | dbSNP/ClinVar 4 (rs2234582) | Benign | Current data set | ||
| c.594C > T | p.Asn98Asn | Heterozygous | HT-93, M-07e, NB-4, SKNO-1, UOC-M1 | dbSNP/ClinVar (rs2234583) | Benign | Current data set | ||
| c.294C > A | p.Gly98Gly | Heterozygous | KG-1 | LOVD (WT1_000161) | Benign | Current data set | ||
| exon 7 | c.1202_1203delGA | p.Arg401fsTer3 | Heterozygous | CTS | Not reported | Pathogenic | PMID 18591546 | |
| KASUMI-6 | CCLE 5 (ACH-000166) | |||||||
| c.1107A > G | p.Arg369Arg | Heterozygous | CMK, HL-60, KG-1, ME-1, MOLM-1, NOMO-1, THP-1 | dbSNP/ClinVar (rs16754) | Benign | Current data set | ||
| Homozygous | HT-93, KASUMI-6, ML-2, SKNO-1, TF-1, YNH-1 | |||||||
| c.1105C > T | p.Arg369Ter | Heterozygous | U-937 | dbSNP/ClinVar (rs1423753702) | Pathogenic | Cellosaurus (CVCL_0007) | ||
| exon 9 | c.1385G > A | p.Arg462Gln | Heterozygous | MOLM-1 | LOVD (WT1_000144) | Likely pathogenic | CCLE (ACH-001573) | |
| exon 10 | c.1484G > A | p.Arg495Gln | Heterozygous | ME-1 | Not reported | VUS | CCLE (ACH-000439) | |
| Intronic variants | Upstream to 5′UTR | c.-883C > T | Intronic variant | Homozygous | All cell lines but CTS | Not reported | VUS | Current data set |
| upstream to 5′UTR | c.-784T > C | Intronic variant | Homozygous | All cell lines but CTS | Not reported | VUS | Current data set | |
| upstream to 5′UTR | c.-654G > C | Intronic variant | Heterozygous | K-562 | Not reported | VUS | Current data set | |
| 5′UTR | c.-247T > C | Intronic variant | Homozygous | all cell lines but CTS | COSV60070502 | VUS | Current data set | |
| intron 3 | c.872 + 16G > A | Intronic variant | Heterozygous | HL-60, K-562, MOLM-1, M-07e, NB-4, UOC-M1 | LOVD (WT1_000123) COSV60066980 | Likely benign | Current data set | |
| intron 6 | c.1099-9T > C | Intronic variant | Heterozygous | HL-60 | LOVD (WT1_000117) COSV60072524 | Likely benign | Current data set | |
| 3′UTR | c.1554 + 88A > G | Intronic variant | Homozygous | UOC-M1 | Not reported | VUS | Current data set | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niktoreh, N.; Weber, L.; Walter, C.; Karimifard, M.; Hoffmeister, L.M.; Breiter, H.; Thivakaran, A.; Soldierer, M.; Drexler, H.G.; Schaal, H.; et al. Understanding WT1 Alterations and Expression Profiles in Hematological Malignancies. Cancers 2023, 15, 3491. https://doi.org/10.3390/cancers15133491
Niktoreh N, Weber L, Walter C, Karimifard M, Hoffmeister LM, Breiter H, Thivakaran A, Soldierer M, Drexler HG, Schaal H, et al. Understanding WT1 Alterations and Expression Profiles in Hematological Malignancies. Cancers. 2023; 15(13):3491. https://doi.org/10.3390/cancers15133491
Chicago/Turabian StyleNiktoreh, Naghmeh, Lisa Weber, Christiane Walter, Mahshad Karimifard, Lina Marie Hoffmeister, Hannah Breiter, Aniththa Thivakaran, Maren Soldierer, Hans Günther Drexler, Heiner Schaal, and et al. 2023. "Understanding WT1 Alterations and Expression Profiles in Hematological Malignancies" Cancers 15, no. 13: 3491. https://doi.org/10.3390/cancers15133491
APA StyleNiktoreh, N., Weber, L., Walter, C., Karimifard, M., Hoffmeister, L. M., Breiter, H., Thivakaran, A., Soldierer, M., Drexler, H. G., Schaal, H., Sendker, S., Reinhardt, D., Schneider, M., & Hanenberg, H. (2023). Understanding WT1 Alterations and Expression Profiles in Hematological Malignancies. Cancers, 15(13), 3491. https://doi.org/10.3390/cancers15133491