
High-Throughput Drug Screening Revealed That Ciclopirox Olamine Can Engender Gastric Cancer Stem-like Cells

 , ,
, ,  , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. High-Throughput Screening of the Prestwick Chemical Library
2.3. Assessment of Ciclopirox Activity and Cytotoxicity
2.4. Protein Extraction and Western Blot
2.5. RNA Extraction and Real-Time PCR
2.6. Evaluation of Cobalt Chloride (CoCl2) Activity and Cytotoxicity
2.7. Clonogenic Assay
2.8. Seahorse Real-Time ATP Rate Assay
2.9. Senescence-Associated β-Galactosidase (SA-β-gal) Assay
2.10. Cell Proliferation Assay
2.11. Statistical Analysis
3. Results
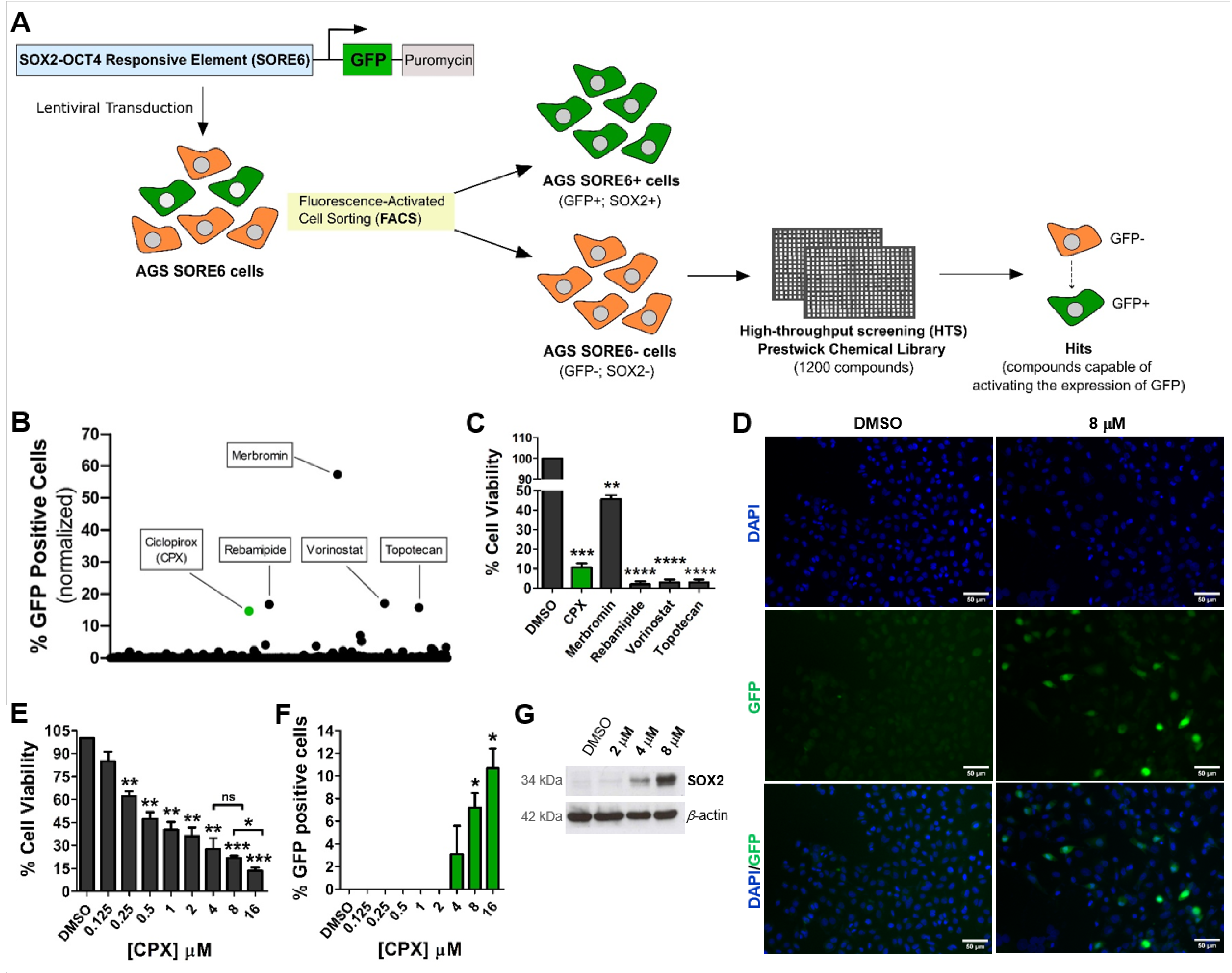
3.1. CPX Activates the SORE6−GFP Reporter System Embedded in Gastric Non-CSCs
3.2. Activation of the SORE6-GFP Reporter System via CPX Seems to Be Strongly Associated with Increased SOX2 Expression
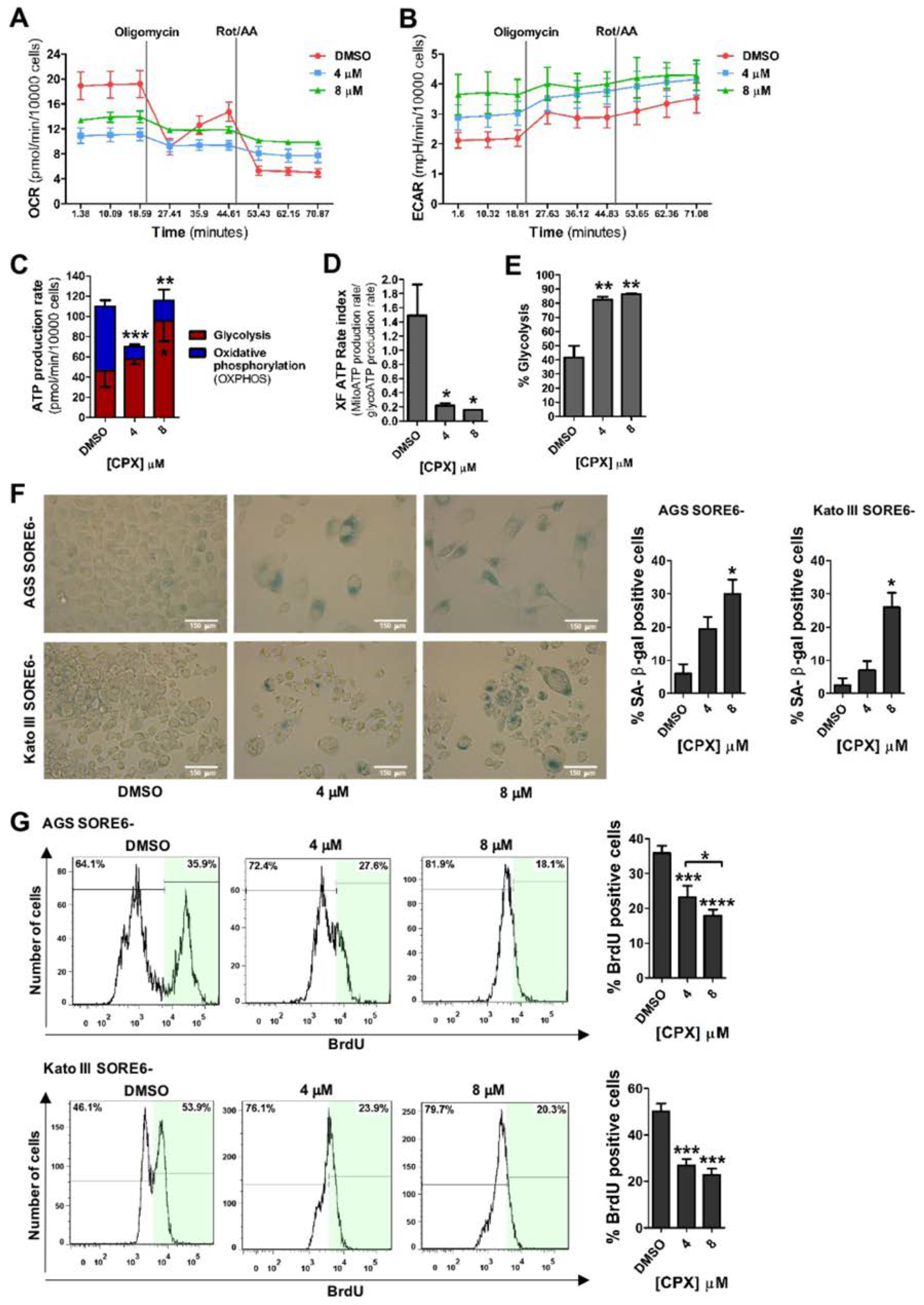
3.3. CPX Promotes a Metabolic Shift from Oxidative Phosphorylation (OXPHOS) to Glycolysis and Induces Cellular Senescence
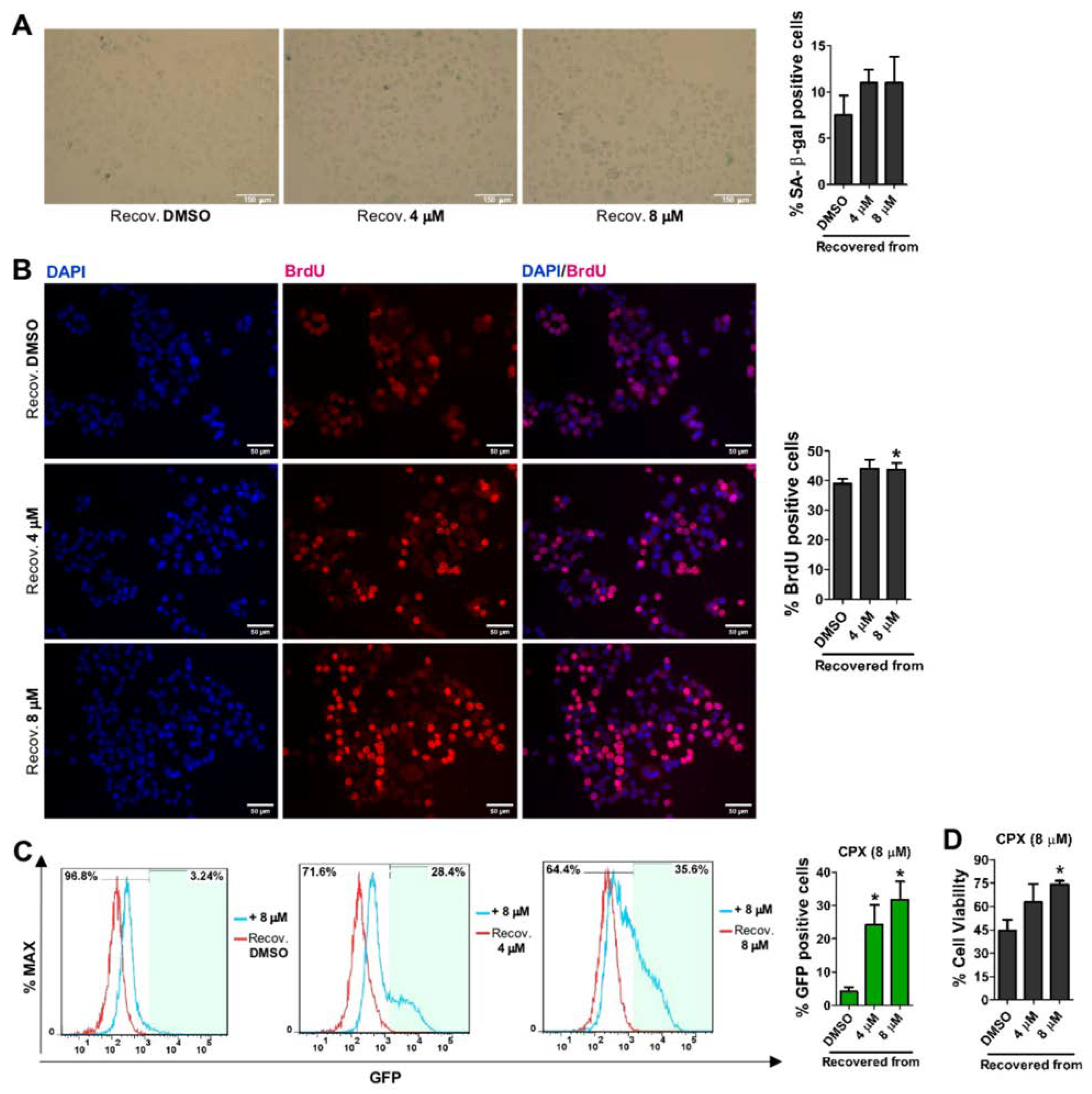
3.4. CPX-Induced Cellular Senescence Is Reversible and Recovered Cells Become More Resistant to CPX and More Prone to SORE6−GFP Reporter System Re-Activation
3.5. Activation of the Reporter System SORE6-GFP by CPX Has a Mechanism Similar to That of the Hypoxia Mimetic CoCl2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-M.; Zhang, J.-G.; Zhang, X.; Li, Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-R.; Wang, S.; Yan, S.-C.; Zhang, Y.; Nelson, P.J.; Jia, H.-L.; Qin, L.-X.; Dong, Q.-Z. Therapeutic Strategies Targeting Cancer Stem Cells and Their Microenvironment. Front. Oncol. 2019, 9, 1104. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Zhang, C.; Luo, H.; Zhang, J.; Zhuang, Z.; Liang, Z.; Wu, X. Targeting Gastric Cancer Stem Cells to Enhance Treatment Response. Cells 2022, 11, 2828. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of Gastric Cancer Stem Cells Using the Cell Surface Marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef]
- Pádua, D.; Barros, R.; Amaral, A.L.; Mesquita, P.; Freire, A.F.; Sousa, M.; Maia, A.F.; Caiado, I.; Fernandes, H.; Pombinho, A.; et al. A SOX2 Reporter System Identifies Gastric Cancer Stem-Like Cells Sensitive to Monensin. Cancers 2020, 12, 495. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal Transduct. Target. Ther. 2020, 5, 228. [Google Scholar] [CrossRef]
- Tang, B.; Raviv, A.; Esposito, D.; Flanders, K.C.; Daniel, C.; Nghiem, B.T.; Garfield, S.; Lim, L.; Mannan, P.; Robles, A.I.; et al. A Flexible Reporter System for Direct Observation and Isolation of Cancer Stem Cells. Stem Cell Rep. 2015, 4, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Feng, Y.; Cheng, L. Cellular Reprogramming as a Therapeutic Target in Cancer. Trends Cell Biol. 2019, 29, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Lin, Y.-C.; Nakamura, Y.; Eckner, R.; Wuputra, K.; Kuo, K.-K.; Lin, C.-S.; Yokoyama, K.K. Potential application of cell reprogramming techniques for cancer research. Cell. Mol. Life Sci. 2019, 76, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Ichida, J.K.; Blanchard, J.; Lam, K.; Son, E.Y.; Chung, J.E.; Egli, D.; Loh, K.M.; Carter, A.C.; Di Giorgio, F.P.; Koszka, K.; et al. A Small-Molecule Inhibitor of Tgf-β Signaling Replaces Sox2 in Reprogramming by Inducing Nanog. Cell Stem Cell 2009, 5, 491–503. [Google Scholar] [CrossRef]
- Kim, Y.; Jeong, J.; Choi, D. Small-molecule-mediated reprogramming: A silver lining for regenerative medicine. Exp. Mol. Med. 2020, 52, 213–226. [Google Scholar] [CrossRef]
- Huangfu, D.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Snitow, M.; Chen, A.E.; Melton, D.A. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 2008, 26, 795–797. [Google Scholar] [CrossRef]
- Mai, T.T.; Hamaï, A.; Hienzsch, A.; Cañeque, T.; Müller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2017, 9, 1025–1033. [Google Scholar] [CrossRef]
- Naujokat, C.; Steinhart, R. Salinomycin as a Drug for Targeting Human Cancer Stem Cells. J. Biomed. Biotechnol. 2012, 2012, 950658. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D.; DeWitt, N. In Vivo Cellular Reprogramming: The Next Generation. Cell 2016, 166, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.-E.; Hong, J.S.; Choi, H.-D.; Kim, Y.-R.; Baik, M.-Y.; Kim, H.-S. Impact of starch granule-associated channel protein on characteristic of and λ-carrageenan entrapment within wheat starch granules. Int. J. Biol. Macromol. 2021, 174, 440–448. [Google Scholar] [CrossRef]
- Kaur, J.; Bhattacharyya, S. Cancer Stem Cells: Metabolic Characterization for Targeted Cancer Therapy. Front. Oncol. 2021, 11, 756888. [Google Scholar] [CrossRef]
- Herrmann, A.L.; Kuhn, B.J.; Holzer, A.; Krijgsveld, J.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Delineating the Switch between Senescence and Apoptosis in Cervical Cancer Cells under Ciclopirox Treatment. Cancers 2021, 13, 4995. [Google Scholar] [CrossRef]
- Braun, J.A.; Herrmann, A.L.; Blase, J.I.; Frensemeier, K.; Bulkescher, J.; Scheffner, M.; Galy, B.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Effects of the antifungal agent ciclopirox in HPV-positive cancer cells: Repression of viral E6/E7 oncogene expression and induction of senescence and apoptosis. Int. J. Cancer 2020, 146, 461–474. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. From Ancient Pathways to Aging Cells—Connecting Metabolism and Cellular Senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef]
- Santos-De-Frutos, K.; Djouder, N. When dormancy fuels tumour relapse. Commun. Biol. 2021, 4, 747. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Li, H.; Huang, R.; Mo, D.; Zeng, T.; Fang, M.; Li, M. Clinicopathological and Prognostic Significance of Cancer Stem Cell Markers in Ovarian Cancer Patients: Evidence from 52 Studies. Cell. Physiol. Biochem. 2018, 46, 1716–1726. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.H.; Ro, E.J.; Yoon, J.S.; Mizutani, T.; Kang, D.W.; Park, J.C.; Il Kim, T.; Clevers, H.; Choi, K.Y. 5-FU promotes stemness of colorectal cancer via p53-mediated WNT/β-catenin pathway activation. Nat. Commun. 2020, 11, 5321. [Google Scholar] [CrossRef] [PubMed]
- Baisiwala, S.; Hall, R.R., 3rd; Saathoff, M.R.; Shireman, J.M.; Park, C.; Budhiraja, S.; Goel, C.; Warnke, L.; Hardiman, C.; Wang, J.Y.; et al. LNX1 Modulates Notch1 Signaling to Promote Expansion of the Glioma Stem Cell Population during Temozolomide Therapy in Glioblastoma. Cancers 2020, 12, 3505. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic Reprogramming in Cancer. Science 2013, 339, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and reprogramming: Origins of cancer stem cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef]
- Hochedlinger, K.; Jaenisch, R. Induced Pluripotency and Epigenetic Reprogramming. Cold Spring Harb. Perspect. Biol. 2015, 7, a019448. [Google Scholar] [CrossRef]
- Muñoz, P.; Iliou, M.S.; Esteller, M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol. Oncol. 2012, 6, 620–636. [Google Scholar] [CrossRef]
- Gupta, A.K.; Plott, T. Ciclopirox: A broad-spectrum antifungal with antibacterial and anti-inflammatory properties. Int. J. Dermatol. 2004, 43 (Suppl. S1), 3–8. [Google Scholar] [CrossRef]
- Schmeel, L.C.; Schmeel, F.C.; Kim, Y.; Endo, T.; Lu, D.; Schmidt-Wolf, I.G.H. Targeting the Wnt/beta-catenin pathway in multiple myeloma. Anticancer Res. 2013, 33, 4719–4726. [Google Scholar]
- Sen, S.; Hassane, D.C.; Corbett, C.; Becker, M.W.; Jordan, C.T.; Guzman, M.L. Novel mTOR inhibitory activity of ciclopirox enhances parthenolide antileukemia activity. Exp. Hematol. 2013, 41, 799–807. [Google Scholar] [CrossRef]
- Zhou, H.; Shen, T.; Luo, Y.; Liu, L.; Chen, W.; Xu, B.; Han, X.; Pang, J.; Rivera, C.A.; Huang, S. The antitumor activity of the fungicide ciclopirox. Int. J. Cancer 2010, 127, 2467–2477. [Google Scholar] [CrossRef] [PubMed]
- Mémin, E.; Hoque, M.; Jain, M.R.; Heller, D.S.; Li, H.; Cracchiolo, B.; Hanauske-Abel, H.M.; Pe’Ery, T.; Mathews, M.B. Blocking eIF5A Modification in Cervical Cancer Cells Alters the Expression of Cancer-Related Genes and Suppresses Cell Proliferation. Cancer Res. 2014, 74, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Zhou, N.; Li, L.; Mo, S.; Zhou, Y.; Deng, Y.; Chen, T.; Shan, C.; Chen, Q.; Lu, B. Correction: Ciclopirox activates PERK-dependent endoplasmic reticulum stress to drive cell death in colorectal cancer. Cell Death Dis. 2020, 11, 719. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Che, G.; Jiang, K.; Zhou, Z.; Wu, L.; Xu, M.; Liu, J.; Yan, S. Ciclopirox Olamine Exerts Tumor-Suppressor Effects via Topoisomerase II Alpha in Lung Adenocarcinoma. Front. Oncol. 2022, 12, 791916. [Google Scholar] [CrossRef]
- Huang, Z.; Huang, S. Reposition of the Fungicide Ciclopirox for Cancer Treatment. Recent Patents Anti-Cancer Drug Discov. 2021, 16, 122–135. [Google Scholar] [CrossRef]
- Minden, M.D.; Hogge, D.E.; Weir, S.J.; Kasper, J.; Webster, D.A.; Patton, L.; Jitkova, Y.; Hurren, R.; Gronda, M.; Goard, C.A.; et al. Oral ciclopirox olamine displays biological activity in a phase I study in patients with advanced hematologic malignancies. Am. J. Hematol. 2014, 89, 363–368. [Google Scholar] [CrossRef]
- Milanovic, M.; Yu, Y.; Schmitt, C.A. The Senescence–Stemness Alliance–A Cancer-Hijacked Regeneration Principle. Trends Cell Biol. 2018, 28, 1049–1061. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, X. Radiochemotherapy-induced DNA repair promotes the biogenesis of gastric cancer stem cells. Stem Cell Res. Ther. 2022, 13, 481. [Google Scholar] [CrossRef]
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. [Google Scholar] [CrossRef]
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.; Battistini, L.; Blandino, R.; et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 2012, 31, 3148–3163. [Google Scholar] [CrossRef] [PubMed]
- Karabicici, M.; Alptekin, S.; Karagonlar, Z.F.; Erdal, E. Doxorubicin-induced senescence promotes stemness and tumorigenicity in EpCAM−/CD133− nonstem cell population in hepatocellular carcinoma cell line, HuH-7. Mol. Oncol. 2021, 15, 2185–2202. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. The use of cobalt chloride as a chemical hypoxia model. J. Appl. Toxicol. 2019, 39, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, Q.; Yang, X.; Yu, X.; Yu, D.; Zhao, W. Effects of cobalt chloride on the stem cell marker expression and osteogenic differentiation of stem cells from human exfoliated deciduous teeth. Cell Stress Chaperones 2019, 24, 527–538. [Google Scholar] [CrossRef]
- Di Mattia, M.; Mauro, A.; Monache, S.D.; Pulcini, F.; Russo, V.; Berardinelli, P.; Citeroni, M.R.; Turriani, M.; Peserico, A.; Barboni, B. Hypoxia-Mimetic CoCl2 Agent Enhances Pro-Angiogenic Activities in Ovine Amniotic Epithelial Cells-Derived Conditioned Medium. Cells 2022, 11, 461. [Google Scholar] [CrossRef]
- Lopez-Sánchez, L.M.; Jimenez, C.; Valverde, A.; Hernandez, V.; Peñarando, J.; Martinez, A.; Lopez-Pedrera, C.; Muñoz-Castañeda, J.R.; De la Haba-Rodríguez, J.R.; Aranda, E.; et al. CoCl2, a Mimic of Hypoxia, Induces Formation of Polyploid Giant Cells with Stem Characteristics in Colon Cancer. PLoS ONE 2014, 9, e99143. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, C.; Ling, S.; Wei, R.; Wang, J.; Xu, X. The metabolic flexibility of quiescent CSC: Implications for chemotherapy resistance. Cell Death Dis. 2021, 12, 835. [Google Scholar] [CrossRef]
- Addeo, M.; Di Paola, G.; Verma, H.K.; Laurino, S.; Russi, S.; Zoppoli, P.; Falco, G.; Mazzone, P. Gastric Cancer Stem Cells: A Glimpse on Metabolic Reprogramming. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K.; Kharazinejad, E.; Majidpoor, J.; Ahadi, R. Hypoxia in solid tumors: A key promoter of cancer stem cell (CSC) resistance. J. Cancer Res. Clin. Oncol. 2020, 146, 19–31. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Bae, K.-M.; Dai, Y.; Vieweg, J.; Siemann, D.W. Hypoxia regulates SOX2 expression to promote prostate cancer cell invasion and sphere formation. Am. J. Cancer Res. 2016, 6, 1078–1088. [Google Scholar] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pádua, D.; Figueira, P.; Pinto, M.; Maia, A.F.; Peixoto, J.; Lima, R.T.; Pombinho, A.; Pereira, C.F.; Almeida, R.; Mesquita, P. High-Throughput Drug Screening Revealed That Ciclopirox Olamine Can Engender Gastric Cancer Stem-like Cells. Cancers 2023, 15, 4406. https://doi.org/10.3390/cancers15174406
Pádua D, Figueira P, Pinto M, Maia AF, Peixoto J, Lima RT, Pombinho A, Pereira CF, Almeida R, Mesquita P. High-Throughput Drug Screening Revealed That Ciclopirox Olamine Can Engender Gastric Cancer Stem-like Cells. Cancers. 2023; 15(17):4406. https://doi.org/10.3390/cancers15174406
Chicago/Turabian StylePádua, Diana, Paula Figueira, Mariana Pinto, André Filipe Maia, Joana Peixoto, Raquel T. Lima, António Pombinho, Carlos Filipe Pereira, Raquel Almeida, and Patrícia Mesquita. 2023. "High-Throughput Drug Screening Revealed That Ciclopirox Olamine Can Engender Gastric Cancer Stem-like Cells" Cancers 15, no. 17: 4406. https://doi.org/10.3390/cancers15174406
APA StylePádua, D., Figueira, P., Pinto, M., Maia, A. F., Peixoto, J., Lima, R. T., Pombinho, A., Pereira, C. F., Almeida, R., & Mesquita, P. (2023). High-Throughput Drug Screening Revealed That Ciclopirox Olamine Can Engender Gastric Cancer Stem-like Cells. Cancers, 15(17), 4406. https://doi.org/10.3390/cancers15174406