
NAFLD-Related HCC: Focus on the Latest Relevant Preclinical Models


Abstract
:Simple Summary
Abstract
1. Introduction
2. Mouse Models of Non-Alcoholic Fatty Liver Disease
2.1. Murine Models of NAFL
2.1.1. High-Fat Diet (HFD)
2.1.2. Lepob/Lepob (ob/ob) Mice
2.1.3. Leptin-Receptor-Deficient (db/db) Mice
2.2. Murine Models of NASH
2.2.1. Methionine- and Choline-Deficient Diet (MCD)
2.2.2. Choline-Deficient L-Amino Acid-Defined (CDAA) Diet
2.2.3. High-Fructose Diet
2.2.4. The American-Lifestyle-Induced Obesity Syndrome (ALIOS) Diet
2.3. Murine NASH-HCC Models
2.3.1. Choline-Deficient L-Amino Acid-Defined, High-Fat Diet (CDA-HFD)
2.3.2. Choline-Deficient, High-Fat Diet (CD-HFD)
2.3.3. Western Diet (WD) + Carbon Tetrachloride (CCl4)
2.3.4. MUP-uPA + HFD
2.3.5. DIAMOND Mice
3. Ex Vivo Models of Non-Alcoholic Fatty Liver Disease
3.1. Spheroids
3.2. Organoids
3.3. Liver-on-a-Chip
3.4. Precision Cut Liver Slices
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current Concepts and Future Challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to Diabetes Mellitus, Cardiovascular Disease or Cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Diehl, A.M.; Farpour-Lambert, N.J.; Zhao, L.; Tilg, H. Why We Need to Curb the Emerging Worldwide Epidemic of Nonalcoholic Fatty Liver Disease. Nat. Metab. 2019, 1, 1027–1029. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; George, J. Genetic Contributions to NAFLD: Leveraging Shared Genetics to Uncover Systems Biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD Disease Burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the Period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The Global Epidemiology of Nonalcoholic Fatty Liver Disease (NAFLD) and Nonalcoholic Steatohepatitis (NASH): A Systematic Review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E. Challenges in the Hepatic Histopathology in Non-Alcoholic Fatty Liver Disease. Gut 2017, 66, 1539–1540. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E.; Carpenter, D.H.; Rinella, M.; Harrison, S.A.; Loomba, R.; Younossi, Z.; Neuschwander-Tetri, B.A.; Sanyal, A.J. American Association for the Study of Liver Diseases NASH Task Force NAFLD: Reporting Histologic Findings in Clinical Practice. Hepatology 2021, 73, 2028–2038. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and Validation of a Histological Scoring System for Nonalcoholic Fatty Liver Disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis Progression in Nonalcoholic Fatty Liver vs Nonalcoholic Steatohepatitis: A Systematic Review and Meta-Analysis of Paired-Biopsy Studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e1–9, quiz e39–40. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.Q.; Singal, A.G.; Kono, Y.; Tan, D.J.H.; El-Serag, H.B.; Loomba, R. Changing Global Epidemiology of Liver Cancer from 2010 to 2019: NASH Is the Fastest Growing Cause of Liver Cancer. Cell Metab. 2022, 34, 969–977.e2. [Google Scholar] [CrossRef] [PubMed]
- Fingas, C.D.; Best, J.; Sowa, J.-P.; Canbay, A. Epidemiology of Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. Clin. Liver Dis. 2016, 8, 119–122. [Google Scholar] [CrossRef]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global Epidemiology of NAFLD-Related HCC: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef]
- Piscaglia, F.; Svegliati-Baroni, G.; Barchetti, A.; Pecorelli, A.; Marinelli, S.; Tiribelli, C.; Bellentani, S. HCC-NAFLD Italian Study Group Clinical Patterns of Hepatocellular Carcinoma in Nonalcoholic Fatty Liver Disease: A Multicenter Prospective Study. Hepatology 2016, 63, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans Is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, V.; Zalinski, S.; Chelbi, E.; Guedj, N.; Degos, F.; Vilgrain, V.; Bedossa, P.; Belghiti, J. Hepatocellular Carcinomas in Patients with Metabolic Syndrome Often Develop without Significant Liver Fibrosis: A Pathological Analysis. Hepatology 2009, 49, 851–859. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: An Emerging Menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, J.; Riaz, D.R.; Shi, G.; Liu, C.; Dai, Y. Outcomes of Liver Transplantation for Nonalcoholic Steatohepatitis: A Systematic Review and Meta-Analysis. Clin. Gastroenterol. Hepatol. 2014, 12, 394–402.e1. [Google Scholar] [CrossRef]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic Steatohepatitis Is the Second Leading Etiology of Liver Disease among Adults Awaiting Liver Transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Haldar, D.; Kern, B.; Hodson, J.; Armstrong, M.J.; Adam, R.; Berlakovich, G.; Fritz, J.; Feurstein, B.; Popp, W.; Karam, V.; et al. Outcomes of Liver Transplantation for Non-Alcoholic Steatohepatitis: A European Liver Transplant Registry Study. J. Hepatol. 2019, 71, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuppalanchi, R.; Noureddin, M.; Alkhouri, N.; Sanyal, A.J. Therapeutic Pipeline in Nonalcoholic Steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 373–392. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A. Update on Nonalcoholic Fatty Liver Disease: Genes Involved in Nonalcoholic Fatty Liver Disease and Associated Inflammation. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 391–396. [Google Scholar] [CrossRef]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Valenti, L.V.C.; Baselli, G.A. Genetics of Nonalcoholic Fatty Liver Disease: A 2018 Update. Curr. Pharm. Des. 2018, 24, 4566–4573. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of Fatty Acids Stored in Liver and Secreted via Lipoproteins in Patients with Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular Mechanisms of Lipotoxicity and Glucotoxicity in Nonalcoholic Fatty Liver Disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Begriche, K.; Massart, J.; Robin, M.-A.; Bonnet, F.; Fromenty, B. Mitochondrial Adaptations and Dysfunctions in Nonalcoholic Fatty Liver Disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Kim, J.Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R.J.; Saltiel, A.R.; Karin, M. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018, 175, 133–145.e15. [Google Scholar] [CrossRef] [Green Version]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic Reticulum Stress Signalling and the Pathogenesis of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Luedde, T. Apoptosis and Necroptosis in the Liver: A Matter of Life and Death. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 738–752. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular Senescence Drives Age-Dependent Hepatic Steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef] [Green Version]
- Meijnikman, A.S.; Herrema, H.; Scheithauer, T.P.M.; Kroon, J.; Nieuwdorp, M.; Groen, A.K. Evaluating Causality of Cellular Senescence in Non-Alcoholic Fatty Liver Disease. JHEP Rep. 2021, 3, 100301. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic Activation of Intrahepatic CD8+ T Cells and NKT Cells Causes Nonalcoholic Steatohepatitis and Liver Cancer via Cross-Talk with Hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef] [Green Version]
- Pinter, M.; Pinato, D.J.; Ramadori, P.; Heikenwalder, M. NASH and Hepatocellular Carcinoma: Immunology and Immunotherapy. Clin. Cancer Res. 2023, 29, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Peiseler, M.; Schwabe, R.; Hampe, J.; Kubes, P.; Heikenwälder, M.; Tacke, F. Immune Mechanisms Linking Metabolic Injury to Inflammation and Fibrosis in Fatty Liver Disease—Novel Insights into Cellular Communication Circuits. J. Hepatol. 2022, 77, 1136–1160. [Google Scholar] [CrossRef]
- Barreby, E.; Chen, P.; Aouadi, M. Macrophage Functional Diversity in NAFLD—More than Inflammation. Nat. Rev. Endocrinol. 2022, 18, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH Limits Anti-Tumour Surveillance in Immunotherapy-Treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Febbraio, M.A.; Reibe, S.; Shalapour, S.; Ooi, G.J.; Watt, M.J.; Karin, M. Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metab. 2019, 29, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Oligschlaeger, Y.; Shiri-Sverdlov, R. NAFLD Preclinical Models: More than a Handful, Less of a Concern? Biomedicines 2020, 8, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical Models of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Gallage, S.; Avila, J.E.B.; Ramadori, P.; Focaccia, E.; Rahbari, M.; Ali, A.; Malek, N.P.; Anstee, Q.M.; Heikenwalder, M. A Researcher’s Guide to Preclinical Mouse NASH Models. Nat. Metab. 2022, 4, 1632–1649. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Suzuki, J.; Tsujioka, S.; Sasaki, M.; Gomori, A.; Shirakura, T.; Hirose, H.; Ito, M.; Ishihara, A.; Iwaasa, H.; et al. Longitudinal Analysis of Murine Steatohepatitis Model Induced by Chronic Exposure to High-Fat Diet. Hepatol. Res. 2007, 37, 50–57. [Google Scholar] [CrossRef]
- Hariri, N.; Thibault, L. High-Fat Diet-Induced Obesity in Animal Models. Nutr. Res. Rev. 2010, 23, 270–299. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.; Chandrasekera, P.C.; Barnard, N.D. You Are What You Eat, or Are You? The Challenges of Translating High-Fat-Fed Rodents to Human Obesity and Diabetes. Nutr. Diabetes 2014, 4, e135. [Google Scholar] [CrossRef] [Green Version]
- Winzell, M.S.; Ahrén, B. The High-Fat Diet-Fed Mouse: A Model for Studying Mechanisms and Treatment of Impaired Glucose Tolerance and Type 2 Diabetes. Diabetes 2004, 53 (Suppl. S3), S215–S219. [Google Scholar] [CrossRef] [Green Version]
- Eccleston, H.B.; Andringa, K.K.; Betancourt, A.M.; King, A.L.; Mantena, S.K.; Swain, T.M.; Tinsley, H.N.; Nolte, R.N.; Nagy, T.R.; Abrams, G.A.; et al. Chronic Exposure to a High-Fat Diet Induces Hepatic Steatosis, Impairs Nitric Oxide Bioavailability, and Modifies the Mitochondrial Proteome in Mice. Antioxid. Redox Signal. 2011, 15, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Flessa, C.-M.; Nasiri-Ansari, N.; Kyrou, I.; Leca, B.M.; Lianou, M.; Chatzigeorgiou, A.; Kaltsas, G.; Kassi, E.; Randeva, H.S. Genetic and Diet-Induced Animal Models for Non-Alcoholic Fatty Liver Disease (NAFLD) Research. Int. J. Mol. Sci. 2022, 23, 15791. [Google Scholar] [CrossRef]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER Stress Cooperates with Hypernutrition to Trigger TNF-Dependent Spontaneous HCC Development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Velázquez, K.T.; Enos, R.T.; Bader, J.E.; Sougiannis, A.T.; Carson, M.S.; Chatzistamou, I.; Carson, J.A.; Nagarkatti, P.S.; Nagarkatti, M.; Murphy, E.A. Prolonged High-Fat-Diet Feeding Promotes Non-Alcoholic Fatty Liver Disease and Alters Gut Microbiota in Mice. World J. Hepatol. 2019, 11, 619–637. [Google Scholar] [CrossRef]
- Moon, H.-S.; Dalamaga, M.; Kim, S.-Y.; Polyzos, S.A.; Hamnvik, O.-P.; Magkos, F.; Paruthi, J.; Mantzoros, C.S. Leptin’s Role in Lipodystrophic and Nonlipodystrophic Insulin-Resistant and Diabetic Individuals. Endocr. Rev. 2013, 34, 377–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Chandrasekera, P.C.; Pippin, J.J. Leptin- and Leptin Receptor-Deficient Rodent Models: Relevance for Human Type 2 Diabetes. Curr. Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, H.H.; Feigh, M.; Veidal, S.S.; Rigbolt, K.T.; Vrang, N.; Fosgerau, K. Mouse Models of Nonalcoholic Steatohepatitis in Preclinical Drug Development. Drug Discov. Today 2017, 22, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.N.B.; Veidal, S.S.; Rigbolt, K.T.G.; Tølbøl, K.S.; Roth, J.D.; Jelsing, J.; Vrang, N.; Feigh, M. Obese Diet-Induced Mouse Models of Nonalcoholic Steatohepatitis-Tracking Disease by Liver Biopsy. World J. Hepatol. 2016, 8, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Goldin, R.D. Mouse Models in Non-Alcoholic Fatty Liver Disease and Steatohepatitis Research. Int. J. Exp. Pathol. 2006, 87, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Imajo, K.; Fujita, K.; Yoneda, M.; Nozaki, Y.; Ogawa, Y.; Shinohara, Y.; Kato, S.; Mawatari, H.; Shibata, W.; Kitani, H.; et al. Hyperresponsivity to Low-Dose Endotoxin during Progression to Nonalcoholic Steatohepatitis Is Regulated by Leptin-Mediated Signaling. Cell Metab. 2012, 16, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.Q.; Lin, H.Z.; Lane, M.D.; Clemens, M.; Diehl, A.M. Obesity Increases Sensitivity to Endotoxin Liver Injury: Implications for the Pathogenesis of Steatohepatitis. Proc. Natl. Acad. Sci. USA 1997, 94, 2557–2562. [Google Scholar] [CrossRef]
- Hummel, K.P.; Dickie, M.M.; Coleman, D.L. Diabetes, a New Mutation in the Mouse. Science 1966, 153, 1127–1128. [Google Scholar] [CrossRef]
- Soret, P.-A.; Magusto, J.; Housset, C.; Gautheron, J. In Vitro and In Vivo Models of Non-Alcoholic Fatty Liver Disease: A Critical Appraisal. J. Clin. Med. 2020, 10, 36. [Google Scholar] [CrossRef]
- Trak-Smayra, V.; Paradis, V.; Massart, J.; Nasser, S.; Jebara, V.; Fromenty, B. Pathology of the Liver in Obese and Diabetic Ob/Ob and Db/Db Mice Fed a Standard or High-Calorie Diet. Int. J. Exp. Pathol. 2011, 92, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Pessayre, D. Inhibition of Mitochondrial Beta-Oxidation as a Mechanism of Hepatotoxicity. Pharmacol. Ther. 1995, 67, 101–154. [Google Scholar] [CrossRef]
- Sahai, A.; Malladi, P.; Pan, X.; Paul, R.; Melin-Aldana, H.; Green, R.M.; Whitington, P.F. Obese and Diabetic Db/Db Mice Develop Marked Liver Fibrosis in a Model of Nonalcoholic Steatohepatitis: Role of Short-Form Leptin Receptors and Osteopontin. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1035–G1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handa, P.; Morgan-Stevenson, V.; Maliken, B.D.; Nelson, J.E.; Washington, S.; Westerman, M.; Yeh, M.M.; Kowdley, K.V. Iron Overload Results in Hepatic Oxidative Stress, Immune Cell Activation, and Hepatocellular Ballooning Injury, Leading to Nonalcoholic Steatohepatitis in Genetically Obese Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G117–G127. [Google Scholar] [CrossRef]
- Caballero, F.; Fernández, A.; Matías, N.; Martínez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernández-Checa, J.C.; García-Ruiz, C. Specific Contribution of Methionine and Choline in Nutritional Nonalcoholic Steatohepatitis: Impact on Mitochondrial S-Adenosyl-L-Methionine and Glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itagaki, H.; Shimizu, K.; Morikawa, S.; Ogawa, K.; Ezaki, T. Morphological and Functional Characterization of Non-Alcoholic Fatty Liver Disease Induced by a Methionine-Choline-Deficient Diet in C57BL/6 Mice. Int. J. Clin. Exp. Pathol. 2013, 6, 2683–2696. [Google Scholar]
- Matsuzawa, N.; Takamura, T.; Kurita, S.; Misu, H.; Ota, T.; Ando, H.; Yokoyama, M.; Honda, M.; Zen, Y.; Nakanuma, Y.; et al. Lipid-Induced Oxidative Stress Causes Steatohepatitis in Mice Fed an Atherogenic Diet. Hepatology 2007, 46, 1392–1403. [Google Scholar] [CrossRef]
- Rinella, M.E.; Green, R.M. The Methionine-Choline Deficient Dietary Model of Steatohepatitis Does Not Exhibit Insulin Resistance. J. Hepatol. 2004, 40, 47–51. [Google Scholar] [CrossRef]
- Kashireddy, P.V.; Rao, M.S. Lack of Peroxisome Proliferator-Activated Receptor Alpha in Mice Enhances Methionine and Choline Deficient Diet-Induced Steatohepatitis. Hepatol. Res. 2004, 30, 104–110. [Google Scholar] [CrossRef]
- Rizki, G.; Arnaboldi, L.; Gabrielli, B.; Yan, J.; Lee, G.S.; Ng, R.K.; Turner, S.M.; Badger, T.M.; Pitas, R.E.; Maher, J.J. Mice Fed a Lipogenic Methionine-Choline-Deficient Diet Develop Hypermetabolism Coincident with Hepatic Suppression of SCD-1. J. Lipid Res. 2006, 47, 2280–2290. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Kakizaki, S.; Takizawa, D.; Ichikawa, T.; Sato, K.; Takagi, H.; Mori, M. Interstrain Differences in Susceptibility to Non-Alcoholic Steatohepatitis. J. Gastroenterol. Hepatol. 2008, 23, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Nakae, D.; Yoshiji, H.; Mizumoto, Y.; Horiguchi, K.; Shiraiwa, K.; Tamura, K.; Denda, A.; Konishi, Y. High Incidence of Hepatocellular Carcinomas Induced by a Choline Deficient L-Amino Acid Defined Diet in Rats. Cancer Res. 1992, 52, 5042–5045. [Google Scholar] [PubMed]
- Yoshiji, H.; Nakae, D.; Mizumoto, Y.; Horiguchi, K.; Tamura, K.; Denda, A.; Tsujii, T.; Konishi, Y. Inhibitory Effect of Dietary Iron Deficiency on Inductions of Putative Preneoplastic Lesions as Well as 8-Hydroxydeoxyguanosine in DNA and Lipid Peroxidation in the Livers of Rats Caused by Exposure to a Choline-Deficient L-Amino Acid Defined Diet. Carcinogenesis 1992, 13, 1227–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodama, Y.; Kisseleva, T.; Iwaisako, K.; Miura, K.; Taura, K.; De Minicis, S.; Osterreicher, C.H.; Schnabl, B.; Seki, E.; Brenner, D.A. C-Jun N-Terminal Kinase-1 from Hematopoietic Cells Mediates Progression from Hepatic Steatosis to Steatohepatitis and Fibrosis in Mice. Gastroenterology 2009, 137, 1467–1477.e5. [Google Scholar] [CrossRef] [Green Version]
- Denda, A.; Kitayama, W.; Kishida, H.; Murata, N.; Tamura, K.; Kusuoka, O.; Tsutsumi, M.; Nishikawa, F.; Kita, E.; Nakae, D.; et al. Expression of Inducible Nitric Oxide (NO) Synthase but Not Prevention by Its Gene Ablation of Hepatocarcinogenesis with Fibrosis Caused by a Choline-Deficient, L-Amino Acid-Defined Diet in Rats and Mice. Nitric Oxide 2007, 16, 164–176. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hada, N.; Sakamaki, Y.; Uno, A.; Shiga, T.; Tanaka, C.; Ito, T.; Katsume, A.; Sudoh, M. An Improved Mouse Model That Rapidly Develops Fibrosis in Non-Alcoholic Steatohepatitis. Int. J. Exp. Pathol. 2013, 94, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose Consumption as a Risk Factor for Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Nonalcoholic Steatohepatitis Clinical Research Network Increased Fructose Consumption Is Associated with Fibrosis Severity in Patients with Nonalcoholic Fatty Liver Disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, S.; Ke, J.-Y.; Pellizzon, M.A. Targeted Nutrient Modifications in Purified Diets Differentially Affect Nonalcoholic Fatty Liver Disease and Metabolic Disease Development in Rodent Models. Curr. Dev. Nutr. 2020, 4, nzaa078. [Google Scholar] [CrossRef]
- Kohli, R.; Kirby, M.; Xanthakos, S.A.; Softic, S.; Feldstein, A.E.; Saxena, V.; Tang, P.H.; Miles, L.; Miles, M.V.; Balistreri, W.F.; et al. High-Fructose, Medium Chain Trans Fat Diet Induces Liver Fibrosis and Elevates Plasma Coenzyme Q9 in a Novel Murine Model of Obesity and Nonalcoholic Steatohepatitis. Hepatology 2010, 52, 934–944. [Google Scholar] [CrossRef] [Green Version]
- Nigro, D.; Menotti, F.; Cento, A.S.; Serpe, L.; Chiazza, F.; Dal Bello, F.; Romaniello, F.; Medana, C.; Collino, M.; Aragno, M.; et al. Chronic Administration of Saturated Fats and Fructose Differently Affect SREBP Activity Resulting in Different Modulation of Nrf2 and Nlrp3 Inflammasome Pathways in Mice Liver. J. Nutr. Biochem. 2017, 42, 160–171. [Google Scholar] [CrossRef]
- Lambertz, J.; Weiskirchen, S.; Landert, S.; Weiskirchen, R. Fructose: A Dietary Sugar in Crosstalk with Microbiota Contributing to the Development and Progression of Non-Alcoholic Liver Disease. Front. Immunol. 2017, 8, 1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose Stimulated de Novo Lipogenesis Is Promoted by Inflammation. Nat. Metab. 2020, 2, 1034–1045. [Google Scholar] [CrossRef]
- Tetri, L.H.; Basaranoglu, M.; Brunt, E.M.; Yerian, L.M.; Neuschwander-Tetri, B.A. Severe NAFLD with Hepatic Necroinflammatory Changes in Mice Fed Trans Fats and a High-Fructose Corn Syrup Equivalent. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G987–G995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevaskis, J.L.; Griffin, P.S.; Wittmer, C.; Neuschwander-Tetri, B.A.; Brunt, E.M.; Dolman, C.S.; Erickson, M.R.; Napora, J.; Parkes, D.G.; Roth, J.D. Glucagon-like Peptide-1 Receptor Agonism Improves Metabolic, Biochemical, and Histopathological Indices of Nonalcoholic Steatohepatitis in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G762–G772. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.E.; Poolman, T.M.; Arvaniti, A.; Cox, R.D.; Gathercole, L.L.; Tomlinson, J.W. The American Lifestyle-Induced Obesity Syndrome Diet in Male and Female Rodents Recapitulates the Clinical and Transcriptomic Features of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G345–G360. [Google Scholar] [CrossRef]
- De Minicis, S.; Agostinelli, L.; Rychlicki, C.; Sorice, G.P.; Saccomanno, S.; Candelaresi, C.; Giaccari, A.; Trozzi, L.; Pierantonelli, I.; Mingarelli, E.; et al. HCC Development Is Associated to Peripheral Insulin Resistance in a Mouse Model of NASH. PLoS ONE 2014, 9, e97136. [Google Scholar] [CrossRef] [PubMed]
- Ikawa-Yoshida, A.; Matsuo, S.; Kato, A.; Ohmori, Y.; Higashida, A.; Kaneko, E.; Matsumoto, M. Hepatocellular Carcinoma in a Mouse Model Fed a Choline-Deficient, L-Amino Acid-Defined, High-Fat Diet. Int. J. Exp. Pathol. 2017, 98, 221–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Kim, J.; Han, J.; Oh, D.; Kim, M.; Jeong, H.; Kim, T.-J.; Kim, S.-W.; Kim, J.N.; Seo, Y.-S.; et al. Formyl Peptide Receptor 2 Determines Sex-Specific Differences in the Progression of Nonalcoholic Fatty Liver Disease and Steatohepatitis. Nat. Commun. 2022, 13, 578. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef]
- Chen, T.; Xiong, M.; Zong, X.; Ge, Y.; Zhang, H.; Wang, M.; Won Han, G.; Yi, C.; Ma, L.; Ye, R.D.; et al. Structural Basis of Ligand Binding Modes at the Human Formyl Peptide Receptor 2. Nat. Commun. 2020, 11, 1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet GPIbα Is a Mediator and Potential Interventional Target for NASH and Subsequent Liver Cancer. Nat. Med. 2019, 25, 641–655. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.-I.; Hoshida, Y.; et al. A Simple Diet- and Chemical-Induced Murine NASH Model with Rapid Progression of Steatohepatitis, Fibrosis and Liver Cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.K.; Bhattacharya, D.; Borgerding, J.N.; Fiel, M.I.; Faith, J.J.; Friedman, S.L. Modeling Dysbiosis of Human NASH in Mice: Loss of Gut Microbiome Diversity and Overgrowth of Erysipelotrichales. PLoS ONE 2021, 16, e0244763. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. P62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalapour, S.; Lin, X.-J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.; Perkins, A.; Matsutani, T.; et al. Inflammation-Induced IgA+ Cells Dismantle Anti-Liver Cancer Immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, F.; Zhang, P.; Liu, J.; Wang, R.; Kaufman, R.J.; Yaden, B.C.; Karin, M. ATF4 Suppresses Hepatocarcinogenesis by Inducing SLC7A11 (XCT) to Block Stress-Related Ferroptosis. J. Hepatol. 2023, 79, 362–377. [Google Scholar] [CrossRef]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.-K.; Mirshahi, F.; et al. A Diet-Induced Animal Model of Non-Alcoholic Fatty Liver Disease and Hepatocellular Cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Hoshida, Y.; Nijman, S.M.B.; Kobayashi, M.; Chan, J.A.; Brunet, J.-P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative Transcriptome Analysis Reveals Common Molecular Subclasses of Human Hepatocellular Carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef] [Green Version]
- Tanner, N.; Kubik, L.; Luckert, C.; Thomas, M.; Hofmann, U.; Zanger, U.M.; Böhmert, L.; Lampen, A.; Braeuning, A. Regulation of Drug Metabolism by the Interplay of Inflammatory Signaling, Steatosis, and Xeno-Sensing Receptors in HepaRG Cells. Drug Metab. Dispos. 2018, 46, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Ramos, M.J.; Bandiera, L.; Menolascina, F.; Fallowfield, J.A. In Vitro Models for Non-Alcoholic Fatty Liver Disease: Emerging Platforms and Their Applications. iScience 2022, 25, 103549. [Google Scholar] [CrossRef]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and Disease Consequences of Nonalcoholic Fatty Liver Disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hirai, Y.; Iida, K.; Ito, S.; Trumm, M.; Terada, S.; Sakai, R.; Tsuchiya, T.; Tabata, O.; Kamei, K. Integrated-Gut-Liver-on-a-Chip Platform as an in Vitro Human Model of Non-Alcoholic Fatty Liver Disease. Commun. Biol. 2023, 6, 310. [Google Scholar] [CrossRef]
- Kaur, S.; Kidambi, S.; Ortega-Ribera, M.; Thuy, L.T.T.; Nieto, N.; Cogger, V.C.; Xie, W.-F.; Tacke, F.; Gracia-Sancho, J. In Vitro Models for the Study of Liver Biology and Diseases: Advances and Limitations. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of Multicell Spheroids in Tissue Culture as a Model of Nodular Carcinomas. J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar] [PubMed]
- Gunti, S.; Hoke, A.T.K.; Vu, K.P.; London, N.R. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers 2021, 13, 874. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Lauschke, V.M. 3D Human Liver Spheroids for Translational Pharmacology and Toxicology. Basic Clin. Pharmacol. Toxicol. 2022, 130 (Suppl. S1), 5–15. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Hendriks, D.F.G.; Moro, S.M.L.; Ellis, E.; Walsh, J.; Renblom, A.; Fredriksson Puigvert, L.; Dankers, A.C.A.; Jacobs, F.; Snoeys, J.; et al. Characterization of Primary Human Hepatocyte Spheroids as a Model System for Drug-Induced Liver Injury, Liver Function and Disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tostões, R.M.; Leite, S.B.; Serra, M.; Jensen, J.; Björquist, P.; Carrondo, M.J.T.; Brito, C.; Alves, P.M. Human Liver Cell Spheroids in Extended Perfusion Bioreactor Culture for Repeated-Dose Drug Testing. Hepatology 2012, 55, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Hartanto, Y.; Zhang, H. Advances in Multicellular Spheroids Formation. J. R. Soc. Interface 2017, 14, 20160877. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Jiang, T.; Chen, D.; Wang, Q.; Zhang, L.W. Three-Dimensional Liver Models: State of the Art and Their Application for Hepatotoxicity Evaluation. Crit. Rev. Toxicol. 2020, 50, 279–309. [Google Scholar] [CrossRef]
- Basu, A.; Dydowiczová, A.; Trosko, J.E.; Bláha, L.; Babica, P. Ready to Go 3D? A Semi-Automated Protocol for Microwell Spheroid Arrays to Increase Scalability and Throughput of 3D Cell Culture Testing. Toxicol. Mech. Methods 2020, 30, 590–604. [Google Scholar] [CrossRef]
- Damelin, L.H.; Coward, S.; Kirwan, M.; Collins, P.; Selden, C.; Hodgson, H.J.F. Fat-Loaded HepG2 Spheroids Exhibit Enhanced Protection from Pro-Oxidant and Cytokine Induced Damage. J. Cell. Biochem. 2007, 101, 723–734. [Google Scholar] [CrossRef]
- Chong, L.; Tsai, C.; Yang, K.; Liao, C.; Hsu, Y. Targeting Protein Palmitoylation Decreases Palmitate-induced Sphere Formation of Human Liver Cancer Cells. Mol. Med. Rep. 2020, 22, 939–947. [Google Scholar] [CrossRef]
- Kozyra, M.; Johansson, I.; Nordling, Å.; Ullah, S.; Lauschke, V.M.; Ingelman-Sundberg, M. Human Hepatic 3D Spheroids as a Model for Steatosis and Insulin Resistance. Sci. Rep. 2018, 8, 14297. [Google Scholar] [CrossRef] [Green Version]
- Eilenberger, C.; Selinger, F.; Rothbauer, M.; Lin, Y.; Limbeck, A.; Schädl, B.; Grillari, J.; Kavok, N.S.; Klochkov, V.K.; Malyukin, Y.V.; et al. Cytotoxicity, Retention, and Anti-Inflammatory Effects of a CeO2 Nanoparticle-Based Supramolecular Complex in a 3D Liver Cell Culture Model. ACS Pharmacol. Transl. Sci. 2021, 4, 101–106. [Google Scholar] [CrossRef]
- Mukherjee, S.; Zhelnin, L.; Sanfiz, A.; Pan, J.; Li, Z.; Yarde, M.; McCarty, J.; Jarai, G. Development and Validation of an in Vitro 3D Model of NASH with Severe Fibrotic Phenotype. Am. J. Transl. Res. 2019, 11, 1531–1540. [Google Scholar] [PubMed]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurrell, T.; Kastrinou-Lampou, V.; Fardellas, A.; Hendriks, D.F.G.; Nordling, Å.; Johansson, I.; Baze, A.; Parmentier, C.; Richert, L.; Ingelman-Sundberg, M. Human Liver Spheroids as a Model to Study Aetiology and Treatment of Hepatic Fibrosis. Cells 2020, 9, 964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pingitore, P.; Sasidharan, K.; Ekstrand, M.; Prill, S.; Lindén, D.; Romeo, S. Human Multilineage 3D Spheroids as a Model of Liver Steatosis and Fibrosis. Int. J. Mol. Sci. 2019, 20, 1629. [Google Scholar] [CrossRef] [Green Version]
- Ströbel, S.; Kostadinova, R.; Fiaschetti-Egli, K.; Rupp, J.; Bieri, M.; Pawlowska, A.; Busler, D.; Hofstetter, T.; Sanchez, K.; Grepper, S.; et al. A 3D Primary Human Cell-Based in Vitro Model of Non-Alcoholic Steatohepatitis for Efficacy Testing of Clinical Drug Candidates. Sci. Rep. 2021, 11, 22765. [Google Scholar] [CrossRef] [PubMed]
- Prill, S.; Caddeo, A.; Baselli, G.; Jamialahmadi, O.; Dongiovanni, P.; Rametta, R.; Kanebratt, K.P.; Pujia, A.; Pingitore, P.; Mancina, R.M.; et al. The TM6SF2 E167K Genetic Variant Induces Lipid Biosynthesis and Reduces Apolipoprotein B Secretion in Human Hepatic 3D Spheroids. Sci. Rep. 2019, 9, 11585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, B.E.; Rajagopal, V.; Smith, C.; Cohick, E.; Whissell, G.; Gamboa, M.; Pai, R.; Sigova, A.; Grossman, I.; Bumcrot, D.; et al. Discovery and Targeting of the Signaling Controls of PNPLA3 to Effectively Reduce Transcription, Expression, and Function in Pre-Clinical NAFLD/NASH Settings. Cells 2020, 9, 2247. [Google Scholar] [CrossRef]
- Tanaka, Y.; Shimanaka, Y.; Caddeo, A.; Kubo, T.; Mao, Y.; Kubota, T.; Kubota, N.; Yamauchi, T.; Mancina, R.M.; Baselli, G.; et al. LPIAT1/MBOAT7 Depletion Increases Triglyceride Synthesis Fueled by High Phosphatidylinositol Turnover. Gut 2021, 70, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Underhill, G.H.; Khetani, S.R. Advances in Engineered Human Liver Platforms for Drug Metabolism Studies. Drug Metab. Dispos. 2018, 46, 1626–1637. [Google Scholar] [CrossRef]
- Simian, M.; Bissell, M.J. Organoids: A Historical Perspective of Thinking in Three Dimensions. J. Cell Biol. 2017, 216, 31–40. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a Dish: Modeling Development and Disease Using Organoid Technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Huch, M.; Koo, B.-K. Modeling Mouse and Human Development Using Organoid Cultures. Development 2015, 142, 3113–3125. [Google Scholar] [CrossRef] [Green Version]
- Cacciamali, A.; Villa, R.; Dotti, S. 3D Cell Cultures: Evolution of an Ancient Tool for New Applications. Front. Physiol. 2022, 13, 836480. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.P.; Baumgarten, S.F.; Verma, R.; Lunov, O.; Dejneka, A.; Sullivan, G.J. Liver Organoids: Recent Developments, Limitations and Potential. Front. Med. 2021, 8, 574047. [Google Scholar] [CrossRef]
- Wei, J.; Zhang, W.; Zhao, B. Human Liver Organoid: Modeling Liver Steatosis and Beyond. Cell Regen. 2023, 12, 17. [Google Scholar] [CrossRef]
- Ramli, M.N.B.; Lim, Y.S.; Koe, C.T.; Demircioglu, D.; Tng, W.; Gonzales, K.A.U.; Tan, C.P.; Szczerbinska, I.; Liang, H.; Soe, E.L.; et al. Human Pluripotent Stem Cell-Derived Organoids as Models of Liver Disease. Gastroenterology 2020, 159, 1471–1486.e12. [Google Scholar] [CrossRef]
- Shinozawa, T.; Kimura, M.; Cai, Y.; Saiki, N.; Yoneyama, Y.; Ouchi, R.; Koike, H.; Maezawa, M.; Zhang, R.-R.; Dunn, A.; et al. High-Fidelity Drug-Induced Liver Injury Screen Using Human Pluripotent Stem Cell-Derived Organoids. Gastroenterology 2021, 160, 831–846.e10. [Google Scholar] [CrossRef]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e6. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, I.; Burton, S.A.; Munn, C.; Ohshima, M.; Goedland, M.E.; Czysz, K.; Rajesh, D. IPSC-Derived Hepatocytes Generated from NASH Donors Provide a Valuable Platform for Disease Modeling and Drug Discovery. Biol. Open 2020, 9, bio055087. [Google Scholar] [CrossRef] [PubMed]
- McCarron, S.; Bathon, B.; Conlon, D.M.; Abbey, D.; Rader, D.J.; Gawronski, K.; Brown, C.D.; Olthoff, K.M.; Shaked, A.; Raabe, T.D. Functional Characterization of Organoids Derived From Irreversibly Damaged Liver of Patients With NASH. Hepatology 2021, 74, 1825–1844. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, D.; Brouwers, J.F.; Hamer, K.; Geurts, M.H.; Luciana, L.; Massalini, S.; López-Iglesias, C.; Peters, P.J.; Rodríguez-Colman, M.J.; Chuva de Sousa Lopes, S.; et al. Engineered Human Hepatocyte Organoids Enable CRISPR-Based Target Discovery and Drug Screening for Steatosis. Nat. Biotechnol. 2023, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, M.T. Organoids Illuminate NAFLD Pathogenesis. Nat. Rev. Drug Discov. 2023, 22, 269. [Google Scholar] [CrossRef]
- Kimura, M.; Iguchi, T.; Iwasawa, K.; Dunn, A.; Thompson, W.L.; Yoneyama, Y.; Chaturvedi, P.; Zorn, A.M.; Wintzinger, M.; Quattrocelli, M.; et al. En Masse Organoid Phenotyping Informs Metabolic-Associated Genetic Susceptibility to NASH. Cell 2022, 185, 4216–4232.e16. [Google Scholar] [CrossRef]
- Ingber, D.E. Human Organs-on-Chips for Disease Modelling, Drug Development and Personalized Medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef] [PubMed]
- Vargas, R.; Egurbide-Sifre, A.; Medina, L. Organ-on-a-Chip Systems for New Drugs Development. ADMET DMPK 2021, 9, 111–141. [Google Scholar] [CrossRef]
- Sun, W.; Luo, Z.; Lee, J.; Kim, H.-J.; Lee, K.; Tebon, P.; Feng, Y.; Dokmeci, M.R.; Sengupta, S.; Khademhosseini, A. Organ-on-a-Chip for Cancer and Immune Organs Modeling. Adv. Healthc. Mater. 2019, 8, e1900754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, A.K.; Scalzo, S.; de Souza, R.T.V.; Santana, P.H.G.; Marques, B.L.; Oliveira, L.F.; Filho, D.M.; Kihara, A.H.; da Costa Santiago, H.; Parreira, R.C.; et al. Strategic Use of Organoids and Organs-on-Chip as Biomimetic Tools. Semin. Cell Dev. Biol. 2023, 144, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Palasantzas, V.E.J.M.; Tamargo-Rubio, I.; Le, K.; Slager, J.; Wijmenga, C.; Jonkers, I.H.; Kumar, V.; Fu, J.; Withoff, S. IPSC-Derived Organ-on-a-Chip Models for Personalized Human Genetics and Pharmacogenomics Studies. Trends Genet. 2023, 39, 268–284. [Google Scholar] [CrossRef]
- Polidoro, M.A.; Ferrari, E.; Marzorati, S.; Lleo, A.; Rasponi, M. Experimental Liver Models: From Cell Culture Techniques to Microfluidic Organs-on-Chip. Liver Int. 2021, 41, 1744–1761. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, J.; Wang, X.; Feng, L.; Wu, J.; Zhu, X.; Wen, W.; Gong, X. Organ-on-a-Chip: Recent Breakthroughs and Future Prospects. BioMed Eng. OnLine 2020, 19, 9. [Google Scholar] [CrossRef] [Green Version]
- Hassan, S.; Sebastian, S.; Maharjan, S.; Lesha, A.; Carpenter, A.-M.; Liu, X.; Xie, X.; Livermore, C.; Zhang, Y.S.; Zarrinpar, A. Liver-on-a-Chip Models of Fatty Liver Disease. Hepatology 2020, 71, 733–740. [Google Scholar] [CrossRef]
- Du, K.; Li, S.; Li, C.; Li, P.; Miao, C.; Luo, T.; Qiu, B.; Ding, W. Modeling Nonalcoholic Fatty Liver Disease on a Liver Lobule Chip with Dual Blood Supply. Acta Biomater. 2021, 134, 228–239. [Google Scholar] [CrossRef]
- Deguchi, S.; Takayama, K. State-of-the-Art Liver Disease Research Using Liver-on-a-Chip. Inflamm. Regen. 2022, 42, 62. [Google Scholar] [CrossRef]
- Freag, M.S.; Namgung, B.; Reyna Fernandez, M.E.; Gherardi, E.; Sengupta, S.; Jang, H.L. Human Nonalcoholic Steatohepatitis on a Chip. Hepatol. Commun. 2021, 5, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wei, W.; Chen, Z.; Lin, B.; Zhao, W.; Luo, Y.; Zhang, X. Engineered Liver-on-a-Chip Platform to Mimic Liver Functions and Its Biomedical Applications: A Review. Micromachines 2019, 10, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gori, M.; Simonelli, M.C.; Giannitelli, S.M.; Businaro, L.; Trombetta, M.; Rainer, A. Investigating Nonalcoholic Fatty Liver Disease in a Liver-on-a-Chip Microfluidic Device. PLoS ONE 2016, 11, e0159729. [Google Scholar] [CrossRef] [Green Version]
- Lasli, S.; Kim, H.-J.; Lee, K.; Suurmond, C.-A.E.; Goudie, M.; Bandaru, P.; Sun, W.; Zhang, S.; Zhang, N.; Ahadian, S.; et al. A Human Liver-on-a-Chip Platform for Modeling Nonalcoholic Fatty Liver Disease. Adv. Biosyst. 2019, 3, e1900104. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Puengel, T.; Loomba, R.; Friedman, S.L. An Integrated View of Anti-Inflammatory and Antifibrotic Targets for the Treatment of NASH. J. Hepatol. 2023, 79, 552–566. [Google Scholar] [CrossRef]
- Tacke, F.; Weiskirchen, R. Non-Alcoholic Fatty Liver Disease (NAFLD)/Non-Alcoholic Steatohepatitis (NASH)-Related Liver Fibrosis: Mechanisms, Treatment and Prevention. Ann. Transl. Med. 2021, 9, 729. [Google Scholar] [CrossRef]
- Perazzo, H.; Dufour, J.-F. The Therapeutic Landscape of Non-Alcoholic Steatohepatitis. Liver Int. 2017, 37, 634–647. [Google Scholar] [CrossRef]
- Sulaiman, S.A.; Dorairaj, V.; Adrus, M.N.H. Genetic Polymorphisms and Diversity in Nonalcoholic Fatty Liver Disease (NAFLD): A Mini Review. Biomedicines 2022, 11, 106. [Google Scholar] [CrossRef]
- Kostrzewski, T.; Maraver, P.; Ouro-Gnao, L.; Levi, A.; Snow, S.; Miedzik, A.; Rombouts, K.; Hughes, D. A Microphysiological System for Studying Nonalcoholic Steatohepatitis. Hepatol. Commun. 2020, 4, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, H. Multiple Organs Involved in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Cell Biosci. 2020, 10, 140. [Google Scholar] [CrossRef]
- Bauer, K.C.; Littlejohn, P.T.; Ayala, V.; Creus-Cuadros, A.; Finlay, B.B. Nonalcoholic Fatty Liver Disease and the Gut-Liver Axis: Exploring an Undernutrition Perspective. Gastroenterology 2022, 162, 1858–1875.e2. [Google Scholar] [CrossRef]
- Lee, S.Y.; Sung, J.H. Gut-Liver on a Chip toward an in Vitro Model of Hepatic Steatosis. Biotechnol. Bioeng. 2018, 115, 2817–2827. [Google Scholar] [CrossRef]
- Jeon, J.-W.; Lee, S.H.; Kim, D.; Sung, J.H. In Vitro Hepatic Steatosis Model Based on Gut-Liver-on-a-Chip. Biotechnol. Prog. 2021, 37, e3121. [Google Scholar] [CrossRef]
- Slaughter, V.L.; Rumsey, J.W.; Boone, R.; Malik, D.; Cai, Y.; Sriram, N.N.; Long, C.J.; McAleer, C.W.; Lambert, S.; Shuler, M.L.; et al. Validation of an Adipose-Liver Human-on-a-Chip Model of NAFLD for Preclinical Therapeutic Efficacy Evaluation. Sci. Rep. 2021, 11, 13159. [Google Scholar] [CrossRef]
- Smith, P.F.; Gandolfi, A.J.; Krumdieck, C.L.; Putnam, C.W.; Zukoski, C.F.; Davis, W.M.; Brendel, K. Dynamic Organ Culture of Precision Liver Slices for in Vitro Toxicology. Life Sci. 1985, 36, 1367–1375. [Google Scholar] [CrossRef]
- Olinga, P.; Schuppan, D. Precision-Cut Liver Slices: A Tool to Model the Liver Ex Vivo. J. Hepatol. 2013, 58, 1252–1253. [Google Scholar] [CrossRef] [Green Version]
- Dewyse, L.; Reynaert, H.; van Grunsven, L.A. Best Practices and Progress in Precision-Cut Liver Slice Cultures. Int. J. Mol. Sci. 2021, 22, 7137. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, I.A.M.; Olinga, P.; de Jager, M.H.; Merema, M.T.; de Kanter, R.; van de Kerkhof, E.G.; Groothuis, G.M.M. Preparation and Incubation of Precision-Cut Liver and Intestinal Slices for Application in Drug Metabolism and Toxicity Studies. Nat. Protoc. 2010, 5, 1540–1551. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, M.; Vallier, L.; Guillot, A. Modeling Nonalcoholic Fatty Liver Disease in the Dish Using Human-Specific Platforms: Strategies and Limitations. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Paish, H.L.; Reed, L.H.; Brown, H.; Bryan, M.C.; Govaere, O.; Leslie, J.; Barksby, B.S.; Garcia Macia, M.; Watson, A.; Xu, X.; et al. A Bioreactor Technology for Modeling Fibrosis in Human and Rodent Precision-Cut Liver Slices. Hepatology 2019, 70, 1377–1391. [Google Scholar] [CrossRef]
- Wu, X.; Roberto, J.B.; Knupp, A.; Kenerson, H.L.; Truong, C.D.; Yuen, S.Y.; Brempelis, K.J.; Tuefferd, M.; Chen, A.; Horton, H.; et al. Precision-Cut Human Liver Slice Cultures as an Immunological Platform. J. Immunol. Methods 2018, 455, 71–79. [Google Scholar] [CrossRef] [PubMed]
- van de Bovenkamp, M.; Groothuis, G.M.M.; Meijer, D.K.F.; Olinga, P. Liver Slices as a Model to Study Fibrogenesis and Test the Effects of Anti-Fibrotic Drugs on Fibrogenic Cells in Human Liver. Toxicol. Vitr. 2008, 22, 771–778. [Google Scholar] [CrossRef] [PubMed]
- van de Bovenkamp, M.; Groothuis, G.M.M.; Draaisma, A.L.; Merema, M.T.; Bezuijen, J.I.; van Gils, M.J.; Meijer, D.K.F.; Friedman, S.L.; Olinga, P. Precision-Cut Liver Slices as a New Model to Study Toxicity-Induced Hepatic Stellate Cell Activation in a Physiologic Milieu. Toxicol. Sci. 2005, 85, 632–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, E.; Doornebal, E.J.; Chokshi, S. Precision-Cut Liver Slices: A Versatile Tool to Advance Liver Research. Hepatol. Int. 2019, 13, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Janssen, A.W.F.; Betzel, B.; Stoopen, G.; Berends, F.J.; Janssen, I.M.; Peijnenburg, A.A.; Kersten, S. The Impact of PPARα Activation on Whole Genome Gene Expression in Human Precision Cut Liver Slices. BMC Genom. 2015, 16, 760. [Google Scholar] [CrossRef] [Green Version]
- Westra, I.M.; Mutsaers, H.A.M.; Luangmonkong, T.; Hadi, M.; Oosterhuis, D.; de Jong, K.P.; Groothuis, G.M.M.; Olinga, P. Human Precision-Cut Liver Slices as a Model to Test Antifibrotic Drugs in the Early Onset of Liver Fibrosis. Toxicol. Vitr. 2016, 35, 77–85. [Google Scholar] [CrossRef]
- Westra, I.M.; Oosterhuis, D.; Groothuis, G.M.M.; Olinga, P. Precision-Cut Liver Slices as a Model for the Early Onset of Liver Fibrosis to Test Antifibrotic Drugs. Toxicol. Appl. Pharmacol. 2014, 274, 328–338. [Google Scholar] [CrossRef]
- Aoudjehane, L.; Gautheron, J.; Le Goff, W.; Goumard, C.; Gilaizeau, J.; Nget, C.S.; Savier, E.; Atif, M.; Lesnik, P.; Morichon, R.; et al. Novel Defatting Strategies Reduce Lipid Accumulation in Primary Human Culture Models of Liver Steatosis. Dis. Model. Mech. 2020, 13, dmm042663. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, D.; Becker, C.; Readhead, B.; Goossens, N.; Novik, J.; Fiel, M.I.; Cousens, L.P.; Magnusson, B.; Backmark, A.; Hicks, R.; et al. Repositioning of a Novel GABA-B Receptor Agonist, AZD3355 (Lesogaberan), for the Treatment of Non-Alcoholic Steatohepatitis. Sci. Rep. 2021, 11, 20827. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
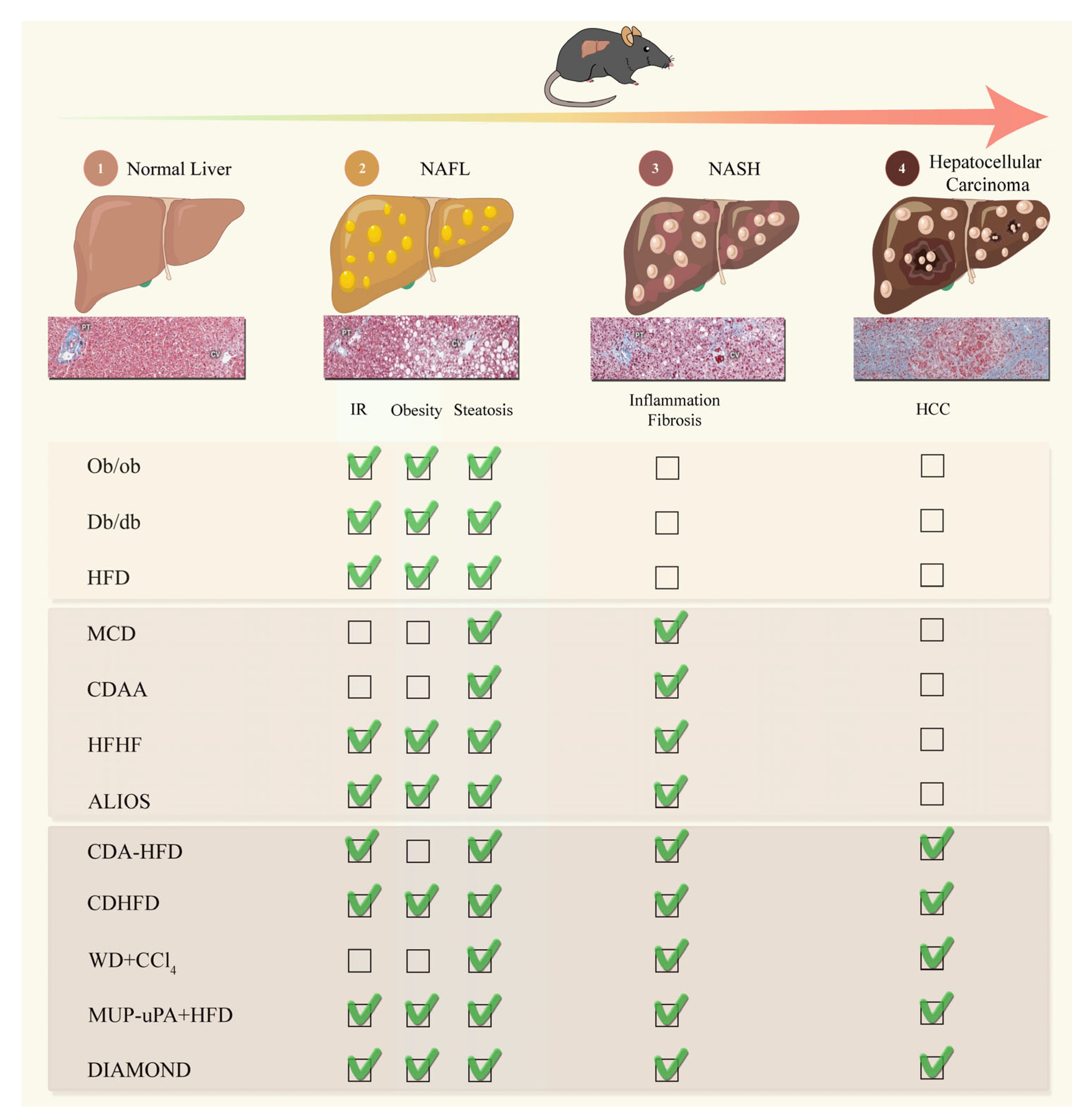
| Model Type or Name | Phenotype | Fibrosis | NASH | Human NASH Gene Signature | Time to Disease | Subsequent Development of HCC | Advantage | Disadvantage | References | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Insulin Resistance | Obesity | Steatosis | Inflammation/ER Stress | Ballooning | |||||||||
| NAFLD | |||||||||||||
| HFD | YES | YES | Strong | Weak | NO | Slight | NO | NO | Translocation of bacteria in 1 week, fibrosis after 9 weeks, hepatic inflammation after 19 weeks | No; requires additional insult | Low costs; easy to operate | Requires large sample size; difficult comparison between groups and protocols related to various dietary compositions | Ito et al., 2007; Nakagawa et al., 2014; Flessa et al., 2022 [44,49,50] |
| ob/ob (point mutation in leptin gene) | YES | YES | Strong | Weak | NO | Second insult | NO | ND | First signs of obesity recognizable at 4 weeks of age | No; requires additional insult | Features similar to human NAFLD | Leptin deficiency does not seem to play a role in NAFLD and NASH development in humans; Do not develop NASH or fibrosis without second insult | Kristiansen et al., 2016 [55] |
| db/db (point mutation in leptin receptor) | YES | YES | Strong | Weak | NO | Second insult | NO | ND | Develop NASH after the addition of MCD diet for 2 weeks | No; requires additional insult | Features similar to human NAFLD | Leptin deficiency does not seem to play a role in NAFLD and NASH development in humans; Do not develop NASH or fibrosis without second insult | Trak-Smayra et al., 2011 [61] |
| NASH | |||||||||||||
| MCD | NO | Weight loss | Strong | Strong | YES | YES | YES | ND | Steatohepatitis after 10 days and fibrosis after 8–10 weeks | No; requires additional insult | Short period; Easy to operate; High reproducibility, appropriate for the study of fibrosis mechanisms | No NAFLD-related metabolic syndrome; severe weight loss and liver atrophy | Caballero et al., 2010; Itagaki et al., 2013 [65,66] |
| CDAA | NO | Weight loss | Strong | Medium | YES | YES | YES | ND | Mild to moderate fibrosis ~22 weeks | ~25% prevalence of HCC 84 weeks | Robust; reproducible | Long-period; high costs; age-related systemic changes, No advanced fibrosis; Disease etiology does not mimic humans | Kodama et al., 2009; Denda et al., 2007; Matsumoto et al., 2013 [76,77,78] |
| High-Fructose Diet | YES | YES | Strong | Medium | YES | YES | YES | YES | 12~16 weeks can have fibrosis and NASH | Need other diet | Recapitulates histopathological characteristics of human from NAFLD to NASH | Do not develop HCC without second insult | Kohli et al., 2010; Nigro et al., 2017 [80,81] |
| ALIOS | YES | YES | Strong | Medium | YES | YES | YES | ND | NASH 12–16 weeks versus late NASH 24–30 weeks | Not relevant | Recapitulates histopathological characteristics of human NASH | No longer available as trans fats cannot be included in food | Tetri et al., 2008; Trevaskis et al., 2012; Gallage et al., 2022 [43,84,85] |
| NASH-HCC | |||||||||||||
| CDA-HFD | NO | NO | Strong | Weak | YES | YES | YES | ND | Lipid droplets with infiltration of inflammatory cells after 1 week;enlarged fatty liver with fibrosis in 6 week; NASH ~12 weeks | 24–36 weeks: 100% of mice | Accelerated model of NASH with severe fibrosis. High induction of hepatocellular adenomas and carcinomas | Does not induce obesity or metabolic syndrome | Matsumoto et al., 2013; De Minicis et al., 2014; Ikawa-Yoshida et al., 2017 [76,87,88] |
| CDHFD | YES | YES | Strong | Weak | YES | YES | YES | ND | NASH 16~18 weeks; HCA~20 weeks | 12 months: ~25–30% of mice; 15 months: ~50–70% of mice | ROS production, lipid peroxidation, and mitochondrial dysfunction. Fibrosis around central vein. Overexpression of cytokines TNF-α and IL-6 | Lack of choline is less physiological. Lower degree of fibrosis compared to WDs | Wolf et al., 2014; Pfister et al., 2021; Malehmir et al., 2019 [35,39,92] |
| WD+CCl4 | NO | NO | Strong | Strong | YES | YES | YES | YES | Microbiome remodeling within 12 weeks; F4 fibrosis and HCC ~24 weeks, | 24 weeks: 100% of mice | Recapitulates histopathological and transcriptional characteristics of human NASH | Does not induce obesity or metabolic syndrome. | Tsuchida et al., 2018 [93] |
| Mup-upA +HFD | YES | YES | Strong | Medium | YES | YES | YES | YES | Ballooning hepatocytes, pericellular and bridging fibrosis ~24 weeks; small HCC ~32 weeks | ~32–40 weeks: ~80% of mice | Can spontaneously mimic Human NASH to HCC; does not rely on the administration of liver toxins or carcinogens | Genetic background | Nakagawa et al., 2014; Febbraio et al., 2019 [40,50] |
| DIAMOND | YES | YES | Strong | Weak | YES | YES | YES | YES | Mice develop steatosis within 16 weeks; NASH ~16–24 weeks and nodule formation by 52 weeks (90%) | 52 weeks: 90% of mice | Histology and transcriptome mirror human NASH | Genetic background of DIAMOND mice is unique, making it difficult to cross them with other gene targeted mice | Asgharpour et al., 2016 [98] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, J.; Celton-Morizur, S.; Desdouets, C. NAFLD-Related HCC: Focus on the Latest Relevant Preclinical Models. Cancers 2023, 15, 3723. https://doi.org/10.3390/cancers15143723
Fang J, Celton-Morizur S, Desdouets C. NAFLD-Related HCC: Focus on the Latest Relevant Preclinical Models. Cancers. 2023; 15(14):3723. https://doi.org/10.3390/cancers15143723
Chicago/Turabian StyleFang, Jing, Séverine Celton-Morizur, and Chantal Desdouets. 2023. "NAFLD-Related HCC: Focus on the Latest Relevant Preclinical Models" Cancers 15, no. 14: 3723. https://doi.org/10.3390/cancers15143723
APA StyleFang, J., Celton-Morizur, S., & Desdouets, C. (2023). NAFLD-Related HCC: Focus on the Latest Relevant Preclinical Models. Cancers, 15(14), 3723. https://doi.org/10.3390/cancers15143723