

Lack of Influence of Non-Overlapping Mutations in BRAF, NRAS, or NF1 on 12-Month Best Objective Response and Long-Term Survival after Checkpoint Inhibitor-Based Treatment for Metastatic Melanoma

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects and Design
2.2. Study Methods
2.3. Treatment Regimens
2.4. Response Assessment
2.5. Statistical Analysis
3. Results
3.1. Patient Characteristics
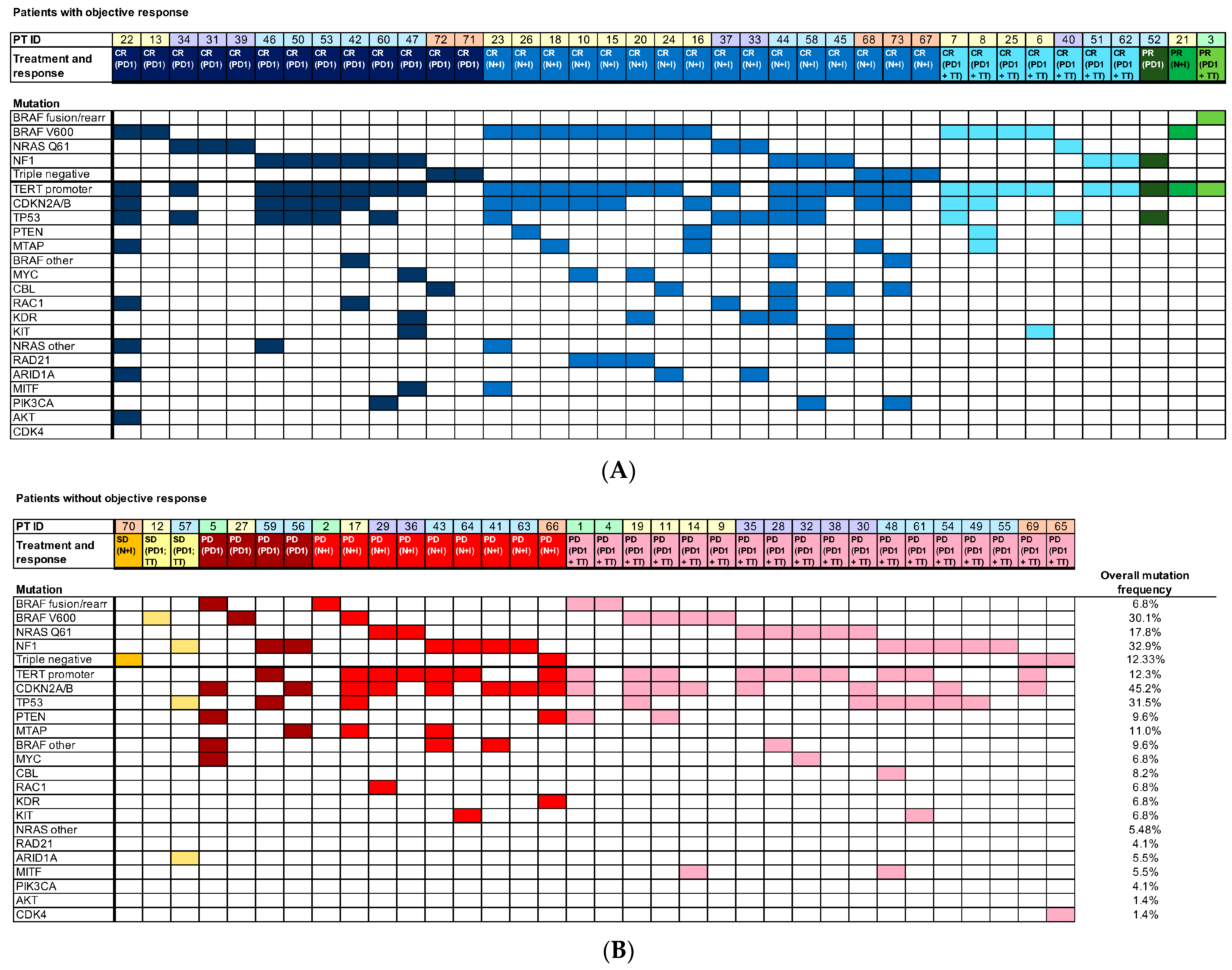
3.2. Mutation Frequency
3.3. Analysis of Treatment Outcome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J Clin 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Curti, B.D.; Faries, M.B. Recent Advances in the Treatment of Melanoma. N. Engl. J. Med. 2021, 384, 2229–2240. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Korman, A.J.; Peggs, K.S.; Allison, J.P. Checkpoint blockade in cancer immunotherapy. Adv. Immunol. 2006, 90, 297–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliot, T.A.E.; Jennings, E.K.; Lecky, D.A.J.; Thawait, N.; Flores-Langarica, A.; Copland, A.; Maslowski, K.M.; Wraith, D.C.; Bending, D. Antigen and checkpoint receptor engagement recalibrates T cell receptor signal strength. Immunity 2021, 54, 2481–2496.e6. [Google Scholar] [CrossRef]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef] [Green Version]
- Korn, E.L.; Liu, P.Y.; Lee, S.J.; Chapman, J.A.; Niedzwiecki, D.; Suman, V.J.; Moon, J.; Sondak, V.K.; Atkins, M.B.; Eisenhauer, E.A.; et al. Meta-analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future phase II trials. J. Clin. Oncol. 2008, 26, 527–534. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes With Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients With Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef]
- Consortium, I.T.P.-C.A.o.W.G. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Amann, V.C.; Ramelyte, E.; Thurneysen, S.; Pitocco, R.; Bentele-Jaberg, N.; Goldinger, S.M.; Dummer, R.; Mangana, J. Developments in targeted therapy in melanoma. Eur. J. Surg. Oncol. 2017, 43, 581–593. [Google Scholar] [CrossRef]
- Shaughnessy, M.; Klebanov, N.; Tsao, H. Clinical and therapeutic implications of melanoma genomics. J. Trans. Genet. Genom. 2018, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas, N. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.Y.; Miller, D.M.; Tsao, H. Somatic driver mutations in melanoma. Cancer 2017, 123, 2104–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franken, M.G.; Leeneman, B.; Gheorghe, M.; Uyl-de Groot, C.A.; Haanen, J.; van Baal, P.H.M. A systematic literature review and network meta-analysis of effectiveness and safety outcomes in advanced melanoma. Eur. J. Cancer 2019, 123, 58–71. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Flaherty, K.T.; Robert, C.; Arance, A.; de Groot, J.W.B.; Garbe, C.; Gogas, H.J.; Gutzmer, R.; Krajsova, I.; Liszkay, G.; et al. COLUMBUS 5-Year Update: A Randomized, Open-Label, Phase III Trial of Encorafenib Plus Binimetinib Versus Vemurafenib or Encorafenib in Patients With BRAF V600-Mutant Melanoma. J. Clin. Oncol. 2022, 40, 4178–4188. [Google Scholar] [CrossRef] [PubMed]
- Sarkisian, S.; Davar, D. MEK inhibitors for the treatment of NRAS mutant melanoma. Drug Des. Devel. Ther. 2018, 12, 2553–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.B.; Puzanov, I. Treatment of NRAS-mutant melanoma. Curr. Treat. Options Oncol. 2015, 16, 15. [Google Scholar] [CrossRef]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Mason, R.; Dearden, H.C.; Nguyen, B.; Soon, J.A.; Smith, J.L.; Randhawa, M.; Mant, A.; Warburton, L.; Lo, S.; Meniawy, T.; et al. Combined ipilimumab and nivolumab first-line and after BRAF-targeted therapy in advanced melanoma. Pigment Cell Melanoma Res. 2020, 33, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Ribas, A.; Robert, C.; Schachter, J.; Nyakas, M.; Daud, A.; Arance, A.; Carlino, M.S.; O’Day, S.J.; Long, G.V.; et al. Association of BRAF V600E/K Mutation Status and Prior BRAF/MEK Inhibition With Pembrolizumab Outcomes in Advanced Melanoma: Pooled Analysis of 3 Clinical Trials. JAMA Oncol. 2020, 6, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Lee, S.J.; Chmielowski, B.; Ribas, A.; Tarhini, A.A.; Truong, T.-G.; Davar, D.; O’Rourke, M.A.; Curti, B.D.; Brell, J.M.; et al. DREAMseq (Doublet, Randomized Evaluation in Advanced Melanoma Sequencing): A phase III trial—ECOG-ACRIN EA6134. J. Clin. Oncol. 2021, 39, 356154. [Google Scholar] [CrossRef]
- Adler, N.R.; Wolfe, R.; Kelly, J.W.; Haydon, A.; McArthur, G.A.; McLean, C.A.; Mar, V.J. Tumour mutation status and sites of metastasis in patients with cutaneous melanoma. Br. J. Cancer 2017, 117, 1026–1035. [Google Scholar] [CrossRef]
- Barbour, A.P.; Tang, Y.H.; Armour, N.; Dutton-Regester, K.; Krause, L.; Loffler, K.A.; Lambie, D.; Burmeister, B.; Thomas, J.; Smithers, B.M.; et al. BRAF mutation status is an independent prognostic factor for resected stage IIIB and IIIC melanoma: Implications for melanoma staging and adjuvant therapy. Eur. J. Cancer 2014, 50, 2668–2676. [Google Scholar] [CrossRef]
- Heppt, M.V.; Siepmann, T.; Engel, J.; Schubert-Fritschle, G.; Eckel, R.; Mirlach, L.; Kirchner, T.; Jung, A.; Gesierich, A.; Ruzicka, T.; et al. Prognostic significance of BRAF and NRAS mutations in melanoma: A German study from routine care. BMC Cancer 2017, 17, 536. [Google Scholar] [CrossRef] [Green Version]
- Lebbe, C.; Meyer, N.; Mortier, L.; Marquez-Rodas, I.; Robert, C.; Rutkowski, P.; Menzies, A.M.; Eigentler, T.; Ascierto, P.A.; Smylie, M.; et al. Evaluation of Two Dosing Regimens for Nivolumab in Combination With Ipilimumab in Patients With Advanced Melanoma: Results From the Phase IIIb/IV CheckMate 511 Trial. J. Clin. Oncol. 2019, 37, 867–875. [Google Scholar] [CrossRef]
- Samlowski, W.; Adajar, C. Cautious addition of targeted therapy to PD-1 inhibitors after initial progression of BRAF mutant metastatic melanoma on checkpoint inhibitor therapy. BMC Cancer 2021, 21, 1187–1199. [Google Scholar] [CrossRef]
- Hilts, A.; Samlowski, W. Cautious Addition of MEK Inhibitors to PD-1 Antibody Treatment in Patients with NRAS or NF1 Mutant Metastatic Melanoma Failing Initial Immunotherapy. Ann. Case Rep. 2022, 7, 795–805. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Kaplan, E.L.; Meier, P. Nonparametric Estimation from Incomplete Observations. J. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- Bland, J.M.; Altman, D.G. The logrank test. BMJ 2004, 328, 1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Symanowski, J.; Paul, S.; Qu, Y.; Zagar, A.; Hong, S. The type I error and power of non-parametric logrank and Wilcoxon tests with adjustment for covariates-a simulation study. Stat. Med. 2008, 27, 5850–5860. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.; Samlowski, W.; Lopez-Flores, R. Outcome of Elective Checkpoint Inhibitor Discontinuation in Patients with Metastatic Melanoma Who Achieved a Complete Remission: Real-World Data. Biomedicines 2022, 10, 1144. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Wu, X.; Yu, J.; Yu, H.; Xu, T.; Brown, K.M.; Bai, X.; Dai, J.; Ma, M.; Tang, H.; et al. Analysis of NRAS gain in 657 patients with melanoma and evaluation of its sensitivity to a MEK inhibitor. Eur. J. Cancer 2018, 89, 90–101. [Google Scholar] [CrossRef]
- Adler, N.R.; Wolfe, R.; McArthur, G.A.; Kelly, J.W.; Haydon, A.; McLean, C.A.; Mar, V.J. Tumour mutation status and melanoma recurrence following a negative sentinel lymph node biopsy. Br. J. Cancer 2018, 118, 1289–1295. [Google Scholar] [CrossRef] [Green Version]
- van Not, O.J.; Blokx, W.A.M.; van den Eertwegh, A.J.M.; de Meza, M.M.; Haanen, J.B.; Blank, C.U.; Aarts, M.J.B.; van den Berkmortel, F.W.P.J.; de Groot, J.W.B.; Hospers, G.A.P.; et al. BRAF and NRAS Mutation Status and Response to Checkpoint Inhibition in Advanced Melanoma. JCO Precis. Oncol. 2022, 6, e2200018. [Google Scholar] [CrossRef]
- Möller, T.; Schulze, H.J. The Influence of c-Kit and NRAS Mutation on Patients’ Survival in Metastatic Melanoma Receiving Immune Checkpoint Inhibitors and Chemotherapy. Dermato 2022, 2, 6. [Google Scholar] [CrossRef]
- Ramon-Patino, J.L.; Schmid, S.; Lau, S.; Seymour, L.; Gaudreau, P.O.; Li, J.J.N.; Bradbury, P.A.; Calvo, E. iRECIST and atypical patterns of response to immuno-oncology drugs. J. Immunother. Cancer 2022, 10, e004849. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Overall Survival in Days (OS) | Progression-Free Survival in Days (PFS) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Driver Mutation | N | Mean | SD | Median | Min | Max | p | Mean | SD | Median | Min | Max | p |
| All Patients | 73 | 818.9 | 487.38 | 720 | 61 | 1950 | -- | 633.1 | 521.31 | 524 | 43 | 1950 | -- |
| BRAF fusion/rearr. | 5 | 744 | 584.6 | 606 | 259 | 1749 | 0.724 | 316.2 | 237.14 | 182 | 125 | 625 | 0.161 |
| BRAF V600 | 22 | 841 | 414 | 776 | 61 | 1642 | 0.801 | 726.5 | 483.97 | 731.5 | 46 | 1642 | 0.318 |
| NF1 | 13 | 820.15 | 309.9 | 778 | 258 | 1386 | 0.992 | 611.15 | 439 | 620 | 87 | 1386 | 0.869 |
| NRAS | 24 | 831.25 | 606.37 | 704 | 82 | 1772 | 0.895 | 624.42 | 596.76 | 464 | 43 | 1772 | 0.922 |
| “Triple negative” | 9 | 771.78 | 555.03 | 722 | 69 | 1950 | 0.759 | 635.33 | 634.25 | 665 | 54 | 1950 | 0.989 |
| CDKN2AB | 33 | 843.7 | 454.57 | 732 | 61 | 1749 | 0.696 | 636.53 | 485.63 | 524 | 46 | 1570 | 0.951 |
| TERT promoter | 47 | 862.6 | 491.49 | 732 | 61 | 1772 | 0.306 | 684.28 | 520.02 | 589 | 43 | 1772 | 0.262 |
| PTEN | 7 | 812.43 | 500.99 | 650 | 360 | 1749 | 0.971 | 504.71 | 353.97 | 477 | 182 | 1170 | 0.497 |
| TP53 | 23 | 851.17 | 521.43 | 778 | 61 | 1570 | 0.704 | 683.17 | 545.98 | 524 | 43 | 1570 | 0.581 |
| MTAP | 8 | 625.5 | 426.69 | 647 | 61 | 1245 | 0.237 | 536.75 | 451.48 | 533 | 61 | 1170 | 0.583 |
| BRAF passenger | 7 | 728 | 507.38 | 675 | 83 | 1455 | 0.607 | 655.14 | 531.6 | 625 | 61 | 1455 | 0.907 |
| MYC | 5 | 925 | 466.91 | 675 | 563 | 1606 | 0.615 | 762 | 545.1 | 625 | 128 | 1606 | 0.570 |
| CBL | 6 | 986.5 | 642.02 | 981 | 82 | 1950 | 0.383 | 982.17 | 649.39 | 981 | 56 | 1950 | 0.087 |
| RAC1 | 5 | 1132.4 | 247.69 | 1112 | 778 | 1455 | 0.137 | 1128.4 | 254.9 | 1112 | 758 | 1455 | 0.027 |
| KDR | 5 | 1031.4 | 540.93 | 1242 | 360 | 1606 | 0.316 | 1004.4 | 584.44 | 1242 | 225 | 1606 | 0.099 |
| KIT | 5 | 999.2 | 650.88 | 909 | 102 | 1659 | 0.395 | 671.6 | 647.48 | 720 | 43 | 1606 | 0.865 |
| NRAS passenger | 6 | 856 | 467.73 | 916 | 161 | 1353 | 0.847 | 845.33 | 487.08 | 916 | 97 | 1353 | 0.301 |
| RAD21 | 3 | 864.67 | 330.54 | 813 | 563 | 1218 | 0.869 | 754.67 | 170.17 | 813 | 563 | 888 | 0.683 |
| ARID1A | 4 | 1115.25 | 296.77 | 1186 | 703 | 1386 | 0.213 | 1115.25 | 296.77 | 1186 | 703 | 1386 | 0.056 |
| MITF | 4 | 923.5 | 664.82 | 1003 | 82 | 1606 | 0.662 | 796.75 | 756.08 | 762.5 | 56 | 1606 | 0.522 |
| Overall Survival in Days (OS) | Progression-Free Survival in Days (PFS) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Response Type | N | Mean | SD | Median | Min | Max | Mean | SD | Median | Min | Max |
| Complete response | 36 | 1062.6 | 411.3 | 1095.5 | 447 | 1950 | 1002.2 | 443.9 | 898.5 | 93 | 1950 |
| Partial response | 3 | 517.6 | 204.3 | 606 | 284 | 663 | 356.6 | 141.4 | 298 | 254 | 518 |
| Stable disease | 3 | 566.3 | 445.0 | 703 | 69 | 927 | 537.3 | 420.7 | 703 | 59 | 850 |
| Progressive disease | 31 | 589.4 | 464.2 | 489 | 61 | 1749 | 240.3 | 278.8 | 131 | 43 | 1218 |
| Overall Survival in Days (OS) | Progression-Free Survival in Days (PFS) | ||||||
|---|---|---|---|---|---|---|---|
| BORR | Comparator | Mean Difference | Std. Error | p | Mean Difference | Std. Error | p |
| CR | PR | 545 | 259.5 | 0.236 | 645.556 | 224.427 | 0.032 |
| SD | 496.333 | 259.5 | 0.36 | 464.889 | 224.427 | 0.252 | |
| PD | 473.247 | 105.809 | <0.001 | 761.867 | 105.809 | <0.001 | |
| PR | SD | −48.667 | 352.591 | 1.000 | −180.667 | 352.591 | 1.000 |
| PD | −71.753 | 261.105 | 1.000 | 116.312 | 261.105 | 1.000 | |
| SD | PD | −23.086 | 261.105 | 1.000 | −296.978 | 225.815 | 1.000 |
 Statistically significant (p < 0.0083), All genetic mutations with a p value approaching statistical significance were included in the multivariate ANOVA analysis, but none reached significance.
Statistically significant (p < 0.0083), All genetic mutations with a p value approaching statistical significance were included in the multivariate ANOVA analysis, but none reached significance.Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panning, A.; Samlowski, W.; Allred, G. Lack of Influence of Non-Overlapping Mutations in BRAF, NRAS, or NF1 on 12-Month Best Objective Response and Long-Term Survival after Checkpoint Inhibitor-Based Treatment for Metastatic Melanoma. Cancers 2023, 15, 3527. https://doi.org/10.3390/cancers15133527
Panning A, Samlowski W, Allred G. Lack of Influence of Non-Overlapping Mutations in BRAF, NRAS, or NF1 on 12-Month Best Objective Response and Long-Term Survival after Checkpoint Inhibitor-Based Treatment for Metastatic Melanoma. Cancers. 2023; 15(13):3527. https://doi.org/10.3390/cancers15133527
Chicago/Turabian StylePanning, Alyssa, Wolfram Samlowski, and Gabriel Allred. 2023. "Lack of Influence of Non-Overlapping Mutations in BRAF, NRAS, or NF1 on 12-Month Best Objective Response and Long-Term Survival after Checkpoint Inhibitor-Based Treatment for Metastatic Melanoma" Cancers 15, no. 13: 3527. https://doi.org/10.3390/cancers15133527
APA StylePanning, A., Samlowski, W., & Allred, G. (2023). Lack of Influence of Non-Overlapping Mutations in BRAF, NRAS, or NF1 on 12-Month Best Objective Response and Long-Term Survival after Checkpoint Inhibitor-Based Treatment for Metastatic Melanoma. Cancers, 15(13), 3527. https://doi.org/10.3390/cancers15133527