
Breast Sarcomas, Phyllodes Tumors, and Desmoid Tumors: Turning the Magnifying Glass on Rare and Aggressive Entities
 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Epidemiology and Risk Factors
3. Prevention and Screening
3.1. Prevention
3.2. Screening
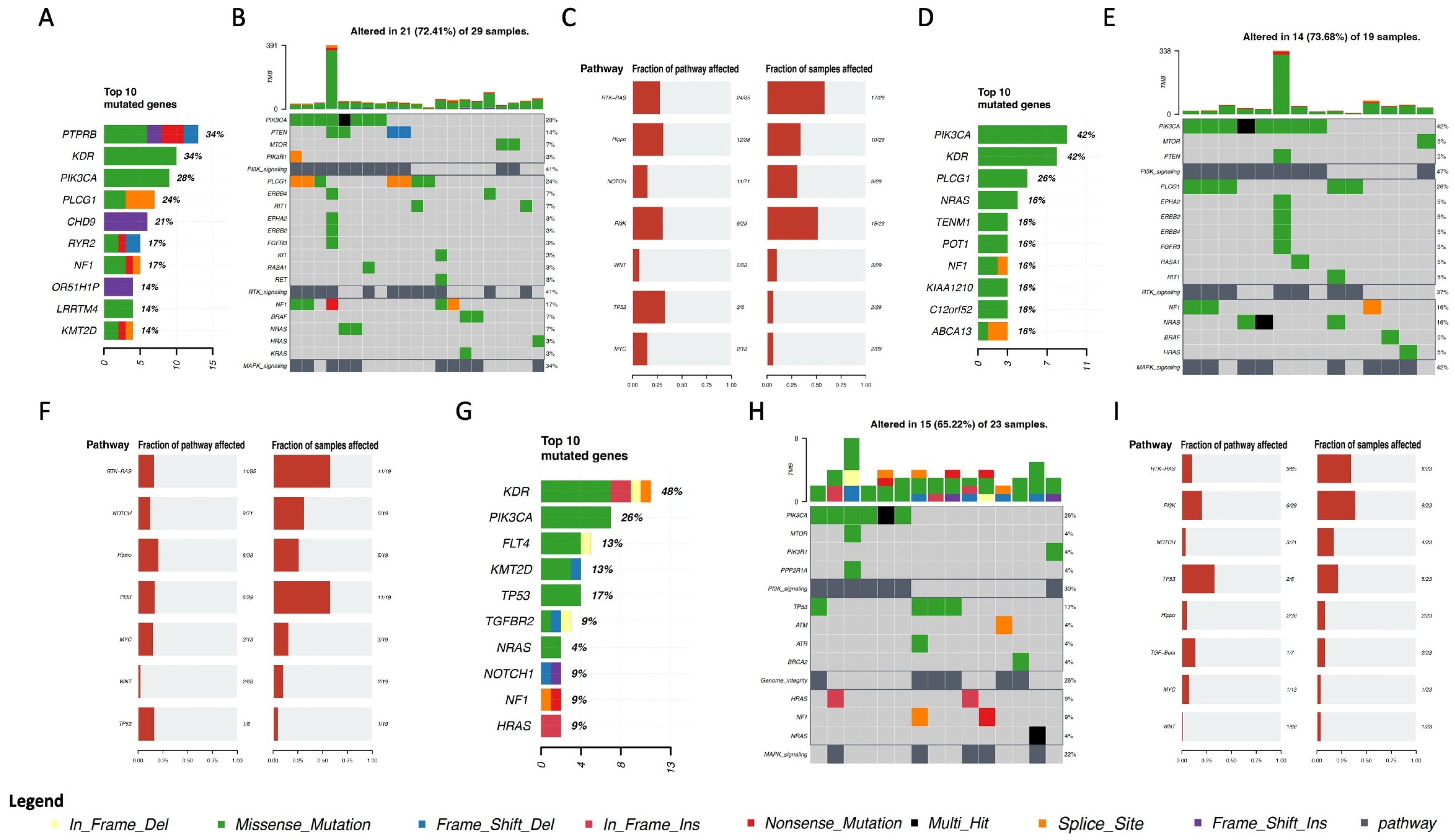
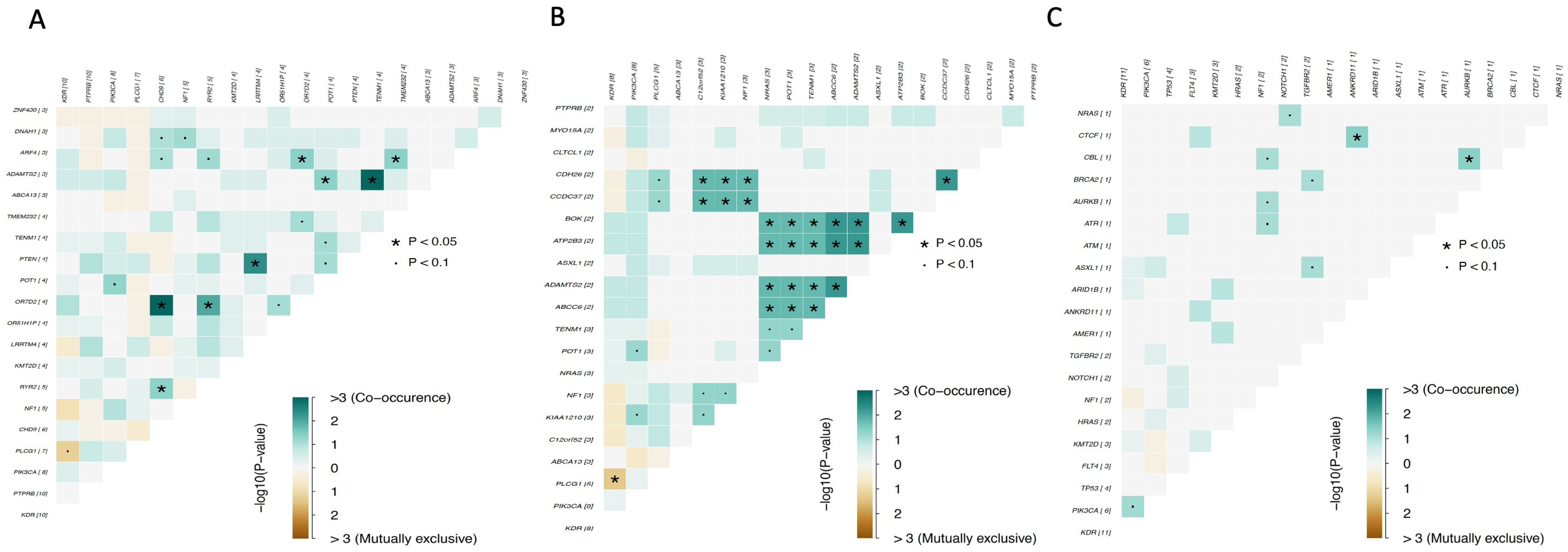
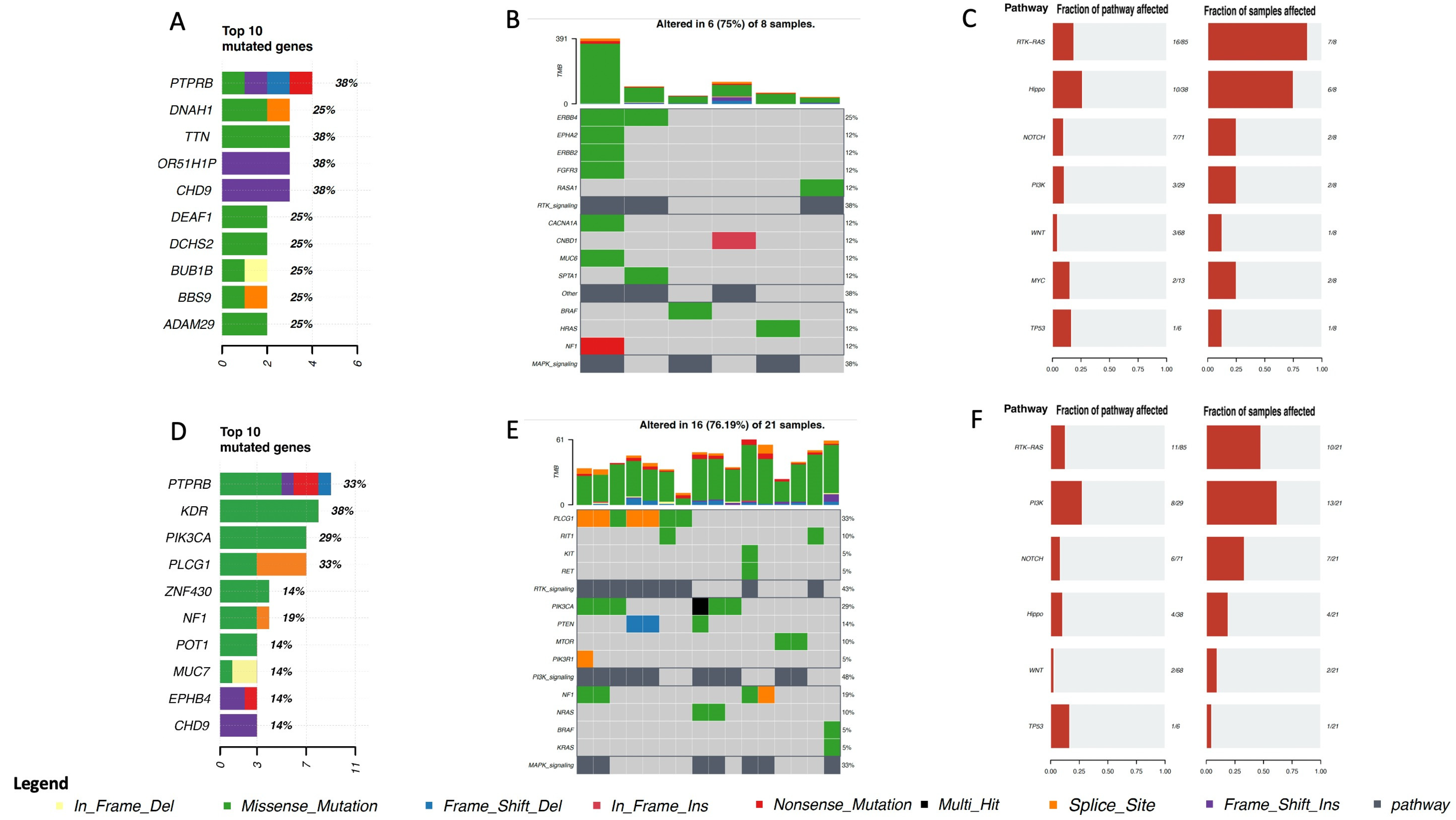
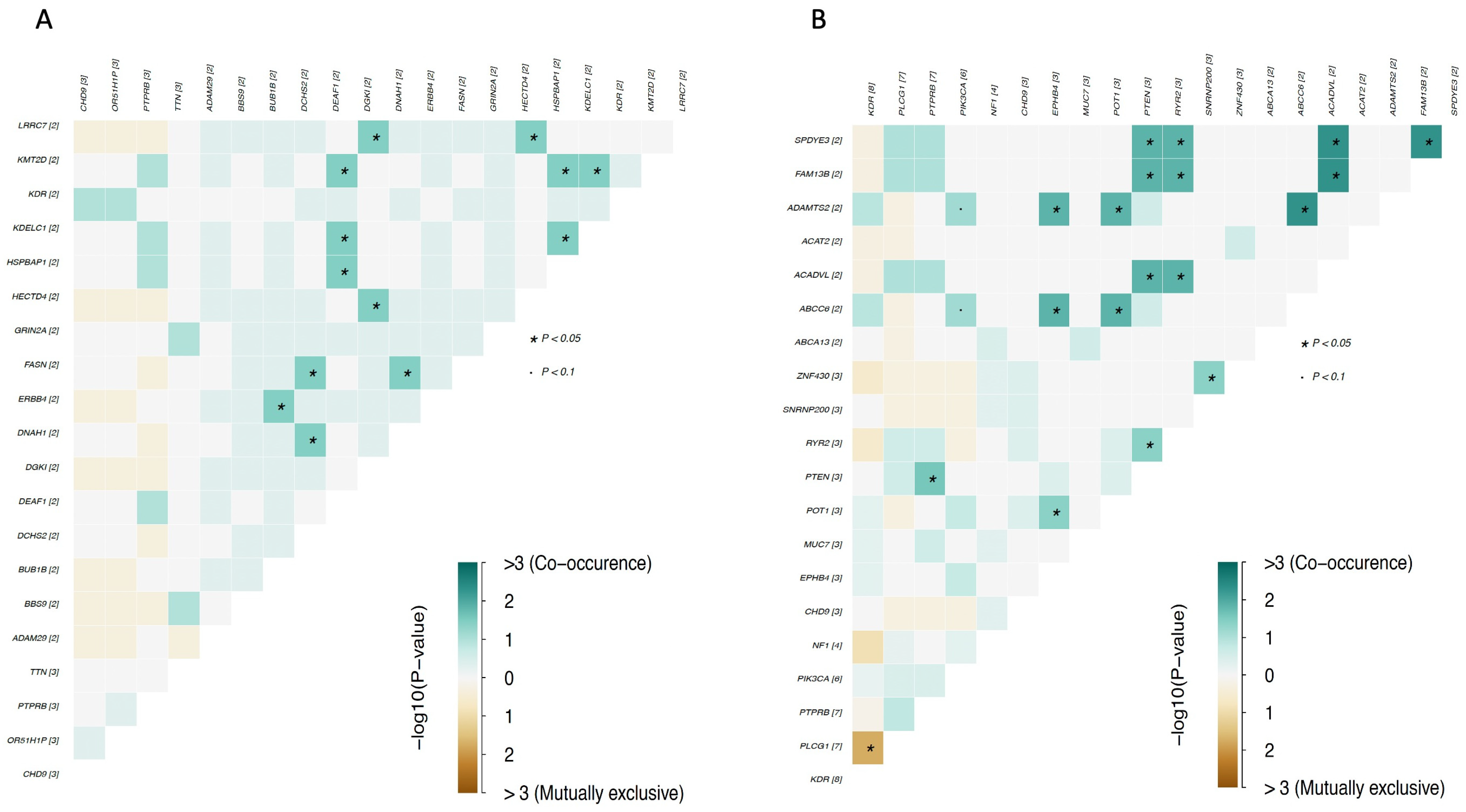
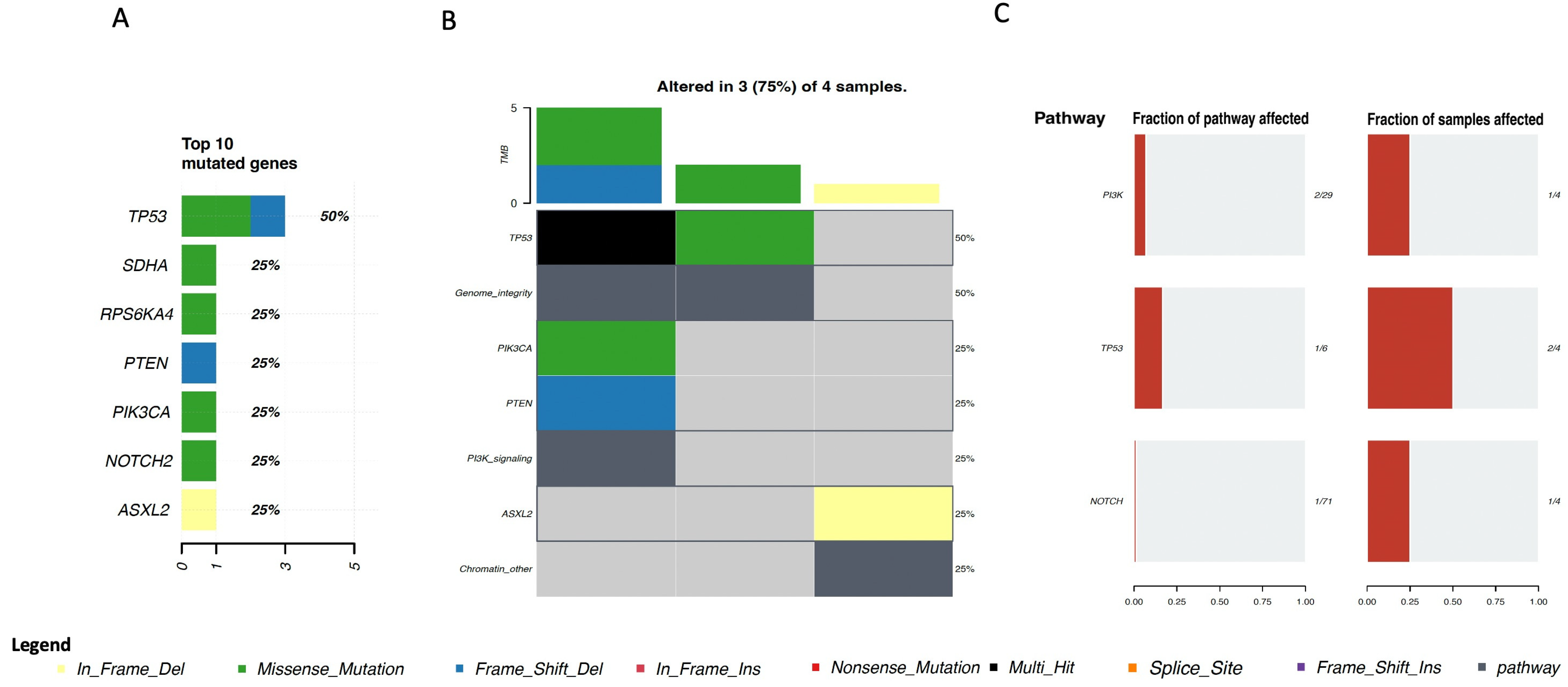
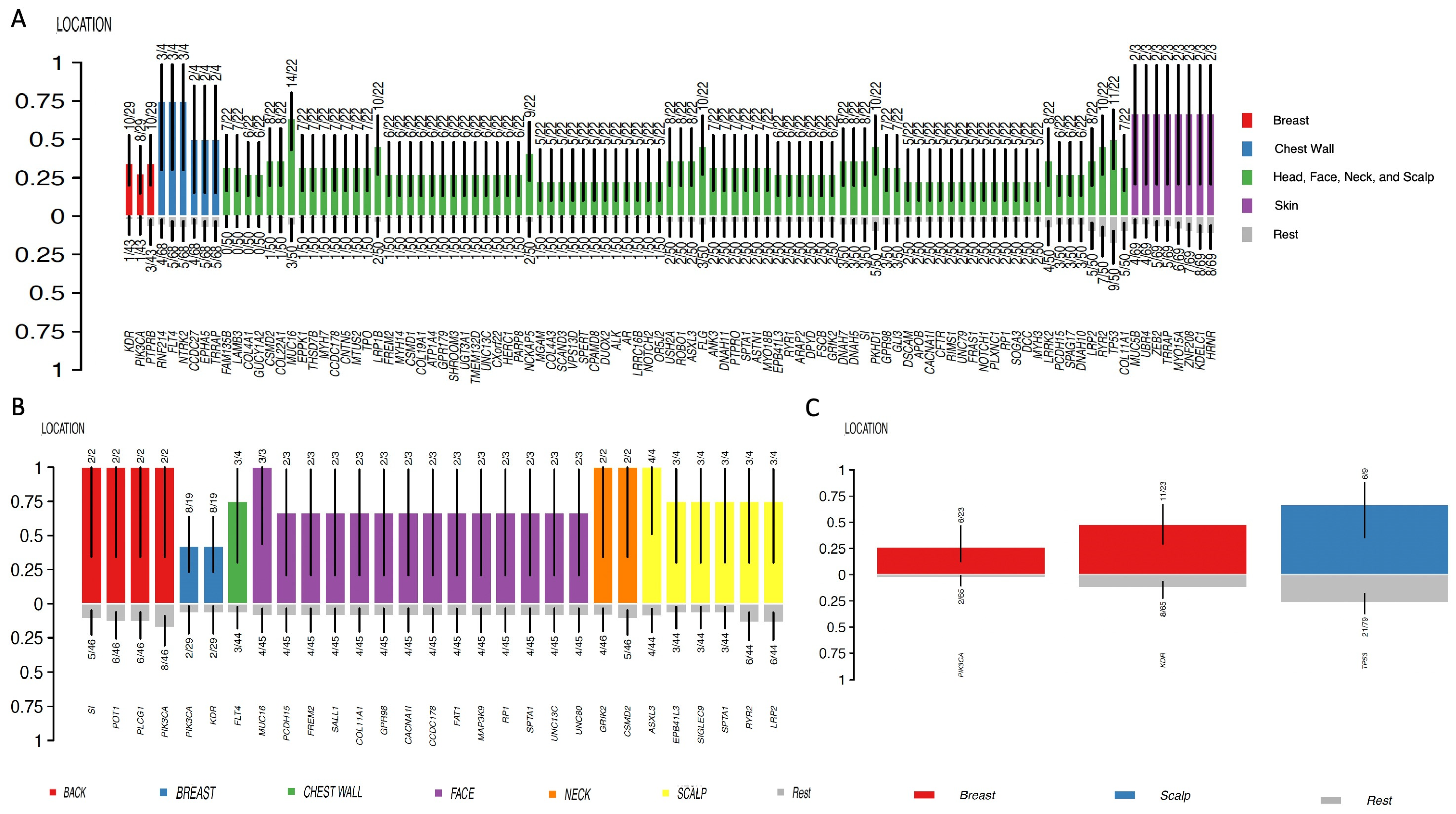
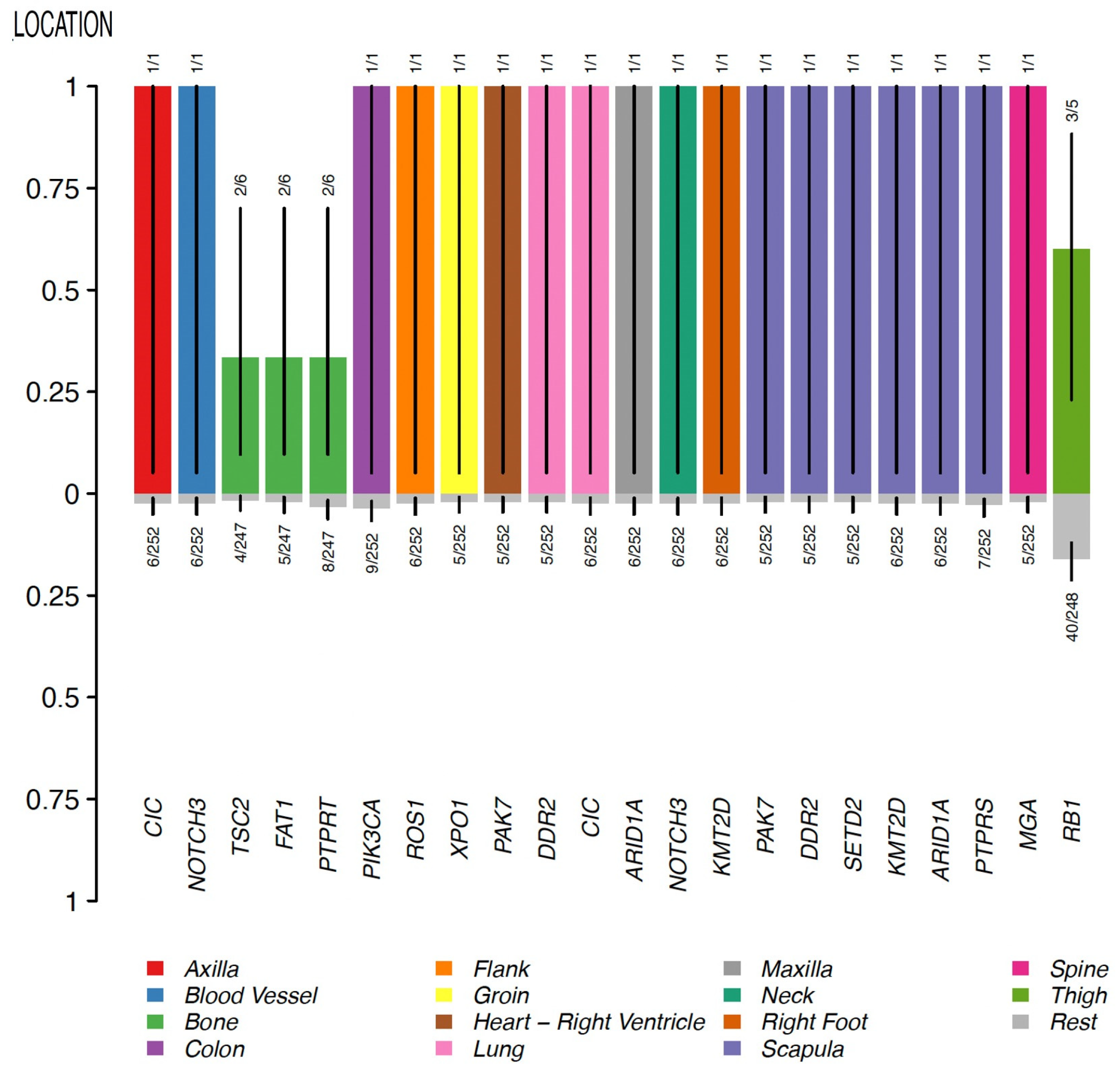
4. Genomic Profile and Molecular Landscape of Breast Sarcomas
- -
- Cohort A, from the Angiosarcoma Project [13], included data on whole exome sequencing (WES), medical records (radiation exposure prior to sarcoma diagnosis, type and duration of adjuvant treatments, etc.), and pathology results and patient-reported data [14]. Although this cohort included a total of 83 samples, only 29 were from the breast.
- ○
- Cohort A.1 included samples from patients who had been exposed to radiation prior to a sarcoma diagnosis.
- ○
- Cohort A.2 included samples from patients who had not been exposed to radiation prior to a sarcoma diagnosis.
- -
- -
- Cohort C, from the Memorial Sloan Kettering Cancer Center (MSKCC) [16] MSK-IMPACT, included genomic data obtained through a panel of 505 genes (MSK-IMPACT gene panel) and clinical data, including the origin of each sample (primary or metastatic lesion). This cohort comprised a total of 2138 samples, of which 31 were from breasts. The cohort included samples from angiosarcoma, undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma/high-grade spindle cell sarcoma, desmoid/aggressive fibromatosis, leiomyosarcoma, and round cell sarcoma.
- ○
- Cohort C.1 included samples from patients with a diagnosis of breast angiosarcoma.
- ○
- Cohort C.2 included samples from patients with a diagnosis of other types of breast sarcoma.
5. Clinical Characteristics
- -
- -
- A rare case of breast angiosarcoma in a 17-year-old girl with an unusual clinical presentation [38]; this breast angiosarcoma initially mimicked an inflammatory breast carcinoma and then grew quickly as a mass, occupying the entire right breast with indistinct borders and poor mobility [38]. In addition, two erythematous patches on the skin and a cystic lesion of approximately 3 by 4 cm surrounded by indurated tissue underneath the larger erythematous patch were also observed [38].
- -
- A review of twenty-two cases of primary and secondary breast angiosarcomas and an additional meta-analysis from Roswell Park Comprehensive Cancer Center described primary breast angiosarcomas (i.e., those that develop de novo, without prior breast radiation exposure) as typically occurring in 30-to-50 year-old women and presenting as a large mass without skin changes [36]. On the other hand, secondary breast angiosarcomas (typically associated with radiation exposure to the breast and/or chest wall or with chronic lymphedema following breast surgery and lymph node dissection) are typically reported in 60–70-year-old women and present as ecchymosis with or without ulceration [36].
- -
- Radiation-induced breast angiosarcoma may also present as skin discoloration or purplish-red nodules, as thickening or elevation of the skin without color change, and/or as diffuse skin extension of lesions of various morphologies [31]. Another common feature is the presence of multifocality with microsatellite lesions [31].
6. Diagnosis and Staging
7. Prognostic and Predictive Factors
7.1. Prognostic Factors
7.2. Predictive Factors
8. Treatment
8.1. General Management
8.2. Angiosarcoma
8.3. Other Breast Sarcomas
9. Specificities and Management of Breast Phyllodes and Desmoid Tumors
9.1. Phyllodes Tumors of the Breast
9.2. Breast Desmoid Tumors
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karlin, N.J.; Wong, D.A. Mesenchymal Neoplasms and Primary Lymphomas of the Breast. In The Breast; Elsevier: Amsterdam, The Netherlands, 2018; pp. 156–168.e6. [Google Scholar] [CrossRef]
- Li, G.Z.; Raut, C.P.; Hunt, K.K.; Feng, M.; Chugh, R. Breast Sarcomas, Phyllodes Tumors, and Desmoid Tumors: Epidemiology, Diagnosis, Staging, and Histology-Specific Management Considerations. Am. Soc. Clin. Oncol. Educ. Book 2021, 41, 390–404. [Google Scholar] [CrossRef]
- Adem, C.; Reynolds, C.; Ingle, J.N.; Nascimento, A.G. Primary breast sarcoma: Clinicopathologic series from the Mayo Clinic and review of the literature. Br. J. Cancer 2004, 91, 237–241. [Google Scholar] [CrossRef]
- Zelek, L.; Llombart-Cussac, A.; Terrier, P.; Pivot, X.; Guinebretiere, J.; Le Pechoux, C.; Tursz, T.; Rochard, F.; Spielmann, M.; Le Cesne, A. Prognostic Factors in Primary Breast Sarcomas: A Series of Patients with Long-Term Follow-Up. J. Clin. Oncol. 2003, 21, 2583–2588. [Google Scholar] [CrossRef]
- Bassett, L.W.; Mahoney, M.C.; Apple, S.; D’Orsi, C. Other Malignant Breast Diseases. In Breast Imaging Expert Radiology; ELSEVIER Saunders: Amsterdam, The Netherlands, 2011; pp. 502–523. [Google Scholar]
- Mesli, S.N.; Ghouali, A.K.; Benamara, F.; Taleb, F.A.; Tahraoui, H.; Abi-Ayad, C. Stewart-Treves Syndrome Involving Chronic Lymphedema after Mastectomy of Breast Cancer. Case Rep. Surg. 2017, 2017, 4056459. [Google Scholar] [CrossRef]
- Veiga, L.H.S.; Vo, J.B.; Curtis, R.E.; Mille, M.M.; Lee, C.; Ramin, C.; Bodelon, C.; Bowles, E.J.A.; Buist, D.S.M.; Weinmann, S.; et al. Treatment-related thoracic soft tissue sarcomas in US breast cancer survivors: A retrospective cohort study. Lancet Oncol. 2022, 23, 1451–1464. [Google Scholar] [CrossRef]
- Sánchez-Heras, A.B.; Cajal, T.R.Y.; Pineda, M.; Aguirre, E.; Graña, B.; Chirivella, I.; Balmaña, J.; Brunet, J. SEOM Clinical Guideline on Heritable TP53-Related Cancer Syndrome (2022). Clin. Transl. Oncol. 2023, 1–7. [Google Scholar] [CrossRef]
- Helvie, M.A.; Bevers, T.B. Screening Mammography for Average-Risk Women: The Controversy and NCCN’s Position. J. Natl. Compr. Cancer Netw. 2018, 16, 1398–1404. [Google Scholar] [CrossRef] [Green Version]
- Screening Ages and Frequencies|Cancer Screening, Diagnosis and Care. Available online: https://healthcare-quality.jrc.ec.europa.eu/ecibc/european-breast-cancer-guidelines/screening-ages-and-frequencies (accessed on 20 July 2023).
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Angiosarcoma Project. Available online: https://ascproject.org/ (accessed on 23 June 2023).
- CBioPortal for Cancer Genomics. Available online: https://www.cbioportal.org/study/summary?id=angs_painter_2020 (accessed on 23 June 2023).
- Painter, C.A.; Jain, E.; Tomson, B.N.; Dunphy, M.; Stoddard, R.E.; Thomas, B.S.; Damon, A.L.; Shah, S.; Kim, D.; Gómez Tejeda Zañudo, J.; et al. The Angiosarcoma Project: Enabling genomic and clinical discoveries in a rare cancer through patient-partnered research. Nat. Med. 2020, 26, 181–187. [Google Scholar] [CrossRef]
- Nacev, B.A.; Sanchez-Vega, F.; Smith, S.A.; Antonescu, C.R.; Rosenbaum, E.; Shi, H.; Tang, C.; Socci, N.D.; Rana, S.; Gularte-Mérida, R.; et al. Clinical sequencing of soft tissue and bone sarcomas delineates diverse genomic landscapes and potential therapeutic targets. Nat. Commun. 2022, 13, 340. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA, 2020; Available online: http://www.rstudio.com/ (accessed on 29 June 2023).
- Wickham, H.; François, R.; Henry, L.; Müller, K.; Vaughan, D. Dplyr: A Grammar of Data Manipulation. 2023. Available online: https://github.com/tidyverse/dplyr (accessed on 29 June 2023).
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Voutsadakis, I.A. Cell line models for drug discovery in PIK3CA-mutated colorectal cancers. Med. Oncol. 2022, 39, 89. [Google Scholar] [CrossRef]
- Cai, Y.; Yousef, A.; Grandis, J.R.; Johnson, D.E. NSAID therapy for PIK3CA-Altered colorectal, breast, and head and neck cancer. Adv. Biol. Regul. 2020, 75, 100653. [Google Scholar] [CrossRef]
- Chan, L.; Yue, P.Y.; Wong, Y.; Wong, R.N. MicroRNA-15b contributes to ginsenoside-Rg1-induced angiogenesis through increased expression of VEGFR-2. Biochem. Pharmacol. 2013, 86, 392–400. [Google Scholar] [CrossRef]
- Xu, X.; Wu, L.; Zhou, X.; Zhou, N.; Zhuang, Q.; Yang, J.; Dai, J.; Wang, H.; Chen, S.; Mao, W. Cryptotanshinone inhibits VEGF-induced angiogenesis by targeting the VEGFR2 signaling pathway. Microvasc. Res. 2017, 111, 25–31. [Google Scholar] [CrossRef]
- Munchhof, A.M.; Li, F.; White, H.A.; Mead, L.E.; Krier, T.R.; Fenoglio, A.; Li, X.; Yuan, J.; Yang, F.-C.; Ingram, D.A. Neurofibroma-associated growth factors activate a distinct signaling network to alter the function of neurofibromin-deficient endothelial cells. Hum. Mol. Genet. 2006, 15, 1858–1869. [Google Scholar] [CrossRef]
- Issaeva, I.; Zonis, Y.; Rozovskaia, T.; Orlovsky, K.; Croce, C.M.; Nakamura, T.; Mazo, A.; Eisenbach, L.; Canaani, E. Knockdown of ALR (MLL2) Reveals ALR Target Genes and Leads to Alterations in Cell Adhesion and Growth. Mol. Cell. Biol. 2007, 27, 1889–1903. [Google Scholar] [CrossRef] [Green Version]
- Yin, C.; Zhu, B.; Zhang, T.; Liu, T.; Chen, S.; Liu, Y.; Li, X.; Miao, X.; Li, S.; Mi, X.; et al. Pharmacological Targeting of STK19 Inhibits Oncogenic NRAS-Driven Melanomagenesis. Cell 2019, 176, 1113–1127.e16. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Chen, Y.; Liao, E.-Y.; Jiang, Y.; Liu, F.-Y.; Pennypacker, S.D. Phospholipase C-γ1 is required for the epidermal growth factor receptor-induced squamous cell carcinoma cell mitogenesis. Biochem. Biophys. Res. Commun. 2010, 397, 296–300. [Google Scholar] [CrossRef] [Green Version]
- Behjati, S.; Tarpey, P.S.; Sheldon, H.; Martincorena, I.; Van Loo, P.; Gundem, G.; Wedge, D.C.; Ramakrishna, M.; Cooke, S.L.; Pillay, N.; et al. Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat. Genet. 2014, 46, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; La, H.; Lee, E.J.; Choi, H.-J.; Oh, J.; Thang, N.X.; Hong, K. ATP-Dependent Chromatin Remodeler CHD9 Controls the Proliferation of Embryonic Stem Cells in a Cell Culture Condition-Dependent Manner. Biology 2020, 9, 428. [Google Scholar] [CrossRef]
- Bansal, A.; Kaur, M.; Dalal, V. Pleomorphic Sarcoma of Breast: A Report of Two Cases and Review of Literature. Acta Medica Iran. 2017, 55, 272–276. [Google Scholar]
- Kokkali, S.; Moreno, J.D.; Klijanienko, J.; Theocharis, S. Clinical and Molecular Insights of Radiation-Induced Breast Sarcomas: Is There Hope on the Horizon for Effective Treatment of This Aggressive Disease? Int. J. Mol. Sci. 2022, 23, 4125. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Hou, L. Clinicopathologic characteristics of mixed epithelial/mesenchymal metaplastic breast carcinoma (carcinosarcoma): A meta-analysis of Chinese patients. Pol. J. Pathol. 2019, 70, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Yoon, K.; Onyshchenko, M. Sarcoma of the Breast: Clinical Characteristics and Outcomes of 991 Patients from the National Cancer Database. Sarcoma 2021, 2021, 8828158. [Google Scholar] [CrossRef] [PubMed]
- Gambichler, T.; Horny, K.; Mentzel, T.; Stricker, I.; Tannapfel, A.; Scheel, C.H.; Behle, B.; Quast, D.R.; Lee, Y.-P.; Stücker, M.; et al. Undifferentiated pleomorphic sarcoma of the breast with neoplastic fever: Case report and genomic characterization. J. Cancer Res. Clin. Oncol. 2023, 149, 1465–1471. [Google Scholar] [CrossRef]
- Radu, I.; Scripcariu, V.; Panuța, A.; Rusu, A.; Afrăsânie, V.-A.; Cojocaru, E.; Aniței, M.G.; Alexa-Stratulat, T.; Terinte, C.; Șerban, C.F.; et al. Breast Sarcomas—How Different Are They from Breast Carcinomas? Clinical, Pathological, Imaging and Treatment Insights. Diagnostics 2023, 13, 1370. [Google Scholar] [CrossRef]
- Abdou, Y.; Elkhanany, A.; Attwood, K.; Ji, W.; Takabe, K.; Opyrchal, M. Primary and secondary breast angiosarcoma: Single center report and a meta-analysis. Breast Cancer Res. Treat. 2019, 178, 523–533. [Google Scholar] [CrossRef]
- Zghal, M.; Triki, M.; Khanfir, F.; Bouhamed, M.; Mallouli, I.; Louati, D.; Boudawara, T.; Saguem, I. Concomitant primary angiosarcoma and invasive carcinoma of the breast: A case report. Pan Afr. Med. J. 2022, 42, 70. [Google Scholar] [CrossRef]
- Meng, T.; Zhou, Y.; Ye, M.-N.; Wei, J.-J.; Zhao, Q.-F.; Zhang, X.-Y. Primary Highly Differentiated Breast Angiosarcoma in an Adolescent Girl. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- McGowan, T.; Cummings, B.; O’sullivan, B.; Catton, C.; Miller, N.; Panzarella, T. An analysis of 78 breast sarcoma patients without distant metastases at presentation. Int. J. Radiat. Oncol. 2000, 46, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Pollard, S.G.; Marks, P.V.; Temple, L.N.; Thompson, H.H. Breast Sarcoma. A Clinicopathologic Review of 25 Cases. Cancer 1990, 66, 941–944. [Google Scholar] [CrossRef] [PubMed]
- Liberman, L.; Dershaw, D.D.; Kaufman, R.J.; Rosen, P.P. Angiosarcoma of the breast. Radiology 1992, 183, 649–654. [Google Scholar] [CrossRef]
- Smith, T.B.; Gilcrease, M.Z.; Santiago, L.; Hunt, K.K.; Yang, W.T. Imaging Features of Primary Breast Sarcoma. Am. J. Roentgenol. 2012, 198, W386–W393. [Google Scholar] [CrossRef]
- Shabahang, M.; Franceschi, D.; Sundaram, M.; Castillo, M.H.; Moffat, F.L.; Frank, D.S.; Rosenberg, E.R.; Bullock, K.E.; Livingstone, A.S. Surgical management of primary breast sarcoma. Am. Surg. 2002, 68, 673–677; discussion 677. [Google Scholar] [CrossRef]
- Yang, W.T.; Hennessy, B.T.J.; Dryden, M.J.; Valero, V.; Hunt, K.K.; Krishnamurthy, S. Mammary Angiosarcomas: Imaging Findings in 24 Patients. Radiology 2007, 242, 725–734. [Google Scholar] [CrossRef]
- Elson, B.C.; Ikeda, D.M.; Andersson, I.; Wattsgård, C.; Elson, D.M.I.B.C.; Wurdinger, S.; Herzog, A.B.; Fischer, D.R.; Marx, C.; Raabe, G.; et al. Fibrosarcoma of the breast: Mammographic findings in five cases. Am. J. Roentgenol. 1992, 158, 993–995. [Google Scholar] [CrossRef] [Green Version]
- McGregor, G.I.; Knowling, M.A.; Este, F.A. Sarcoma and cystosarcoma phyllodes tumors of the breast—A retrospective review of 58 cases. Am. J. Surg. 1994, 167, 477–480. [Google Scholar] [CrossRef]
- Foxcroft, L.; Evans, E.; Porter, A. Difficulties in the pre-operative diagnosis of phyllodes tumours of the breast: A study of 84 cases. Breast 2007, 16, 27–37. [Google Scholar] [CrossRef]
- Lim, S.Z.; Ong, K.W.; Tan, B.K.T.; Selvarajan, S.; Tan, P.H. Sarcoma of the breast: An update on a rare entity. J. Clin. Pathol. 2016, 69, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Osman, M.H.; Rabie, N.A.; Elmehrath, A.O.; Bedair, H.M.; Fala, S.Y.; Ghaith, H.S.; Refaat, M.A. Primary and Secondary Breast Sarcoma: Clinical and Pathological Characteristics, Prognostic Factors, and Nomograms for Predicting Survival. Clin. Breast Cancer 2022, 22, e753–e763. [Google Scholar] [CrossRef] [PubMed]
- Hui, A.; Henderson, M.; Speakman, D.; Skandarajah, A. Angiosarcoma of the breast: A difficult surgical challenge. Breast 2012, 21, 584–589. [Google Scholar] [CrossRef]
- Scow, J.S.; Reynolds, C.A.; Degnim, A.C.; Petersen, I.A.; Jakub, J.W.; Boughey, J.C. Primary and secondary angiosarcoma of the breast: The Mayo Clinic experience. J. Surg. Oncol. 2010, 101, 401–407. [Google Scholar] [CrossRef]
- Yin, M.; Mackley, H.B.; Drabick, J.J.; Harvey, H.A. Primary female breast sarcoma: Clinicopathological features, treatment and prognosis. Sci. Rep. 2016, 6, 31497. [Google Scholar] [CrossRef]
- Gronchi, A.; Miah, A.B.; Dei Tos, A.; Abecassis, N.; Bajpai, J.; Bauer, S.; Biagini, R.; Bielack, S.; Blay, J.Y.; Bolle, S.; et al. Soft tissue and visceral sarcomas: ESMO–EURACAN–GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2021, 32, 1348–1365. [Google Scholar] [CrossRef]
- Wiklund, T.; Huuhtanen, R.; Blomqvist, C.; Tukiainen, E.; Virolainen, M.; Virkkunen, P.; Asko-Seljavaara, S.; Björkenheim, J.; Elomaa, I. The importance of a multidisciplinary group in the treatment of soft tissue sarcomas. Eur. J. Cancer 1996, 32, 269–273. [Google Scholar] [CrossRef]
- Pandey, M.; Mathew, A.; Abraham, E.K.; Rajan, B. Primary sarcoma of the breast. J. Surg. Oncol. 2004, 87, 121–125. [Google Scholar] [CrossRef]
- North, J.H.; McPhee, M.; Arredondo, M.; Edge, S.B. Sarcoma of the breast: Implications of the extent of local therapy. Am. Surg. 1998, 64, 1059–1061. [Google Scholar]
- Gullett, N.P.; Delman, K.; Folpe, A.L.; Johnstone, P.A.S. National Surgical Patterns of Care: Regional Lymphadenectomy of Breast Sarcomas. Am. J. Clin. Oncol. 2007, 30, 461–465. [Google Scholar] [CrossRef]
- Blanchard, D.; Reynolds, C.A.; Grant, C.S.; Donohue, J.H. Primary nonphylloides breast sarcomas. Am. J. Surg. 2003, 186, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.P.; Kinne, D.W. BREAST SARCOMA. Surg. Clin. N. Am. 1996, 76, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Barrow, B.J.; Janjan, N.A.; Gutman, H.; Benjamin, R.S.; Allen, P.; Romsdahl, M.M.; Ross, M.I.; Pollock, R.E. Role of radiotherapy in sarcoma of the breast—A retrospective review of the M.D. Anderson experience. Radiother. Oncol. 1999, 52, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, P.A.; Pierce, L.J.; Merino, M.J.; Yang, J.C.; Epstein, A.H.; Delaney, T.F. Primary soft tissue sarcomas of the breast: Local-regional control with post-operative radiotherapy. Int. J. Radiat. Oncol. 1993, 27, 671–675. [Google Scholar] [CrossRef]
- Fury, M.G.; Antonescu, C.R.; Van Zee, K.J.; Brennan, M.E.; Maki, R.G. A 14-Year Retrospective Review of Angiosarcoma. Cancer J. 2005, 11, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Jallali, N.; James, S.; Searle, A.; Ghattaura, A.; Hayes, A.; Harris, P. Surgical management of radiation-induced angiosarcoma after breast conservation therapy. Am. J. Surg. 2012, 203, 156–161. [Google Scholar] [CrossRef]
- Seinen, J.M.; Styring, E.; Verstappen, V.; von Steyern, F.V.; Rydholm, A.; Suurmeijer, A.J.H.; Hoekstra, H.J. Radiation-Associated Angiosarcoma After Breast Cancer: High Recurrence Rate and Poor Survival Despite Surgical Treatment with R0 Resection. Ann. Surg. Oncol. 2012, 19, 2700–2706. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-J.; Ryu, J.-M.; Lee, S.-K.; Chae, B.-J.; Kim, S.-W.; Nam, S.-J.; Yu, J.-H.; Lee, J.-E. Primary Angiosarcoma of the Breast: A Single-Center Retrospective Study in Korea. Curr. Oncol. 2022, 29, 3272–3281. [Google Scholar] [CrossRef]
- Modesto, A.; Filleron, T.; Chevreau, C.; Le Pechoux, C.; Rochaix, P.; Le Guellec, S.; Ducassou, A.; Gangloff, D.; Ferron, G.; Delannes, M. Role of radiation therapy in the conservative management of sarcoma within an irradiated field. Eur. J. Surg. Oncol. 2014, 40, 187–192. [Google Scholar] [CrossRef]
- McClelland, S.; Hatfield, J.; Degnin, C.; Chen, Y.; Mitin, T. Extent of resection and role of adjuvant treatment in resected localized breast angiosarcoma. Breast Cancer Res. Treat. 2019, 175, 409–418. [Google Scholar] [CrossRef]
- Young, R.J.; Brown, N.J.; Reed, M.W.; Hughes, D.; Woll, P.J. Angiosarcoma. Lancet Oncol. 2010, 11, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Florou, V.; Wilky, B.A. Current and Future Directions for Angiosarcoma Therapy. Curr. Treat. Options Oncol. 2018, 19, 14. [Google Scholar] [CrossRef]
- Young, R.; Natukunda, A.; Litière, S.; Woll, P.; Wardelmann, E.; van der Graaf, W. First-line anthracycline-based chemotherapy for angiosarcoma and other soft tissue sarcoma subtypes: Pooled analysis of eleven European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group trials. Eur. J. Cancer 2014, 50, 3178–3186. [Google Scholar] [CrossRef] [PubMed]
- Sher, T.; Hennessy, B.T.; Valero, V.; Broglio, K.; Woodward, W.A.; Trent, J.; Hunt, K.K.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Primary angiosarcomas of the breast. Cancer 2007, 110, 173–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penel, N.; Bui, B.N.; Bay, J.-O.; Cupissol, D.; Ray-Coquard, I.; Piperno-Neumann, S.; Kerbrat, P.; Fournier, C.; Taieb, S.; Jimenez, M.; et al. Phase II Trial of Weekly Paclitaxel for Unresectable Angiosarcoma: The ANGIOTAX Study. J. Clin. Oncol. 2008, 26, 5269–5274. [Google Scholar] [CrossRef]
- Cao, J.; Wang, J.; He, C.; Fang, M. Angiosarcoma: A review of diagnosis and current treatment. Am. J. Cancer Res. 2019, 9, 2303–2313. [Google Scholar]
- van der Graaf, W.T.; Blay, J.-Y.; Chawla, S.P.; Kim, D.-W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Ravi, V.; Sanford, E.M.; Wang, W.-L.; Ross, J.S.; Ramesh, N.; Futreal, A.; Patel, S.; Stephens, P.J.; Miller, V.A.; Ali, S.M. Antitumor Response of VEGFR2- and VEGFR3-Amplified Angiosarcoma to Pazopanib. J. Natl. Compr. Cancer Netw. 2016, 14, 499–502. [Google Scholar] [CrossRef]
- Kollár, A.; Jones, R.L.; Stacchiotti, S.; Gelderblom, H.; Guida, M.; Grignani, G.; Steeghs, N.; Safwat, A.; Katz, D.; Duffaud, F.; et al. Pazopanib in advanced vascular sarcomas: An EORTC Soft Tissue and Bone Sarcoma Group (STBSG) retrospective analysis. Acta Oncol. 2017, 56, 88–92. [Google Scholar] [CrossRef] [Green Version]
- Maki, R.G.; D’Adamo, D.R.; Keohan, M.L.; Saulle, M.; Schuetze, S.M.; Undevia, S.D.; Livingston, M.B.; Cooney, M.M.; Hensley, M.L.; Mita, M.M.; et al. Phase II Study of Sorafenib in Patients with Metastatic or Recurrent Sarcomas. J. Clin. Oncol. 2009, 27, 3133–3140. [Google Scholar] [CrossRef] [Green Version]
- Penel, N.; Ray-Coquard, I.; Bal-Mahieu, C.; Chevreau, C.; Le Cesne, A.; Italiano, A.; Bompas, E.; Clisant, S.; Baldeyrou, B.; Lansiaux, A.; et al. Low level of baseline circulating VEGF-A is associated with better outcome in patients with vascular sarcomas receiving sorafenib: An ancillary study from a phase II trial. Target. Oncol. 2014, 9, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Kharel, S.; Shrestha, A.K.; Khadayat, R.; Singh, M.; Shakya, P. Primary dedifferentiated liposarcoma of the breast: A case report. Clin. Case Rep. 2022, 10, e6275. [Google Scholar] [CrossRef] [PubMed]
- Austin, R.M.; Dupree, W.B. Liposarcoma of the breast: A clinicopathologic study of 20 cases. Hum. Pathol. 1986, 17, 906–913. [Google Scholar] [CrossRef]
- Ilyas, M.I.M.; Nazir, S.; Xiao, P.Q. Breast Leiomyosarcoma: A Systematic Review and Recommendations for Management. Int. Surg. 2019, 104, 196–202. [Google Scholar] [CrossRef]
- Cheikh, T.E.; Hamza, K.; Hicham, B.; Fatiha, E.M.; Hajar, E.O.; Mustapha, B.; Mohamed, E.; Mohamed, E. Leiomyosarcoma of the male breast: Case report. Ann. Med. Surg. 2021, 67, 102495. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Kimura, R.; Yamamura, J.; Akazawa, K.; Kasugai, T.; Tsukamoto, F. Leiomyosarcoma of the breast: A case report and review of the literature about therapeutic management. Breast 2011, 20, 389–393. [Google Scholar] [CrossRef]
- Mance, M.; Smuđ-Orehovec, S.; Vrbanović-Mijatović, V.; Mijatović, D. Primary Alveolar Rhabdomyosarcoma of the Breast in a 17-Year-Old Girl. JCO Oncol. Pract. 2020, 16, 93–95. [Google Scholar] [CrossRef]
- Bayramoglu, Z.; Kebudi, R.; Yilmaz, R.; Bay, S.B.; Kebudi, A.; Karanlik, H.; Iribas, A.; Darendeliler, E.; Onder, S.; Bilgic, B.; et al. Primary Rhabdomyosarcoma of the Breast: Imaging Findings and Literature Review. Breast Care 2018, 13, 293–297. [Google Scholar] [CrossRef]
- Hays, D.M.; Donaldson, S.S.; Shimada, H.; Crist, W.M.; Newton, W.A.; Andrassy, R.J.; Wiener, E.; Green, J.; Triche, T.; Maurer, H.M. Primary and Metastatic Rhabdomyosarcoma in the Breast: Neoplasms of Adolescent Females, a Report from the Intergroup Rhabdomyosarcoma Study. Med. Pediatr. Oncol. 1997, 29, 181–189. [Google Scholar] [CrossRef]
- Amadu, A.M.; Soro, D.; Marras, V.; Satta, G.; Crivelli, P.; Conti, M.; Meloni, G.B. Primary breast chondrosarcoma: Imaging and pathological findings. Eur. J. Radiol. Open 2017, 4, 138–140. [Google Scholar] [CrossRef]
- Limaiem, F.; Kashyap, S. Phyllodes Tumor of the Breast. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Zhang, Y.; Kleer, C.G. Phyllodes Tumor of the Breast: Histopathologic Features, Differential Diagnosis, and Molecular/Genetic Updates. Arch. Pathol. Lab. Med. 2016, 140, 665–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, X.; Chen, K.; Zeng, J.; Bi, Z.; Guo, M.; Chen, Y.; Yao, Y.; Wu, W.; Liang, S.; Nie, Y. Adjuvant radiotherapy and chemotherapy for patients with breast phyllodes tumors: A systematic review and meta-analysis. BMC Cancer 2019, 19, 372. [Google Scholar] [CrossRef] [PubMed]
- Bielsen, V.T.; Andreasen, C. Phyllodes Tumour of the Male Breast. Histopathology 1987, 11, 761–762. [Google Scholar] [CrossRef]
- Mishra, S.P.; Tiwary, S.K.; Mishra, M.; Khanna, A.K. Phyllodes Tumor of Breast: A Review Article. ISRN Surg. 2013, 2013, 361469. [Google Scholar] [CrossRef] [Green Version]
- Mitus, J.; Blecharz, P.; Jakubowicz, J.; Reinfuss, M.; Walasek, T.; Wysocki, W. Phyllodes tumors of the breast. The treatment results for 340 patients from a single cancer centre. Breast 2019, 43, 85–90. [Google Scholar] [CrossRef]
- Matei, R.-A.; Mehedinţu-Ionescu, M.; Paitici, S.; Georgescu, E.F.; Donoiu, A.; Ghemigian, A.M.; Popescu, M.; Totolici, B.D.; Neamţu, C.; Mogoantă, S. Diagnostic difficulties in giant benign phyllodes tumor. Rom. J. Morphol. Embryol. 2022, 62, 1035–1044. [Google Scholar] [CrossRef]
- Laé, M.; Vincent-Salomon, A.; Savignoni, A.; Huon, I.; Fréneaux, P.; Sigal-Zafrani, B.; Aurias, A.; Sastre-Garau, X.; Couturier, J. Phyllodes tumors of the breast segregate in two groups according to genetic criteria. Mod. Pathol. 2007, 20, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Yonemori, K.; Hasegawa, T.; Shimizu, C.; Shibata, T.; Matsumoto, K.; Kouno, T.; Ando, M.; Katsumata, N.; Fujiwara, Y. Correlation of p53 and MIB-1 expression with both the systemic recurrence and survival in cases of phyllodes tumors of the breast. Pathol.-Res. Pract. 2006, 202, 705–712. [Google Scholar] [CrossRef]
- Tan, W.J.; Thike, A.A.; Bay, B.H.; Tan, P.H. Immunohistochemical expression of homeoproteins Six1 and Pax3 in breast phyllodes tumours correlates with histological grade and clinical outcome. Histopathology 2014, 64, 807–817. [Google Scholar] [CrossRef]
- Tse, G.M.; Lee, C.S.; Kung, F.Y.; Scolyer, R.A.; Law, B.K.; Lau, T.-S.; Putti, T.C. Hormonal Receptors Expression in Epithelial Cells of Mammary Phyllodes Tumors Correlates with Pathologic Grade of the Tumor. Am. J. Clin. Pathol. 2002, 118, 522–526. [Google Scholar] [CrossRef]
- Desmoid Tumor-Symptoms, Causes, Treatment|NORD. Available online: https://rarediseases.org/rare-diseases/desmoid-tumor/ (accessed on 20 July 2023).
- Lorenzen, J.; Cramer, M.; Buck, N.; Friedrichs, K.; Graubner, K.; Lühr, C.S.; Lindner, C.; Niendorf, A. Desmoid Type Fibromatosis of the Breast: Ten-Year Institutional Results of Imaging, Histopathology, and Surgery. Breast Care 2020, 16, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Federman, N. Molecular pathogenesis of desmoid tumor and the role of γ-secretase inhibition. NPJ Precis. Oncol. 2022, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Hennuy, C.; Defrère, P.; Maweja, S.; Thiry, A.; Gennigens, C. Bilateral breast desmoid-type fibromatosis, case report and literature review. Gland. Surg. 2022, 11, 1832–1841. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.M.; Mahoney, M.R.; Van Tine, B.A.; Ravi, V.; Attia, S.; Deshpande, H.A.; Gupta, A.A.; Milhem, M.; Conry, R.M.; Movva, S.; et al. Sorafenib for Advanced and Refractory Desmoid Tumors. N. Engl. J. Med. 2018, 379, 2417–2428. [Google Scholar] [CrossRef]
- Kasper, B.; Gruenwald, V.; Reichardt, P.; Bauer, S.; Rauch, G.; Limprecht, R.; Sommer, M.; Dimitrakopoulou-Strauss, A.; Pilz, L.; Haller, F.; et al. Imatinib induces sustained progression arrest in RECIST progressive desmoid tumours: Final results of a phase II study of the German Interdisciplinary Sarcoma Group (GISG). Eur. J. Cancer 2017, 76, 60–67. [Google Scholar] [CrossRef]
- Toulmonde, M.; Pulido, M.; Ray-Coquard, I.; Andre, T.; Isambert, N.; Chevreau, C.; Penel, N.; Bompas, E.; Saada, E.; Bertucci, F.; et al. Pazopanib or methotrexate–vinblastine combination chemotherapy in adult patients with progressive desmoid tumours (DESMOPAZ): A non-comparative, randomised, open-label, multicentre, phase 2 study. Lancet Oncol. 2019, 20, 1263–1272. [Google Scholar] [CrossRef]
- Gounder, M.; Ratan, R.; Alcindor, T.; Schöffski, P.; van der Graaf, W.T.; Wilky, B.A.; Riedel, R.F.; Lim, A.; Smith, L.M.; Moody, S.; et al. Nirogacestat, a γ-Secretase Inhibitor for Desmoid Tumors. N. Engl. J. Med. 2023, 388, 898–912. [Google Scholar] [CrossRef]
- Gounder, M.; Jones, R.; Chugh, R.; Agulnik, M.; Singh, A.; Van Tine, B.; Andelkovic, V.; Choy, E.; Lewin, J.; Ratan, R.; et al. 1488MO Initial results of phase II/III trial of AL102 for treatment of desmoid tumors (DT). Ann. Oncol. 2022, 33, S1227–S1228. [Google Scholar] [CrossRef]
- Duazo-Cassin, L.; Le Guellec, S.; Lusque, A.; Chantalat, E.; Laé, M.; Terrier, P.; Coindre, J.-M.; Boulet, B.; Le Boulc’h, M.; Gangloff, D.; et al. Breast desmoid tumor management in France: Toward a new strategy. Breast Cancer Res. Treat. 2019, 176, 329–335. [Google Scholar] [CrossRef]
- Povoski, S.P.; Marsh, W.L.; Spigos, D.G.; Abbas, A.E.; Buchele, B.A. Management of a patient with multiple recurrences of fibromatosis (desmoid tumor) of the breast involving the chest wall musculature. World J. Surg. Oncol. 2006, 4, 32. [Google Scholar] [CrossRef] [Green Version]
- Laakom, O.; Bergaoui, H.; Ben Hammouda, S.; Khalfalli, A.; Njim, L.; Faleh, R. Fibromatose desmoïde mammaire: À propos de deux cas et revue de la littérature. Pan Afr. Med. J. 2022, 41, 184. [Google Scholar] [CrossRef] [PubMed]
- Neuman, H.B.; Brogi, E.; Ebrahim, A.; Brennan, M.F.; Van Zee, K.J. Desmoid Tumors (Fibromatoses) of the Breast: A 25-Year Experience. Ann. Surg. Oncol. 2007, 15, 274–280. [Google Scholar] [CrossRef]
- Salas, S.; Dufresne, A.; Bui, B.; Blay, J.-Y.; Terrier, P.; Ranchere-Vince, D.; Bonvalot, S.; Stoeckle, E.; Guillou, L.; Le Cesne, A.; et al. Prognostic Factors Influencing Progression-Free Survival Determined from a Series of Sporadic Desmoid Tumors: A Wait-and-See Policy According to Tumor Presentation. J. Clin. Oncol. 2011, 29, 3553–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, J.T.; DeLaney, T.F.; Kobayashi, W.K.; Szymonifka, J.; Yeap, B.Y.; Chen, Y.-L.; Rosenberg, A.E.; Harmon, D.C.; Choy, E.; Yoon, S.S.; et al. Desmoid Tumor: Analysis of Prognostic Factors and Outcomes in a Surgical Series. Ann. Surg. Oncol. 2012, 19, 4028–4035. [Google Scholar] [CrossRef]
- Ballo, M.T.; Zagars, G.K.; Pollack, A.; Pisters, P.W.; Pollock, R.A. Desmoid Tumor: Prognostic Factors and Outcome After Surgery, Radiation Therapy, or Combined Surgery and Radiation Therapy. J. Clin. Oncol. 1999, 17, 158. [Google Scholar] [CrossRef] [PubMed]
- Merchant, N.B.; Lewis, J.J.; Woodruff, J.M.; Leung, D.H.; Brennan, M.F. Extremity and trunk desmoid tumors: A multifactorial analysis of outcome. Cancer 1999, 86, 2045–2052. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Initiation Age | Frequency | |
|---|---|---|
| National Comprehensive Cancer Network (NCCN) | 40 years old (yo) | Annual |
| American Cancer Society (ACS) | 40–44 yo: “Qualified” 45 yo: Strong | Annual: age 40–54 yo Biennial or annual option: age > 54 yo |
| US Preventive Services Task Force (USPSTF) | 50 yo: Grade B 40–49 yo: Grade C | Biennial |
| European Breast Cancer Guidelines | 45–49 yo 50–69 yo 70–74 yo | Screening every 2–3 years Screening every 2 years Screening every 4 years |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esperança-Martins, M.; Melo-Alvim, C.; Dâmaso, S.; Lopes-Brás, R.; Peniche, T.; Nogueira-Costa, G.; Abreu, C.; Luna Pais, H.; de Sousa, R.T.; Torres, S.; et al. Breast Sarcomas, Phyllodes Tumors, and Desmoid Tumors: Turning the Magnifying Glass on Rare and Aggressive Entities. Cancers 2023, 15, 3933. https://doi.org/10.3390/cancers15153933
Esperança-Martins M, Melo-Alvim C, Dâmaso S, Lopes-Brás R, Peniche T, Nogueira-Costa G, Abreu C, Luna Pais H, de Sousa RT, Torres S, et al. Breast Sarcomas, Phyllodes Tumors, and Desmoid Tumors: Turning the Magnifying Glass on Rare and Aggressive Entities. Cancers. 2023; 15(15):3933. https://doi.org/10.3390/cancers15153933
Chicago/Turabian StyleEsperança-Martins, Miguel, Cecília Melo-Alvim, Sara Dâmaso, Raquel Lopes-Brás, Tânia Peniche, Gonçalo Nogueira-Costa, Catarina Abreu, Helena Luna Pais, Rita Teixeira de Sousa, Sofia Torres, and et al. 2023. "Breast Sarcomas, Phyllodes Tumors, and Desmoid Tumors: Turning the Magnifying Glass on Rare and Aggressive Entities" Cancers 15, no. 15: 3933. https://doi.org/10.3390/cancers15153933
APA StyleEsperança-Martins, M., Melo-Alvim, C., Dâmaso, S., Lopes-Brás, R., Peniche, T., Nogueira-Costa, G., Abreu, C., Luna Pais, H., de Sousa, R. T., Torres, S., Gallego-Paez, L. M., Martins, M., Ribeiro, L., & Costa, L. (2023). Breast Sarcomas, Phyllodes Tumors, and Desmoid Tumors: Turning the Magnifying Glass on Rare and Aggressive Entities. Cancers, 15(15), 3933. https://doi.org/10.3390/cancers15153933