
Bispecific Antibodies in Hematological Malignancies: A Scoping Review

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. The Biology of Bispecific Antibodies
2.1. Mechanism of Action
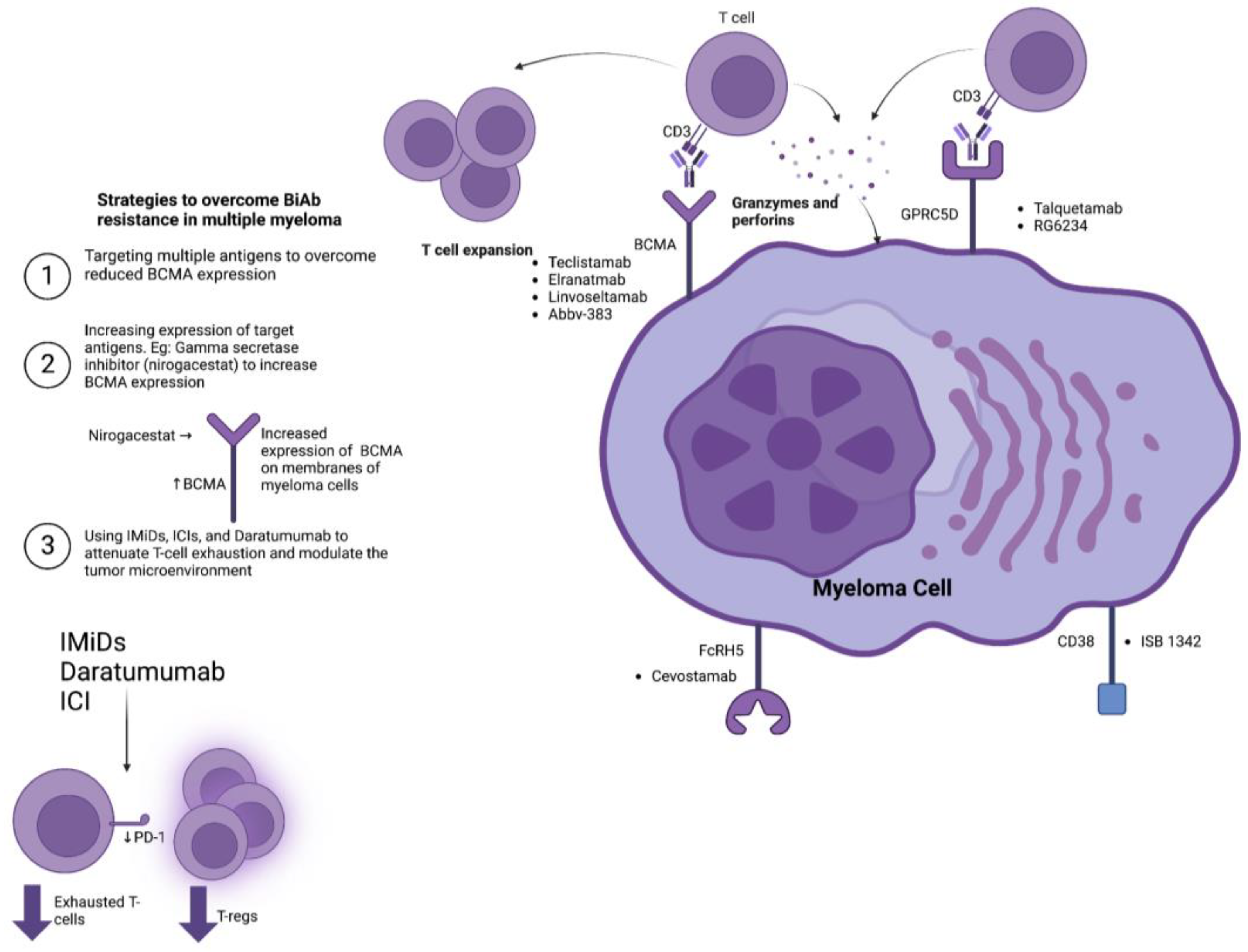
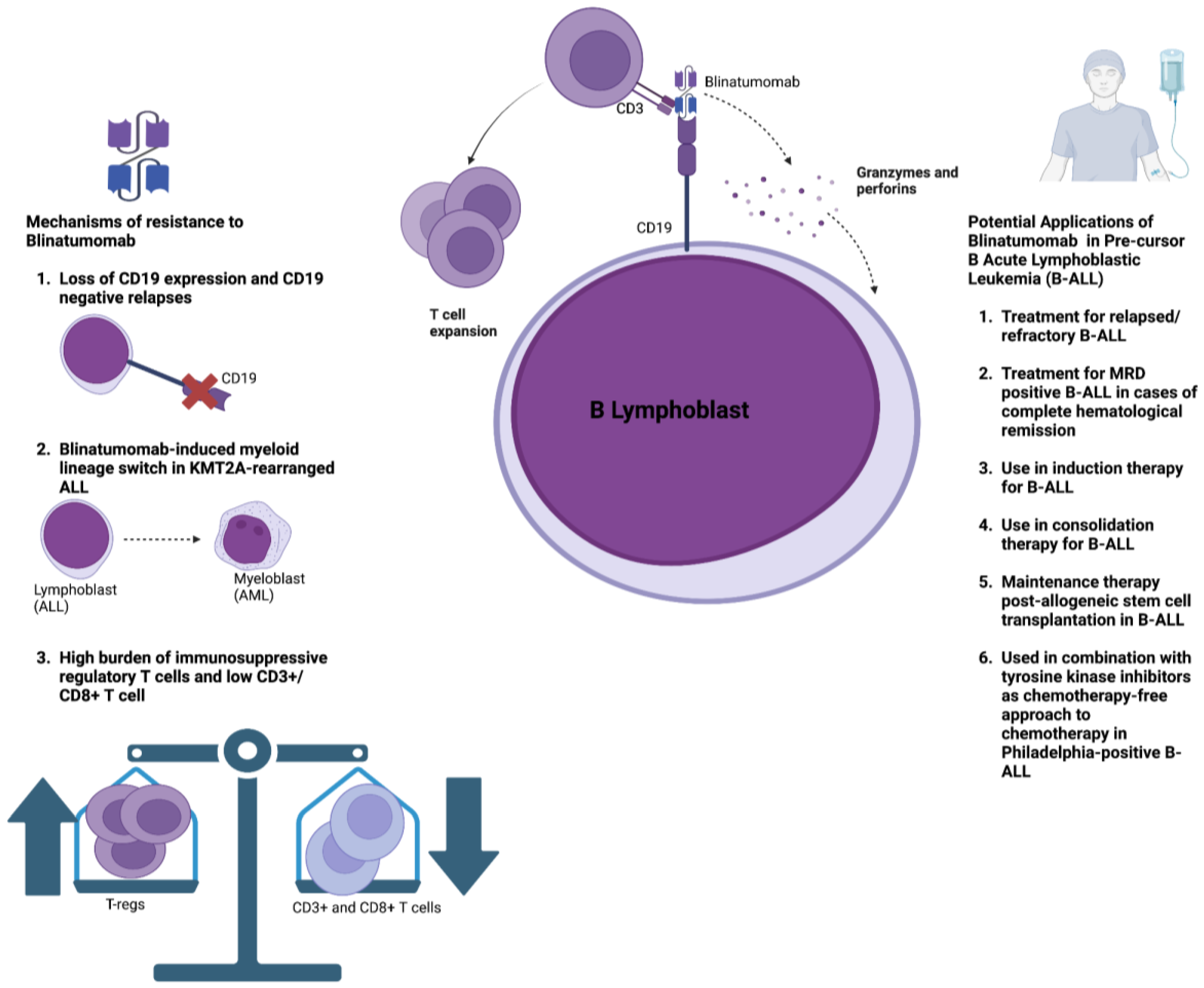
2.2. Resistance Mechanisms
3. Bispecific T-Cell Engagers and Antibodies in the Treatment of Multiple Myeloma
4. Bispecific T-Cell Engagers and Antibodies in the Treatment of Acute Lymphoblastic Leukemia
5. Bispecific T-Cell Engagers and Antibodies in the Treatment of Non-Hodgkin’s Lymphoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BiAB, Trial | BiAB Structure | N | Design | ORR, CR (%) | CRS (All Grade, ≥Grade 3) % | ICANS % | Infections % |
|---|---|---|---|---|---|---|---|
| Mosunetuzumab (Ph2, NCT02500407) [107] | IgG1, humanized | 90 | IV, 21-day cycles, step-up dosing (1/2/60/60 mg) then 30 mg onwards. Pts achieving a CR by cycle 8 completed treatment; those with a partial response or stable disease received 17 cycles total | RRFL 77.8, 60.0 | 44.0, 2.0 | NR | NR |
| Odronextamab (Ph2, NCT03888105) [108] | Fully human IgG4-based | 96 | IV, 21-day cycles, step-up dosing in two regimens (1/20 mg or 0.7/4/20 mg) then 80 mg till cycle 4. Followed by 160 mg maintenance every 2 weeks till disease progression or unacceptable toxicity | RRFL 81.0, 75.0 | 51.0, 0.0 | 0.0 in 0.7/4/20 regimen 3.0 in 1/20 | NR |
| Odronextamab (Ph2, NCT03888105) [112] | Fully human IgG4-based | 121 | IV, 21-day cycles, step-up dosing in two regimens (1/20 mg or 0.7/4/20 mg) then 160 mg till cycle 4. Followed by 320 mg maintenance every 2 weeks till disease progression or unacceptable toxicity | RR DLBCL 53.0, 37.0 | 53.0, 0.0 | 4.0 in 0.7/4/20 regimen 6.0 in 1/20 | NR |
| Epcoritamab (Ph2, NCT03625037) [111] | IgG1, humanized | 157 | SQ, 28-day cycles, once weekly step-up doses in weeks 1–3 of cycle 1, then full doses once weekly through cycle 3, once every 2 weeks in cycles 4–9, and once every 4 weeks in cycle 10 and thereafter, until disease progression or unacceptable toxicity | RR DLBCL 63.0, 39.0 | 49.7, 2.5 | 6.4 (one death) | 45.2 |
| Glofitamab (Ph2, NCT03075696) [109] | 2:1 configuration with bivalency to CD20 | 154 | Pre-treatment with 1000 mg obinutuzumab, followed by IV glofitamab 7 days later, 21-day cycles, two step-up doses (2.5/10 mg) then 30 mg for 12 cycles. | RR DLBCL 58.0, 38.0 | 64.0, 4.0 | 8.0 | 59.0 |
| Mosunetuzumab (Ph1/2, NCT02500407) [116] | IgG1, humanized | 89 | SQ, 21-day cycles, step-up dosing, 3 groups (5/15/45 mg, 5/45/45 mg, 5/45/90/90/45 mg) then 45 mg onwards. Pts achieving a CR by cycle 8 completed treatment; those with a partial response or stable disease received 17 cycles total | iNHL 82.0, 64.0 aNHL 36.0, 20.0 | 27.0, 0.0 | 3.0 | 14.0 grade 3/4 |
| Igm-2323 (Ph1, NCT04082936) [117] | Ten binding domains for CD20; one binding domain for CD3 | 29 | IV on days 1, 8, and 15 of 21-day cycles until disease progression | (FL n = 11) (DLBCL n = 13) (MCL n = 3) (MZL n = 2) 34.8, 21.7 | 20.7, NR | 0.0 | NR |
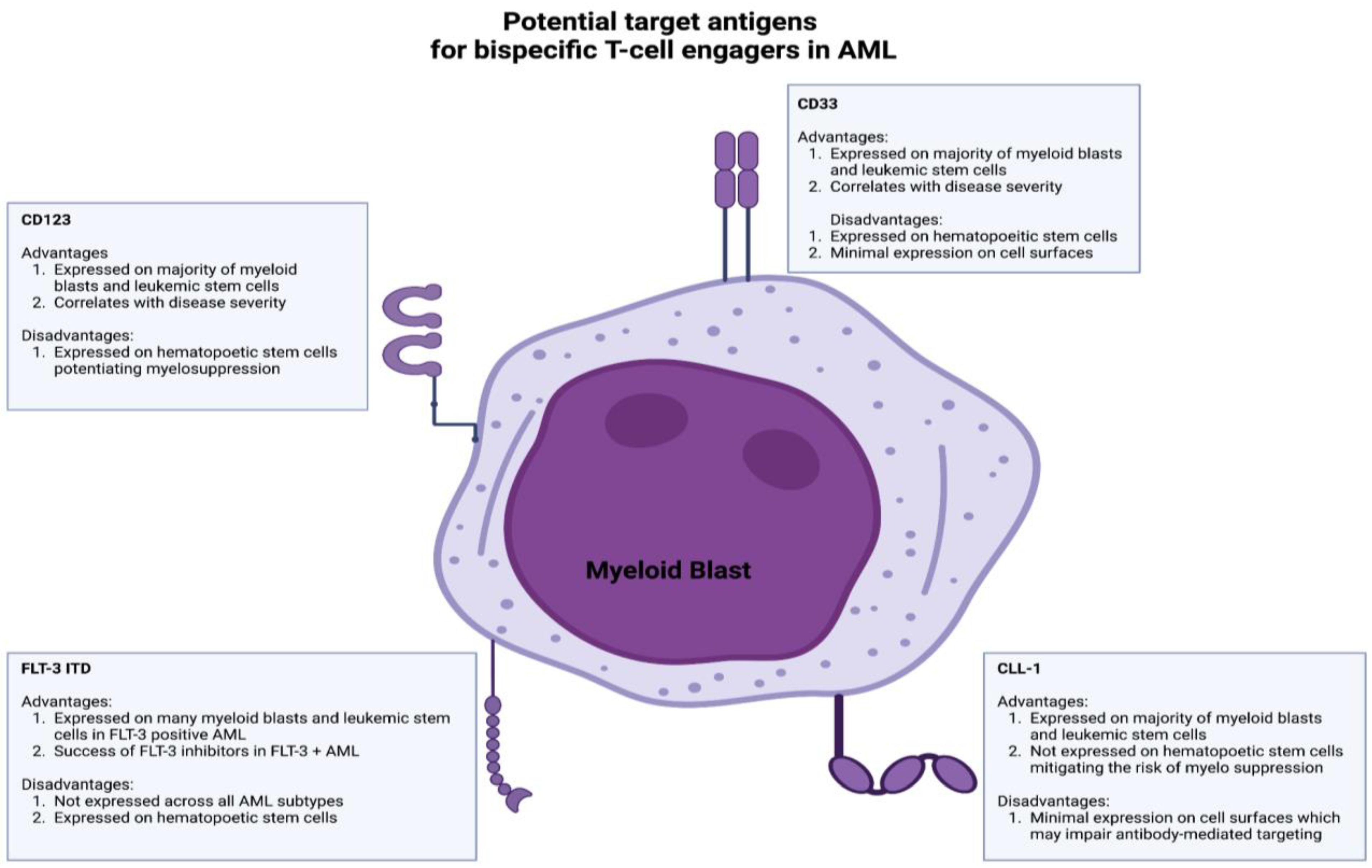
6. Bispecific T-Cell Engagers and Antibodies in the Treatment of Acute Myelogenous Leukemia
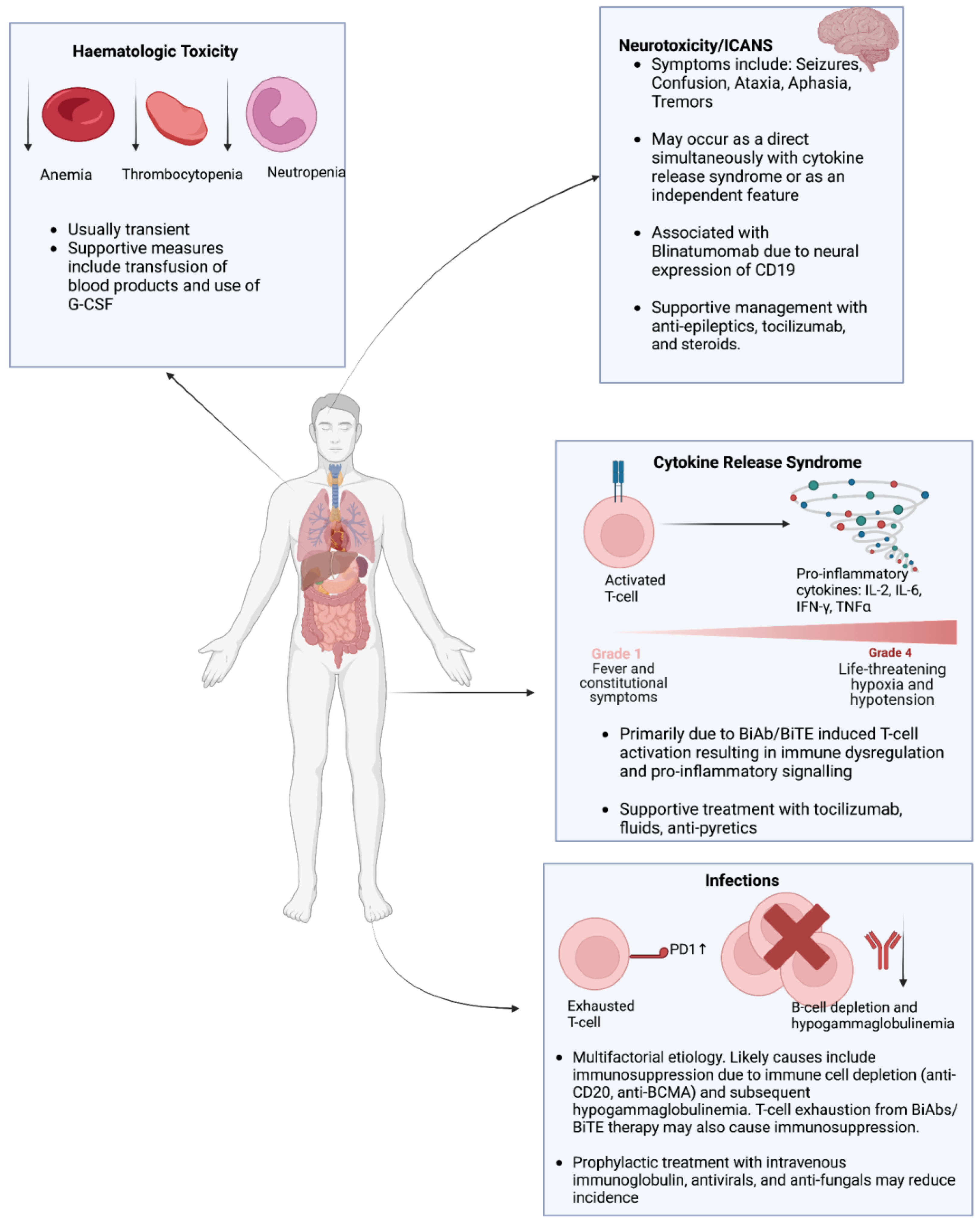
7. Overview of the Toxicities Associated with the Use of Bispecific Antibodies in the Treatment of Hematologic Malignancies
7.1. Cytokine Release Syndrome (CRS)
7.2. Infections
7.3. Hematologic Toxicity
7.4. Neurotoxicity
8. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Tully, S.; Feng, Z.; Grindrod, K.; McFarlane, T.; Chan, K.K.W.; Wong, W.W.L. Impact of Increasing Wait Times on Overall Mortality of Chimeric Antigen Receptor T-Cell Therapy in Large B-Cell Lymphoma: A Discrete Event Simulation Model. JCO Clin. Cancer Inform. 2019, 3, 1–9. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Subklewe, M. BiTEs better than CAR T cells. Blood Adv. 2021, 5, 607–612. [Google Scholar] [CrossRef]
- Topp, M.S.; Kufer, P.; Gökbuget, N.; Goebeler, M.; Klinger, M.; Neumann, S.; Horst, H.A.; Raff, T.; Viardot, A.; Schmid, M.; et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J. Clin. Oncol. 2011, 29, 2493–2498. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Zeidan, A.M.; Bewersdorf, J.P. BiTEs, DARTS, BiKEs and TriKEs—Are Antibody Based Therapies Changing the Future Treatment of AML? Life 2021, 11, 465. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef]
- Thakur, A.; Huang, M.; Lum, L.G. Bispecific antibody based therapeutics: Strengths and challenges. Blood Rev. 2018, 32, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Belmontes, B.; Sawant, D.V.; Zhong, W.; Tan, H.; Kaul, A.; Aeffner, F.; O’Brien, S.A.; Chun, M.; Noubade, R.; Eng, J. Immunotherapy combinations overcome resistance to bispecific T cell engager treatment in T cell–cold solid tumors. Sci. Transl. Med. 2021, 13, eabd1524. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, S.; Wei, X.; Qi, X.; Liu, D.; Liu, L.; Wen, F.; Zhang, J.S.; Wang, F.; Liu, Z.L.; et al. A novel CD19/CD22/CD3 trispecific antibody enhances therapeutic efficacy and overcomes immune escape against B-ALL. Blood 2022, 140, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Foà, R.; Bassan, R.; Vitale, A.; Elia, L.; Piciocchi, A.; Puzzolo, M.C.; Canichella, M.; Viero, P.; Ferrara, F.; Lunghi, M.; et al. Dasatinib-Blinatumomab for Ph-Positive Acute Lymphoblastic Leukemia in Adults. N. Engl. J. Med. 2020, 383, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Bröske, A.E.; Korfi, K.; Belousov, A.; Wilson, S.; Ooi, C.H.; Bolen, C.R.; Canamero, M.; Alcaide, E.G.; James, I.; Piccione, E.C.; et al. Pharmacodynamics and molecular correlates of response to glofitamab in relapsed/refractory non-Hodgkin lymphoma. Blood Adv. 2022, 6, 1025–1037. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Arenas, E.J.; Martínez-Sabadell, A.; Rius Ruiz, I.; Román Alonso, M.; Escorihuela, M.; Luque, A.; Fajardo, C.A.; Gros, A.; Klein, C.; Arribas, J. Acquired cancer cell resistance to T cell bispecific antibodies and CAR T targeting HER2 through JAK2 down-modulation. Nat. Commun. 2021, 12, 1237. [Google Scholar] [CrossRef]
- Verkleij, C.P.M.; Broekmans, M.E.C.; van Duin, M.; Frerichs, K.A.; Kuiper, R.; de Jonge, A.V.; Kaiser, M.; Morgan, G.; Axel, A.; Boominathan, R.; et al. Preclinical activity and determinants of response of the GPRC5DxCD3 bispecific antibody talquetamab in multiple myeloma. Blood Adv. 2021, 5, 2196–2215. [Google Scholar] [CrossRef]
- van de Donk, N.W.; Bahlis, N.; Mateos, M.V.; Weisel, K.; Dholaria, B.; Garfall, A.L.; Goldschmidt, H.; Martin, T.G.; Morillo, D.; Reece, D.E.; et al. S183: Novel Combination Immunotherapy for the Treatment of Relapsed/Refractory Multiple Myeloma: Updated Phase 1B Results for Talquetamab (A GPRC5D X CD3 Bispecific Antibody) in Combination with Daratumumab. HemaSphere 2022, 6, 84–85. [Google Scholar] [CrossRef]
- Duell, J.; Dittrich, M.; Bedke, T.; Mueller, T.; Eisele, F.; Rosenwald, A.; Rasche, L.; Hartmann, E.; Dandekar, T.; Einsele, H.; et al. Frequency of regulatory T cells determines the outcome of the T-cell-engaging antibody blinatumomab in patients with B-precursor ALL. Leukemia 2017, 31, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Selva, D.; Casneuf, T.; Vishwamitra, D.; Stein, S.; Perova, T.; Skerget, S.; Ramos, E.; van Steenbergen, L.; De Maeyer, D.; Boominathan, R.; et al. Teclistamab, a B-Cell Maturation Antigen (BCMA) x CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma (RRMM): Correlative Analyses from MajesTEC-1. Blood 2022, 140, 241–243. [Google Scholar] [CrossRef]
- van de Donk, N.; Zweegman, S. T-cell-engaging bispecific antibodies in cancer. Lancet 2023, 402, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Philipp, N.; Kazerani, M.; Nicholls, A.; Vick, B.; Wulf, J.; Straub, T.; Scheurer, M.; Muth, A.; Hänel, G.; Nixdorf, D.; et al. T-cell exhaustion induced by continuous bispecific molecule exposure is ameliorated by treatment-free intervals. Blood 2022, 140, 1104–1118. [Google Scholar] [CrossRef]
- Meermeier, E.W.; Welsh, S.J.; Sharik, M.E.; Du, M.T.; Garbitt, V.M.; Riggs, D.L.; Shi, C.X.; Stein, C.K.; Bergsagel, M.; Chau, B.; et al. Tumor burden limits bispecific antibody efficacy through T cell exhaustion averted by concurrent cytotoxic therapy. Blood Cancer Discov. 2021, 2, 354–369. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report from International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- Marcon, C.; Simeon, V.; Deias, P.; Facchin, G.; Corso, A.; Derudas, D.; Montefusco, V.; Offidani, M.; Petrucci, M.T.; Zambello, R.; et al. Experts’ consensus on the definition and management of high risk multiple myeloma. Front. Oncol. 2022, 12, 1096852. [Google Scholar] [CrossRef]
- Mateos, M.V.; Weisel, K.; De Stefano, V.; Goldschmidt, H.; Delforge, M.; Mohty, M.; Cavo, M.; Vij, R.; Lindsey-Hill, J.; Dytfeld, D.; et al. LocoMMotion: A prospective, non-interventional, multinational study of real-life current standards of care in patients with relapsed and/or refractory multiple myeloma. Leukemia 2022, 36, 1371–1376. [Google Scholar] [CrossRef]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef]
- Ghermezi, M.; Li, M.; Vardanyan, S.; Harutyunyan, N.M.; Gottlieb, J.; Berenson, A.; Spektor, T.M.; Andreu-Vieyra, C.; Petraki, S.; Sanchez, E.; et al. Serum B-cell maturation antigen: A novel biomarker to predict outcomes for multiple myeloma patients. Haematologica 2017, 102, 785–795. [Google Scholar] [CrossRef]
- Swan, D.; Murphy, P.; Glavey, S.; Quinn, J. Bispecific Antibodies in Multiple Myeloma: Opportunities to Enhance Efficacy and Improve Safety. Cancers 2023, 15, 1819. [Google Scholar] [CrossRef]
- Moreau, P.; Garfall, A.L.; van de Donk, N.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Nooka, A.K.; Martin, T.; Rosinol, L.; Chari, A.; et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2022, 387, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Bahlis, N.J.; Tomasson, M.H.; Mohty, M.; Niesvizky, R.; Nooka, A.K.; Manier, S.; Maisel, C.; Jethava, Y.; Martinez-Lopez, J.; Prince, H.M.; et al. Efficacy and Safety of Elranatamab in Patients with Relapsed/Refractory Multiple Myeloma Naïve to B-Cell Maturation Antigen (BCMA)-Directed Therapies: Results from Cohort a of the Magnetismm-3 Study. Blood 2022, 140, 391–393. [Google Scholar] [CrossRef]
- Lee, H.C.; Bumma, N.; Richter, J.R.; Dhodapkar, M.V.; Hoffman, J.E.; Suvannasankha, A.; Zonder, J.A.; Shah, M.R.; Lentzsch, S.; Maly, J.J.; et al. LINKER-MM1 study: Linvoseltamab (REGN5458) in patients with relapsed/refractory multiple myeloma. J. Clin. Oncol. 2023, 41, 8006. [Google Scholar] [CrossRef]
- D’Souza, A.; Shah, N.; Rodriguez, C.; Voorhees, P.M.; Weisel, K.; Bueno, O.F.; Pothacamury, R.K.; Freise, K.J.; Yue, S.; Ross, J.A.; et al. A Phase I First-in-Human Study of ABBV-383, a B-Cell Maturation Antigen × CD3 Bispecific T-Cell Redirecting Antibody, in Patients with Relapsed/Refractory Multiple Myeloma. J. Clin. Oncol. 2022, 40, 3576–3586. [Google Scholar] [CrossRef]
- Truger, M.S.; Duell, J.; Zhou, X.; Heimeshoff, L.; Ruckdeschel, A.; John, M.; Riedel, A.; Hüper, S.; Peter, J.; Walter, W.; et al. Single- and double-hit events in genes encoding immune targets before and after T cell-engaging antibody therapy in MM. Blood Adv. 2021, 5, 3794–3798. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.; Costa, B.A.; Mailankody, S. GPRC5D as a novel immunotherapeutic target in multiple myeloma. Nat. Rev. Clin. Oncol. 2023, 20, 281–282. [Google Scholar] [CrossRef]
- Chari, A.; Minnema, M.C.; Berdeja, J.G.; Oriol, A.; van de Donk, N.; Rodríguez-Otero, P.; Askari, E.; Mateos, M.V.; Costa, L.J.; Caers, J.; et al. Talquetamab, a T-Cell-Redirecting GPRC5D Bispecific Antibody for Multiple Myeloma. N. Engl. J. Med. 2022, 387, 2232–2244. [Google Scholar] [CrossRef] [PubMed]
- Carlo-Stella, C.; Mazza, R.; Manier, S.; Facon, T.; Yoon, S.-S.; Koh, Y.; Harrison, S.J.; Er, J.; Pinto, A.; Volzone, F.; et al. RG6234, a GPRC5DxCD3 T-Cell Engaging Bispecific Antibody, Is Highly Active in Patients (pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Updated Intravenous (IV) and First Subcutaneous (SC) Results from a Phase I Dose-Escalation Study. Blood 2022, 140, 397–399. [Google Scholar] [CrossRef]
- Li, J.; Stagg, N.J.; Johnston, J.; Harris, M.J.; Menzies, S.A.; DiCara, D.; Clark, V.; Hristopoulos, M.; Cook, R.; Slaga, D.; et al. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer Cell 2017, 31, 383–395. [Google Scholar] [CrossRef]
- Trudel, S.; Cohen, A.D.; Krishnan, A.Y.; Fonseca, R.; Spencer, A.; Berdeja, J.G.; Lesokhin, A.; Forsberg, P.A.; Laubach, J.P.; Costa, L.J.; et al. Cevostamab Monotherapy Continues to Show Clinically Meaningful Activity and Manageable Safety in Patients with Heavily Pre-Treated Relapsed/Refractory Multiple Myeloma (RRMM): Updated Results from an Ongoing Phase I Study. Blood 2021, 138, 157. [Google Scholar] [CrossRef]
- van de Donk, N.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef]
- Mohan, S.R.; Costa Chase, C.; Berdeja, J.G.; Karlin, L.; Belhadj, K.; Perrot, A.; Moreau, P.; Touzeau, C.; Chalopin, T.; Lesokhin, A.M.; et al. Initial Results of Dose Escalation of ISB 1342, a Novel CD3xCD38 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2022, 140, 7264–7266. [Google Scholar] [CrossRef]
- Akhmetzyanova, I.; McCarron, M.J.; Parekh, S.; Chesi, M.; Bergsagel, P.L.; Fooksman, D.R. Dynamic CD138 surface expression regulates switch between myeloma growth and dissemination. Leukemia 2020, 34, 245–256. [Google Scholar] [CrossRef]
- Morillo, D.; Gatt, M.E.; Sebag, M.; Kim, K.; Min, C.-K.; Oriol, A.; Ocio, E.M.; Yoon, S.-S.; Mateos, M.-V.; Chu, M.; et al. First results from the RedirecTT-1 study with teclistamab (tec) + talquetamab (tal) simultaneously targeting BCMA and GPRC5D in patients (pts) with relapsed/refractory multiple myeloma (RRMM). J. Clin. Oncol. 2023, 41, 8002. [Google Scholar] [CrossRef]
- Shearer, T.; Williams, R.L., Jr.; Johnson, M.; Cendrowicz, E.; Leonowens, C.; Smith, M.; Baughman, T.; Breitbach, C.J.; Smith, L.M.; Burgess, M.; et al. Pharmacodynamic Effects of Nirogacestat, a Gamma Secretase Inhibitor, on B-Cell Maturation Antigen in Healthy Participants. Blood 2022, 140, 3080–3081. [Google Scholar] [CrossRef]
- Uckun, F.M. Overcoming the Immunosuppressive Tumor Microenvironment in Multiple Myeloma. Cancers 2021, 13, 2018. [Google Scholar] [CrossRef] [PubMed]
- Visram, A.; Dasari, S.; Anderson, E.; Kumar, S.; Kourelis, T.V. Relapsed multiple myeloma demonstrates distinct patterns of immune microenvironment and malignant cell-mediated immunosuppression. Blood Cancer J. 2021, 11, 45. [Google Scholar] [CrossRef]
- Feyler, S.; Scott, G.B.; Parrish, C.; Jarmin, S.; Evans, P.; Short, M.; McKinley, K.; Selby, P.J.; Cook, G. Tumour cell generation of inducible regulatory T-cells in multiple myeloma is contact-dependent and antigen-presenting cell-independent. PLoS ONE 2012, 7, e35981. [Google Scholar] [CrossRef]
- Soekojo, C.Y.; Chng, W.J. The evolution of immune dysfunction in multiple myeloma. Eur. J. Haematol. 2022, 109, 415–424. [Google Scholar] [CrossRef]
- Krämer, I.; Engelhardt, M.; Fichtner, S.; Neuber, B.; Medenhoff, S.; Bertsch, U.; Hillengass, J.; Raab, M.S.; Hose, D.; Ho, A.D.; et al. Lenalidomide enhances myeloma-specific T-cell responses in vivo and in vitro. Oncoimmunology 2016, 5, e1139662. [Google Scholar] [CrossRef]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Searle, E.; Quach, H.; Wong, S.W.; Costa, L.J.; Hulin, C.; Janowski, W.; Berdeja, J.; Anguille, S.; Matous, J.V.; Touzeau, C.; et al. Teclistamab in Combination with Subcutaneous Daratumumab and Lenalidomide in Patients with Multiple Myeloma: Results from One Cohort of MajesTEC-2, a Phase1b, Multicohort Study. Blood 2022, 140, 394–396. [Google Scholar] [CrossRef]
- Rodríguez-Otero, P.; D’Souza, A.; Reece, D.E.; van de Donk, N.W.C.J.; Chari, A.; Krishnan, A.Y.; Martin, T.G.; Mateos, M.-V.; Morillo, D.; Hurd, D.D.; et al. A novel, immunotherapy-based approach for the treatment of relapsed/refractory multiple myeloma (RRMM): Updated phase 1b results for daratumumab in combination with teclistamab (a BCMA x CD3 bispecific antibody). J. Clin. Oncol. 2022, 40, 8032. [Google Scholar] [CrossRef]
- Rosenblatt, J.; Avigan, D. Targeting the PD-1/PD-L1 axis in multiple myeloma: A dream or a reality? Blood 2017, 129, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Duffield, A.S.; Mullighan, C.G.; Borowitz, M.J. International Consensus Classification of acute lymphoblastic leukemia/lymphoma. Virchows Arch. 2023, 482, 11–26. [Google Scholar] [CrossRef]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef]
- DuVall, A.S.; Sheade, J.; Anderson, D.; Yates, S.J.; Stock, W. Updates in the Management of Relapsed and Refractory Acute Lymphoblastic Leukemia: An Urgent Plea for New Treatments Is Being Answered! JCO Oncol. Pract. 2022, 18, 479–487. [Google Scholar] [CrossRef]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Bassan, R.; Spinelli, O.; Oldani, E.; Intermesoli, T.; Tosi, M.; Peruta, B.; Rossi, G.; Borlenghi, E.; Pogliani, E.M.; Terruzzi, E.; et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood 2009, 113, 4153–4162. [Google Scholar] [CrossRef]
- Brüggemann, M.; Raff, T.; Flohr, T.; Gökbuget, N.; Nakao, M.; Droese, J.; Lüschen, S.; Pott, C.; Ritgen, M.; Scheuring, U.; et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood 2006, 107, 1116–1123. [Google Scholar] [CrossRef]
- Gökbuget, N.; Kneba, M.; Raff, T.; Trautmann, H.; Bartram, C.R.; Arnold, R.; Fietkau, R.; Freund, M.; Ganser, A.; Ludwig, W.D.; et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood 2012, 120, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Raff, T.; Gökbuget, N.; Lüschen, S.; Reutzel, R.; Ritgen, M.; Irmer, S.; Böttcher, S.; Horst, H.A.; Kneba, M.; Hoelzer, D.; et al. Molecular relapse in adult standard-risk ALL patients detected by prospective MRD monitoring during and after maintenance treatment: Data from the GMALL 06/99 and 07/03 trials. Blood 2007, 109, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Borowitz, M.J.; Wood, B.L.; Devidas, M.; Loh, M.L.; Raetz, E.A.; Salzer, W.L.; Nachman, J.B.; Carroll, A.J.; Heerema, N.A.; Gastier-Foster, J.M.; et al. Prognostic significance of minimal residual disease in high risk B-ALL: A report from Children’s Oncology Group study AALL0232. Blood 2015, 126, 964–971. [Google Scholar] [CrossRef]
- Berry, D.A.; Zhou, S.; Higley, H.; Mukundan, L.; Fu, S.; Reaman, G.H.; Wood, B.L.; Kelloff, G.J.; Jessup, J.M.; Radich, J.P. Association of Minimal Residual Disease with Clinical Outcome in Pediatric and Adult Acute Lymphoblastic Leukemia: A Meta-analysis. JAMA Oncol. 2017, 3, e170580. [Google Scholar] [CrossRef] [PubMed]
- Gökbuget, N.; Dombret, H.; Bonifacio, M.; Reichle, A.; Graux, C.; Faul, C.; Diedrich, H.; Topp, M.S.; Brüggemann, M.; Horst, H.A.; et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 2018, 131, 1522–1531. [Google Scholar] [CrossRef]
- Locatelli, F.; Zugmaier, G.; Rizzari, C.; Morris, J.D.; Gruhn, B.; Klingebiel, T.; Parasole, R.; Linderkamp, C.; Flotho, C.; Petit, A.; et al. Effect of Blinatumomab vs Chemotherapy on Event-Free Survival Among Children with High-risk First-Relapse B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. Jama 2021, 325, 843–854. [Google Scholar] [CrossRef]
- Brown, P.A.; Ji, L.; Xu, X.; Devidas, M.; Hogan, L.E.; Borowitz, M.J.; Raetz, E.A.; Zugmaier, G.; Sharon, E.; Bernhardt, M.B.; et al. Effect of Postreinduction Therapy Consolidation with Blinatumomab vs Chemotherapy on Disease-Free Survival in Children, Adolescents, and Young Adults with First Relapse of B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. Jama 2021, 325, 833–842. [Google Scholar] [CrossRef]
- Topp, M.S.; Gökbuget, N.; Stein, A.S.; Zugmaier, G.; O’Brien, S.; Bargou, R.C.; Dombret, H.; Fielding, A.K.; Heffner, L.; Larson, R.A.; et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: A multicentre, single-arm, phase 2 study. Lancet Oncol. 2015, 16, 57–66. [Google Scholar] [CrossRef]
- Stein, A.S.; Schiller, G.; Benjamin, R.; Jia, C.; Zhang, A.; Zhu, M.; Zimmerman, Z.; Topp, M.S. Neurologic adverse events in patients with relapsed/refractory acute lymphoblastic leukemia treated with blinatumomab: Management and mitigating factors. Ann. Hematol. 2019, 98, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Jen, E.Y.; Xu, Q.; Schetter, A.; Przepiorka, D.; Shen, Y.L.; Roscoe, D.; Sridhara, R.; Deisseroth, A.; Philip, R.; Farrell, A.T.; et al. FDA Approval: Blinatumomab for Patients with B-cell Precursor Acute Lymphoblastic Leukemia in Morphologic Remission with Minimal Residual Disease. Clin. Cancer Res. 2019, 25, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Litzow, M.R.; Sun, Z.; Paietta, E.; Mattison, R.J.; Lazarus, H.M.; Rowe, J.M.; Arber, D.A.; Mullighan, C.G.; Willman, C.L.; Zhang, Y.; et al. Consolidation Therapy with Blinatumomab Improves Overall Survival in Newly Diagnosed Adult Patients with B-Lineage Acute Lymphoblastic Leukemia in Measurable Residual Disease Negative Remission: Results from the ECOG-ACRIN E1910 Randomized Phase III National Cooperative Clinical Trials Network Trial. Blood 2022, 140, LBA-1. [Google Scholar] [CrossRef]
- Salek, C.; Folber, F.; Hrabovsky, S.; Koristek, Z.; Horacek, J.M.; Fronkova, E.; Soukup, P.; Benkova, K.; Cetkovsky, P.; Trka, J.; et al. Single Cycle of Blinatumomab Followed By High-Dose Chemotherapy in the Induction Therapy for Ph-Negative Acute Lymphoblastic Leukemia in Adults. Primary Endpoint Analysis of the Blina-Cell Trial. Blood 2022, 140, 3258–3259. [Google Scholar] [CrossRef]
- Foà, R.; Chiaretti, S. Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2022, 386, 2399–2411. [Google Scholar] [CrossRef]
- Jabbour, E.; Haddad, F.G.; Short, N.J.; Kantarjian, H. Treatment of Adults with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia-from Intensive Chemotherapy Combinations to Chemotherapy-Free Regimens: A Review. JAMA Oncol. 2022, 8, 1340–1348. [Google Scholar] [CrossRef]
- Jabbour, E.; Short, N.J.; Jain, N.; Huang, X.; Montalban-Bravo, G.; Banerjee, P.; Rezvani, K.; Jiang, X.; Kim, K.H.; Kanagal-Shamanna, R.; et al. Ponatinib and blinatumomab for Philadelphia chromosome-positive acute lymphoblastic leukaemia: A US, single-centre, single-arm, phase 2 trial. Lancet Haematol. 2023, 10, e24–e34. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Gökbuget, N.; Zugmaier, G.; Degenhard, E.; Goebeler, M.E.; Klinger, M.; Neumann, S.A.; Horst, H.A.; Raff, T.; Viardot, A.; et al. Long-term follow-up of hematologic relapse-free survival in a phase 2 study of blinatumomab in patients with MRD in B-lineage ALL. Blood 2012, 120, 5185–5187. [Google Scholar] [CrossRef]
- Lussana, F.; Gritti, G.; Rambaldi, A. Immunotherapy of Acute Lymphoblastic Leukemia and Lymphoma with T Cell-Redirected Bispecific Antibodies. J. Clin. Oncol. 2021, 39, 444–455. [Google Scholar] [CrossRef]
- Zhao, Y.; Aldoss, I.; Qu, C.; Crawford, J.C.; Gu, Z.; Allen, E.K.; Zamora, A.E.; Alexander, T.B.; Wang, J.; Goto, H.; et al. Tumor-intrinsic and -extrinsic determinants of response to blinatumomab in adults with B-ALL. Blood 2021, 137, 471–484. [Google Scholar] [CrossRef]
- Braig, F.; Brandt, A.; Goebeler, M.; Tony, H.P.; Kurze, A.K.; Nollau, P.; Bumm, T.; Böttcher, S.; Bargou, R.C.; Binder, M. Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood 2017, 129, 100–104. [Google Scholar] [CrossRef]
- Modulation of CD19 surface expression in B cell acute lymphoblastic leukemia. Nat. Immunol. 2022, 23, 1410–1411. [CrossRef]
- Lanza, F.; Maffini, E.; Rondoni, M.; Massari, E.; Faini, A.C.; Malavasi, F. CD22 Expression in B-Cell Acute Lymphoblastic Leukemia: Biological Significance and Implications for Inotuzumab Therapy in Adults. Cancers 2020, 12, 303. [Google Scholar] [CrossRef]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef]
- Roddie, C.; Lekakis, L.J.; Marzolini, M.A.V.; Ramakrishnan, A.; Zhang, Y.; Hu, Y.; Peddareddigari, V.G.R.; Khokhar, N.; Chen, R.; Basilico, S.; et al. Dual targeting of CD19 and CD22 with bicistronic CAR-T cells in patients with relapsed/refractory large B-cell lymphoma. Blood 2023, 141, 2470–2482. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Tang, Y.; Cai, J.; Wan, X.; Hu, S.; Lu, X.; Xie, Z.; Qiao, X.; Jiang, H.; Shao, J.; et al. Coadministration of CD19- and CD22-Directed Chimeric Antigen Receptor T-Cell Therapy in Childhood B-Cell Acute Lymphoblastic Leukemia: A Single-Arm, Multicenter, Phase II Trial. J. Clin. Oncol. 2023, 41, 1670–1683. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Jabbour, E.; Short, N.J.; Senapati, J.; Jain, N.; Huang, X.; Daver, N.; DiNardo, C.D.; Pemmaraju, N.; Wierda, W.; Garcia-Manero, G.; et al. Mini-hyper-CVD plus inotuzumab ozogamicin, with or without blinatumomab, in the subgroup of older patients with newly diagnosed Philadelphia chromosome-negative B-cell acute lymphocytic leukaemia: Long-term results of an open-label phase 2 trial. Lancet Haematol. 2023, 10, e433–e444. [Google Scholar] [CrossRef] [PubMed]
- He, R.R.; Nayer, Z.; Hogan, M.; Cuevo, R.S.; Woodward, K.; Heyer, D.; Curtis, C.A.; Peterson, J.F. Immunotherapy- (Blinatumomab-) Related Lineage Switch of KMT2A/AFF1 Rearranged B-Lymphoblastic Leukemia into Acute Myeloid Leukemia/Myeloid Sarcoma and Subsequently into B/Myeloid Mixed Phenotype Acute Leukemia. Case Rep. Hematol. 2019, 2019, 7394619. [Google Scholar] [CrossRef]
- Wölfl, M.; Rasche, M.; Eyrich, M.; Schmid, R.; Reinhardt, D.; Schlegel, P.G. Spontaneous reversion of a lineage switch following an initial blinatumomab-induced ALL-to-AML switch in MLL-rearranged infant ALL. Blood Adv. 2018, 2, 1382–1385. [Google Scholar] [CrossRef] [PubMed]
- Haddox, C.L.; Mangaonkar, A.A.; Chen, D.; Shi, M.; He, R.; Oliveira, J.L.; Litzow, M.R.; Al-Kali, A.; Hogan, W.J.; Elliott, M.A. Blinatumomab-induced lineage switch of B-ALL with t(4:11)(q21;q23) KMT2A/AFF1 into an aggressive AML: Pre- and post-switch phenotypic, cytogenetic and molecular analysis. Blood Cancer J. 2017, 7, e607. [Google Scholar] [CrossRef]
- Fournier, E.; Inchiappa, L.; Delattre, C.; Pignon, J.M.; Danicourt, F.; Bemba, M.; Roche-Lestienne, C.; Daudignon, A.; Decool, G.; Roumier, C.; et al. Increased risk of adverse acute myeloid leukemia after anti-CD19-targeted immunotherapies in KMT2A-rearranged acute lymphoblastic leukemia: A case report and review of the literature. Leuk. Lymphoma 2019, 60, 1827–1830. [Google Scholar] [CrossRef]
- Hoseini, S.S.; Espinosa-Cotton, M.; Guo, H.F.; Cheung, N.V. Overcoming leukemia heterogeneity by combining T cell engaging bispecific antibodies. J. Immunother. Cancer 2020, 8, 1626. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Morales, S.; Aranda-Uribe, I.S.; Pérez-Amado, C.J.; Ramírez-Bello, J.; Hidalgo-Miranda, A. Mechanisms of Immunosuppressive Tumor Evasion: Focus on Acute Lymphoblastic Leukemia. Front. Immunol. 2021, 12, 737340. [Google Scholar] [CrossRef]
- Feucht, J.; Kayser, S.; Gorodezki, D.; Hamieh, M.; Döring, M.; Blaeschke, F.; Schlegel, P.; Bösmüller, H.; Quintanilla-Fend, L.; Ebinger, M.; et al. T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget 2016, 7, 76902–76919. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ubukawa, K.; Fujishima, M.; Takahashi, N. Correlation between increased immune checkpoint molecule expression and refractoriness to blinatumomab evaluated by longitudinal T cell analysis. Int. J. Hematol. 2021, 113, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.A.; Jiang, X.; Banerjee, P.; Basar, R.; Garg, N.; Chen, K.; Kaplan, M.; Nandivada, V.; Cortes, A.K.N.; Ferrajoli, A.; et al. A phase two study of high dose blinatumomab in Richter’s syndrome. Leukemia 2022, 36, 2228–2232. [Google Scholar] [CrossRef]
- Sandhu, K.S.; Macias, A.; Del Real, M.; Beltran, A.L.; Kim, Y.S.; Zhang, J.; Palmer, J.; Robbins, M.; Loomis, R.; Akhtari, M.; et al. Interim Results of a Phase 1/2 Study of Pembrolizumab Combined with Blinatumomab in Patients with Relapsed/Refractory (r/r) ALL. Blood 2022, 140, 8985–8986. [Google Scholar] [CrossRef]
- Webster, J.; Luskin, M.R.; Prince, G.T.; DeZern, A.E.; DeAngelo, D.J.; Levis, M.J.; Blackford, A.; Sharon, E.; Streicher, H.; Luznik, L.; et al. Blinatumomab in Combination with Immune Checkpoint Inhibitors of PD-1 and CTLA-4 in Adult Patients with Relapsed/Refractory (R/R) CD19 Positive B-Cell Acute Lymphoblastic Leukemia (ALL): Preliminary Results of a Phase I Study. Blood 2018, 132, 557. [Google Scholar] [CrossRef]
- Armitage, J.O.; Gascoyne, R.D.; Lunning, M.A.; Cavalli, F. Non-Hodgkin lymphoma. Lancet 2017, 390, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Dotan, E.; Aggarwal, C.; Smith, M.R. Impact of Rituximab (Rituxan) on the Treatment of B-Cell Non-Hodgkin’s Lymphoma. Pharm. Ther. 2010, 35, 148–157. [Google Scholar]
- Marofi, F.; Rahman, H.S.; Achmad, M.H.; Sergeevna, K.N.; Suksatan, W.; Abdelbasset, W.K.; Mikhailova, M.V.; Shomali, N.; Yazdanifar, M.; Hassanzadeh, A.; et al. A Deep Insight into CAR-T Cell Therapy in Non-Hodgkin Lymphoma: Application, Opportunities, and Future Directions. Front. Immunol. 2021, 12, 681984. [Google Scholar] [CrossRef] [PubMed]
- Blanco, B.; Domínguez-Alonso, C.; Alvarez-Vallina, L. Bispecific Immunomodulatory Antibodies for Cancer Immunotherapy. Clin. Cancer Res. 2021, 27, 5457–5464. [Google Scholar] [CrossRef]
- Horna, P.; Nowakowski, G.; Endell, J.; Boxhammer, R. Comparative Assessment of Surface CD19 and CD20 Expression on B-Cell Lymphomas from Clinical Biopsies: Implications for Targeted Therapies. Blood 2019, 134, 5345. [Google Scholar] [CrossRef]
- Pavlasova, G.; Mraz, M. The regulation and function of CD20: An “enigma” of B-cell biology and targeted therapy. Haematologica 2020, 105, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Chung, C. Current targeted therapies in lymphomas. Am. J. Health Syst. Pharm. 2019, 76, 1825–1834. [Google Scholar] [CrossRef]
- Bartlett, N.L.; Sehn, L.H.; Matasar, M.J.; Schuster, S.J.; Assouline, S.; Giri, P.; Kuruvilla, J.; Canales, M.; Dietrich, S.; Fay, K.; et al. Mosunetuzumab Monotherapy Demonstrates Durable Efficacy with a Manageable Safety Profile in Patients with Relapsed/Refractory Follicular Lymphoma Who Received ≥2 Prior Therapies: Updated Results from a Pivotal Phase II Study. Blood 2022, 140, 1467–1470. [Google Scholar] [CrossRef]
- Kim, T.M.; Taszner, M.; Cho, S.-G.; Novelli, S.; Le Gouill, S.; Poon, M.L.; Villasboas, J.C.; Champion, R.; Bachy, E.; Guidez, S.; et al. Odronextamab in Patients with Relapsed/Refractory (R/R) Follicular Lymphoma (FL) Grade 1-3a: Results from a Prespecified Analysis of the Pivotal Phase II Study ELM-2. Blood 2022, 140, 2280–2282. [Google Scholar] [CrossRef]
- Falchi, L.; Carlo-Stella, C.; Morschhauser, F.; Hutchings, M.; Bachy, E.; Cartron, G.; Khan, C.; Tani, M.; Martinez-Lopez, J.; Bartlett, N.L.; et al. Glofitamab monotherapy in pts with relapsed/refractory (R/R) large B-cell lymphoma (LBCL): Extended follow-up and landmark analyses from a pivotal phase II study. J. Clin. Oncol. 2023, 41, 7550. [Google Scholar] [CrossRef]
- Dickinson, M.J.; Carlo-Stella, C.; Morschhauser, F.; Bachy, E.; Corradini, P.; Iacoboni, G.; Khan, C.; Wróbel, T.; Offner, F.; Trněný, M.; et al. Glofitamab for Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 387, 2220–2231. [Google Scholar] [CrossRef]
- Thieblemont, C.; Phillips, T.; Ghesquieres, H.; Cheah, C.Y.; Clausen, M.R.; Cunningham, D.; Do, Y.R.; Feldman, T.; Gasiorowski, R.; Jurczak, W.; et al. Epcoritamab, a Novel, Subcutaneous CD3xCD20 Bispecific T-Cell-Engaging Antibody, in Relapsed or Refractory Large B-Cell Lymphoma: Dose Expansion in a Phase I/II Trial. J. Clin. Oncol. 2023, 41, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-S.; Kim, T.M.; Cho, S.-G.; Jarque, I.; Iskierka-Jażdżewska, E.; Poon, M.L.; Prince, H.M.; Oh, S.Y.; Lim, F.; Carpio, C.; et al. Odronextamab in Patients with Relapsed/Refractory (R/R) Diffuse Large B-Cell Lymphoma (DLBCL): Results from a Prespecified Analysis of the Pivotal Phase II Study ELM-2. Blood 2022, 140, 1070–1071. [Google Scholar] [CrossRef]
- Olszewski, A.J.; Avigdor, A.; Babu, S.; Levi, I.; Eradat, H.; Abadi, U.; Holmes, H.; McKinney, M.; Woszczyk, D.; Giannopoulos, K.; et al. Mosunetuzumab Monotherapy Continues to Demonstrate Promising Efficacy and Durable Complete Responses in Elderly/Unfit Patients with Previously Untreated Diffuse Large B-Cell Lymphoma. Blood 2022, 140, 1778–1780. [Google Scholar] [CrossRef]
- Hutchings, M.; Mous, R.; Clausen, M.R.; Johnson, P.; Linton, K.M.; Chamuleau, M.E.D.; Lewis, D.J.; Sureda Balari, A.; Cunningham, D.; Oliveri, R.S.; et al. Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: An open-label, phase 1/2 study. Lancet 2021, 398, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Bachy, E.; Le Gouill, S.; Di Blasi, R.; Sesques, P.; Manson, G.; Cartron, G.; Beauvais, D.; Roulin, L.; Gros, F.X.; Rubio, M.T.; et al. A real-world comparison of tisagenlecleucel and axicabtagene ciloleucel CAR T cells in relapsed or refractory diffuse large B cell lymphoma. Nat. Med. 2022, 28, 2145–2154. [Google Scholar] [CrossRef]
- Budde, E.L.; Bartlett, N.L.; Giri, P.; Schuster, S.J.; Assouline, S.; Yoon, S.-S.; Fay, K.; Matasar, M.J.; Gutierrez, N.C.; Marlton, P.; et al. Subcutaneous Mosunetuzumab Is Active with a Manageable Safety Profile in Patients (pts) with Relapsed/Refractory (R/R) B-Cell Non-Hodgkin Lymphomas (B-NHLs): Updated Results from a Phase I/II Study. Blood 2022, 140, 3753–3755. [Google Scholar] [CrossRef]
- Budde, E.; Gopal, A.K.; Kim, W.S.; Flinn, I.W.; Cheah, C.Y.Y.; Nastoupil, L.; Matasar, M.J.; Diefenbach, C.S.; Gregory, G.P.; Qazi, I.; et al. A Phase 1 Dose Escalation Study of Igm-2323, a Novel Anti-CD20 x Anti-CD3 IgM T Cell Engager (TCE) in Patients with Advanced B-Cell Malignancies. Blood 2021, 138, 132. [Google Scholar] [CrossRef]
- Sermer, D.; Elavalakanar, P.; Abramson, J.S.; Palomba, M.L.; Salles, G.; Arnason, J. Targeting CD19 for diffuse large B cell lymphoma in the era of CARs: Other modes of transportation. Blood Rev. 2023, 57, 101002. [Google Scholar] [CrossRef]
- Goebeler, M.E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients with Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results from a Phase I Study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef]
- Topp, M.; Dlugosz-Danecka, M.; Skotnicki, A.B.; Salogub, G.; Viardot, A.; Klein, A.K.; Hess, G.; Michel, C.S.; Grosicki, S.; Gural, A.; et al. Safety of AFM11 in the treatment of patients with B-cell malignancies: Findings from two phase 1 studies. Trials 2023, 24, 4. [Google Scholar] [CrossRef]
- Nair, R.; Jacobs, R.; Cho, S.-G.; Devata, S.; Gaballa, S.; Yoon, D.H.; Stevens, D.A.; Kim, J.S.; Shah, N.N.; Brennan, D.M.; et al. High complete response rate with TNB-486 in relapsed/refractory follicular lymphoma: Interim results from an ongoing phase 1 study. J. Clin. Oncol. 2023, 41, 7524. [Google Scholar] [CrossRef]
- Lesch, S.; Gill, S. The promise and perils of immunotherapy. Blood Adv. 2021, 5, 3709–3725. [Google Scholar] [CrossRef] [PubMed]
- Viardot, A.; Goebeler, M.E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef]
- Brody, J.; Wahlin, B.E.; Phillips, T.J.; Costello, R.; Lugtenburg, P.; Cordoba, R.; Wang, L.; Wu, J.; Elliott, B.; Abbas, A.; et al. Epcoritamab (epco) with gemcitabine + oxaliplatin (GemOx) in patients (pts) with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) ineligible for autologous stem cell transplant (ASCT) induces high response rate even in pts failing CAR T therapy. J. Clin. Oncol. 2022, 40, 7527. [Google Scholar] [CrossRef]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142.e117. [Google Scholar] [CrossRef]
- Rezvani, A.R.; Maloney, D.G. Rituximab resistance. Best. Pract. Res. Clin. Haematol. 2011, 24, 203–216. [Google Scholar] [CrossRef]
- Hayashi, K.; Nagasaki, E.; Kan, S.; Ito, M.; Kamata, Y.; Homma, S.; Aiba, K. Gemcitabine enhances rituximab-mediated complement-dependent cytotoxicity to B cell lymphoma by CD20 upregulation. Cancer Sci. 2016, 107, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, M.; Sureda, A.; Terol, M.J.; Bosch Albareda, F.; Corradini, P.; Larsen, T.S.; Rueda Dominguez, A.; Panchal, A.; Bottos, A.; Carlile, D.; et al. Glofitamab (Glofit) in Combination with Polatuzumab Vedotin (Pola): Phase Ib/II Preliminary Data Support Manageable Safety and Encouraging Efficacy in Relapsed/Refractory (R/R) Diffuse Large B-Cell Lymphoma (DLBCL). Blood 2021, 138, 525. [Google Scholar] [CrossRef]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 155–165. [Google Scholar] [CrossRef]
- Bartlett, N.L.; Assouline, S.; Giri, P.; Schuster, S.J.; Cheah, C.Y.Y.; Matasar, M.J.; Gregory, G.P.; Yoon, D.-H.; Shadman, M.; Fay, K.; et al. Mosunetuzumab monotherapy is active and tolerable in patients with relapsed/refractory diffuse large B-cell lymphoma. Blood Adv. 2023, 7, 4926–4935. [Google Scholar] [CrossRef]
- Westin, J.; Olszewski, A.J.; Fogliatto, L.M.; Kim, W.-S.; Shin, H.-J.; Wu, H.; Yin, S.; Pham, S.; Penuel, E.; Jing, J.; et al. SUNMO: A Phase III Trial Evaluating the Efficacy and Safety of Mosunetuzumab in Combination with Polatuzumab Vedotin Versus Rituximab in Combination with Gemcitabine Plus Oxaliplatin in Patients with Relapsed or Refractory Aggressive B-Cell Non-Hodgkin Lymphoma. Blood 2022, 140, 3771–3772. [Google Scholar] [CrossRef]
- Hertzberg, M.; Ku, M.; Catalani, O.; Althaus, B.; Simko, S.; Gregory, G.P. A phase III trial evaluating glofitamab in combination with gemcitabine plus oxaliplatin versus rituximab in combination with gemcitabine and oxaliplatin in patients with relapsed/refractory (R/R) diffuse large B-cell lymphoma (DLBCL). J. Clin. Oncol. 2021, 39, TPS7575. [Google Scholar] [CrossRef]
- Engelberts, P.J.; Hiemstra, I.H.; de Jong, B.; Schuurhuis, D.H.; Meesters, J.; Beltran Hernandez, I.; Oostindie, S.C.; Neijssen, J.; van den Brink, E.N.; Horbach, G.J.; et al. DuoBody-CD3xCD20 induces potent T-cell-mediated killing of malignant B cells in preclinical models and provides opportunities for subcutaneous dosing. EBioMedicine 2020, 52, 102625. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed]
- Piccione, E.C.; Belousov, A.; Hamidi, H.; Carlo-Stella, C.; Dickinson, M.; Morschhauser, F.; Lundberg, L.; Humphrey, K.; Tracy, S.; Hutchings, M. P1210: Immune Correlates of Response to Glofitamab: Biomarker Findings from a Pivotal Phase Ii Expansion Study in Patients with Relapsed or Refractory (R/R) Diffuse Large B-Cell Lymphoma (Dlbcl). Hemasphere 2022, 6, 1096–1097. [Google Scholar] [CrossRef]
- Danhof, S.; Schreder, M.; Knop, S.; Rasche, L.; Strifler, S.; Löffler, C.; Gogishvili, T.; Einsele, H.; Hudecek, M. Expression of programmed death-1 on lymphocytes in myeloma patients is lowered during lenalidomide maintenance. Haematologica 2018, 103, e126–e129. [Google Scholar] [CrossRef]
- Merryman, R.; Belada, D.; Sureda, A.; Leppä, S.; Vermaat, J.S.P.; Holte, H.; Hutchings, M.; Lugtenburg, P.; de Vos, S.; Abrisqueta, P.; et al. Epcoritamab + R2 regimen and responses in high-risk follicular lymphoma, regardless of POD24 status. J. Clin. Oncol. 2023, 41, 7506. [Google Scholar] [CrossRef]
- Morschhauser, F.; Bishton, M.; Eyre, T.A.; Bachy, E.; Cartron, G.; Ysebaert, L.; Bobillo, S.; Gutierrez, N.C.; Budde, L.E.; Fox, C.P.; et al. Mosunetuzumab in Combination with Lenalidomide Has a Manageable Safety Profile and Encouraging Activity in Patients with Relapsed/Refractory Follicular Lymphoma: Initial Results from a Phase Ib Study. Blood 2021, 138, 129. [Google Scholar] [CrossRef]
- Falchi, L.; Morschhauser, F.; Gribben, J.G.; Huang, H.; Dinh, M.; Conlon, R.; Chen, X.; Elliot, B.; Seymour, J.F. Phase 3 Trial of Subcutaneous Epcoritamab in Combination with Rituximab and Lenalidomide (R2) Vs R2 Among Patients with Relapsed or Refractory Follicular Lymphoma (EPCORE FL-1). Blood 2022, 140, 9338–9339. [Google Scholar] [CrossRef]
- Nastoupil, L.; Morschhauser, F.; Scholz, C.W.; Bishton, M.; Yoon, S.S.; Giri, P.; Wei, M.C.; Knapp, A.; Li, C.C.; Bottos, A.; et al. P1125: Celestimo: A Phase Iii Trial Evaluating the Efficacy and Safety of Mosunetuzumab Plus Lenalidomide Versus Rituximab Plus Lenalidomide in Patients with Relapsed or Refractory Follicular Lymphoma. Hemasphere 2022, 6, 1015–1016. [Google Scholar] [CrossRef]
- Nastoupil, L.J. CELESTIMO: A Randomized Phase III Trial Examining the Efficacy and Safety of Mosunetuzumab in Combination with Lenalidomide Versus Rituximab in Combination with Lenalidomide in Relapsed/Refractory Follicular Lymphoma. Hematologist 2022, 19. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef] [PubMed]
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef]
- Wei, J.; Montalvo-Ortiz, W.; Yu, L.; Krasco, A.; Olson, K.; Rizvi, S.; Fiaschi, N.; Coetzee, S.; Wang, F.; Ullman, E.; et al. CD22-targeted CD28 bispecific antibody enhances antitumor efficacy of odronextamab in refractory diffuse large B cell lymphoma models. Sci. Transl. Med. 2022, 14, eabn1082. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, M.; Carlo-Stella, C.; Gritti, G.; Bosch, F.; Morschhauser, F.; Townsend, W.; Offner, F.; Walter, H.S.; Ghesquieres, H.; Houot, R.; et al. CD19 4-1BBL (RO7227166) a Novel Costimulatory Bispecific Antibody Can be Safely Combined with the T-Cell-Engaging Bispecific Antibody Glofitamab in Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma. Blood 2022, 140, 9461–9463. [Google Scholar] [CrossRef]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef]
- Mohamed Jiffry, M.Z.; Kloss, R.; Ahmed-Khan, M.; Carmona-Pires, F.; Okam, N.; Weeraddana, P.; Dharmaratna, D.; Dandwani, M.; Moin, K. A review of treatment options employed in relapsed/refractory AML. Hematology 2023, 28, 2196482. [Google Scholar] [CrossRef]
- Guy, D.G.; Uy, G.L. Bispecific Antibodies for the Treatment of Acute Myeloid Leukemia. Curr. Hematol. Malig. Rep. 2018, 13, 417–425. [Google Scholar] [CrossRef]
- Liu, J.; Tong, J.; Yang, H. Targeting CD33 for acute myeloid leukemia therapy. BMC Cancer 2022, 22, 24. [Google Scholar] [CrossRef]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Bögeholz, J.; Köhnke, T.; Lichtenegger, F.S.; Schneider, S.; Metzeler, K.H.; Fiegl, M.; et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood 2014, 123, 356–365. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Loken, M.; Gerbing, R.B.; Ho, P.A.; Bernstein, I.D.; Raimondi, S.C.; Hirsch, B.; Franklin, J.; Walter, R.B.; et al. Correlation of CD33 expression level with disease characteristics and response to gemtuzumab ozogamicin containing chemotherapy in childhood AML. Blood 2012, 119, 3705–3711. [Google Scholar] [CrossRef] [PubMed]
- Taussig, D.C.; Pearce, D.J.; Simpson, C.; Rohatiner, A.Z.; Lister, T.A.; Kelly, G.; Luongo, J.L.; Danet-Desnoyers, G.A.; Bonnet, D. Hematopoietic stem cells express multiple myeloid markers: Implications for the origin and targeted therapy of acute myeloid leukemia. Blood 2005, 106, 4086–4092. [Google Scholar] [CrossRef]
- Jen, E.Y.; Ko, C.W.; Lee, J.E.; Del Valle, P.L.; Aydanian, A.; Jewell, C.; Norsworthy, K.J.; Przepiorka, D.; Nie, L.; Liu, J.; et al. FDA Approval: Gemtuzumab Ozogamicin for the Treatment of Adults with Newly Diagnosed CD33-Positive Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 3242–3246. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Liu, D. Gemtuzumab ozogamicin and novel antibody-drug conjugates in clinical trials for acute myeloid leukemia. Biomark. Res. 2019, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Gutarra, M.R.; Green, S.D.; Zeidner, J.F.; Konig, H. CD123-targeted therapy in acute myeloid leukemia. Expert. Rev. Hematol. 2021, 14, 561–576. [Google Scholar] [CrossRef]
- Muñoz, L.; Nomdedéu, J.F.; López, O.; Carnicer, M.J.; Bellido, M.; Aventín, A.; Brunet, S.; Sierra, J. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica 2001, 86, 1261–1269. [Google Scholar]
- Testa, U.; Riccioni, R.; Militi, S.; Coccia, E.; Stellacci, E.; Samoggia, P.; Latagliata, R.; Mariani, G.; Rossini, A.; Battistini, A.; et al. Elevated expression of IL-3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood 2002, 100, 2980–2988. [Google Scholar] [CrossRef]
- Ravandi, F.; Bashey, A.; Stock, W.; Foran, J.M.; Mawad, R.; Egan, D.; Blum, W.; Yang, A.; Pastore, A.; Johnson, C.; et al. Complete Responses in Relapsed/Refractory Acute Myeloid Leukemia (AML) Patients on a Weekly Dosing Schedule of Vibecotamab (XmAb14045), a CD123 x CD3 T Cell-Engaging Bispecific Antibody; Initial Results of a Phase 1 Study. Blood 2020, 136, 4–5. [Google Scholar] [CrossRef]
- Watts, J.; Maris, M.; Lin, T.L.; Patel, P.; Madanat, Y.F.; Cogle, C.R.; Borthakur, G.; Huebner, D.; Khaskhely, N.; Bonham, L.; et al. Updated Results from a Phase 1 Study of APVO436, a Novel Bispecific Anti-CD123 x Anti-CD3 Adaptir™ Molecule, in Relapsed/Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome. Blood 2022, 140, 6204–6205. [Google Scholar] [CrossRef]
- Winer, E.S.; Maris, M.; Sharma, M.R.; Kaminker, P.; Zhao, E.; Ward, A.; Sochacki, A.L. A Phase 1, First-in-Human, Dose-Escalation Study of MGD024, a CD123 x CD3 Bispecific Dart® Molecule, in Patients with Relapsed or Refractory CD123-Positive (+) Hematologic Malignancies. Blood 2022, 140, 11753–11754. [Google Scholar] [CrossRef]
- Ma, H.; Padmanabhan, I.S.; Parmar, S.; Gong, Y. Targeting CLL-1 for acute myeloid leukemia therapy. J. Hematol. Oncol. 2019, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Bakker, A.B.; van den Oudenrijn, S.; Bakker, A.Q.; Feller, N.; van Meijer, M.; Bia, J.A.; Jongeneelen, M.A.; Visser, T.J.; Bijl, N.; Geuijen, C.A.; et al. C-type lectin-like molecule-1: A novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004, 64, 8443–8450. [Google Scholar] [CrossRef]
- van Rhenen, A.; van Dongen, G.A.; Kelder, A.; Rombouts, E.J.; Feller, N.; Moshaver, B.; Stigter-van Walsum, M.; Zweegman, S.; Ossenkoppele, G.J.; Jan Schuurhuis, G. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 2007, 110, 2659–2666. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, P.F.; Doornbos, R.; Dolstra, H.; Shamsili, S.; Bakker, L. Preclinical Evaluation of MCLA117, a CLEC12AxCD3 Bispecific Antibody Efficiently Targeting a Novel Leukemic Stem Cell Associated Antigen in AML. Blood 2015, 126, 325. [Google Scholar] [CrossRef]
- van Loo, P.F.; Hangalapura, B.N.; Thordardottir, S.; Gibbins, J.D.; Veninga, H.; Hendriks, L.J.A.; Kramer, A.; Roovers, R.C.; Leenders, M.; de Kruif, J.; et al. MCLA-117, a CLEC12AxCD3 bispecific antibody targeting a leukaemic stem cell antigen, induces T cell-mediated AML blast lysis. Expert. Opin. Biol. Ther. 2019, 19, 721–733. [Google Scholar] [CrossRef]
- Levis, M.; Small, D. FLT3: ITDoes matter in leukemia. Leukemia 2003, 17, 1738–1752. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbauer, F.; Kern, W.; Schoch, C.; Kohlmann, A.; Hiddemann, W.; Haferlach, T.; Schnittger, S. Detailed analysis of FLT3 expression levels in acute myeloid leukemia. Haematologica 2005, 90, 1617–1625. [Google Scholar] [PubMed]
- Durben, M.; Schmiedel, D.; Hofmann, M.; Vogt, F.; Nübling, T.; Pyz, E.; Bühring, H.J.; Rammensee, H.G.; Salih, H.R.; Große-Hovest, L.; et al. Characterization of a bispecific FLT3 X CD3 antibody in an improved, recombinant format for the treatment of leukemia. Mol. Ther. 2015, 23, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.C.; Agarwal, S.; Ahmad, H.; Amin, K.; Bewersdorf, J.P.; Zeidan, A.M. A review of FLT3 inhibitors in acute myeloid leukemia. Blood Rev. 2022, 52, 100905. [Google Scholar] [CrossRef] [PubMed]
- Antar, A.I.; Otrock, Z.K.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 inhibitors in acute myeloid leukemia: Ten frequently asked questions. Leukemia 2020, 34, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.; Große-Hovest, L.; Nübling, T.; Pyż, E.; Bamberg, M.L.; Aulwurm, S.; Bühring, H.J.; Schwartz, K.; Haen, S.P.; Schilbach, K.; et al. Generation, selection and preclinical characterization of an Fc-optimized FLT3 antibody for the treatment of myeloid leukemia. Leukemia 2012, 26, 1228–1237. [Google Scholar] [CrossRef]
- Mehta, N.K.; Pfluegler, M.; Meetze, K.; Li, B.; Sindel, I.; Vogt, F.; Marklin, M.; Heitmann, J.S.; Kauer, J.; Osburg, L.; et al. A novel IgG-based FLT3xCD3 bispecific antibody for the treatment of AML and B-ALL. J. Immunother. Cancer 2022, 10, 3882. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, G.; Steinhoff, N.; Haegel, H.; De Marco, D.; Bacac, M.; Klein, C. Novel strategies for the mitigation of cytokine release syndrome induced by T cell engaging therapies with a focus on the use of kinase inhibitors. Oncoimmunology 2022, 11, 2083479. [Google Scholar] [CrossRef] [PubMed]
- Lamba, J.K.; Chauhan, L.; Shin, M.; Loken, M.R.; Pollard, J.A.; Wang, Y.C.; Ries, R.E.; Aplenc, R.; Hirsch, B.A.; Raimondi, S.C.; et al. CD33 Splicing Polymorphism Determines Gemtuzumab Ozogamicin Response in De Novo Acute Myeloid Leukemia: Report from Randomized Phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2017, 35, 2674–2682. [Google Scholar] [CrossRef] [PubMed]
- Isidori, A.; Salvestrini, V.; Ciciarello, M.; Loscocco, F.; Visani, G.; Parisi, S.; Lecciso, M.; Ocadlikova, D.; Rossi, L.; Gabucci, E.; et al. The role of the immunosuppressive microenvironment in acute myeloid leukemia development and treatment. Expert. Rev. Hematol. 2014, 7, 807–818. [Google Scholar] [CrossRef]
- Lamble, A.J.; Lind, E.F. Targeting the Immune Microenvironment in Acute Myeloid Leukemia: A Focus on T Cell Immunity. Front. Oncol. 2018, 8, 213. [Google Scholar] [CrossRef]
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch me if you can: How AML and its niche escape immunotherapy. Leukemia 2022, 36, 13–22. [Google Scholar] [CrossRef]
- Wang, H.; Tao, Q.; Wang, Z.; Zhang, Q.; Xiao, H.; Zhou, M.; Dong, Y.; Zhai, Z. Circulating Monocytic Myeloid-Derived Suppressor Cells Are Elevated and Associated with Poor Prognosis in Acute Myeloid Leukemia. J. Immunol. Res. 2020, 2020, 7363084. [Google Scholar] [CrossRef]
- Lv, M.; Wang, K.; Huang, X.J. Myeloid-derived suppressor cells in hematological malignancies: Friends or foes. J. Hematol. Oncol. 2019, 12, 105. [Google Scholar] [CrossRef]
- Dong, Y.; Han, Y.; Huang, Y.; Jiang, S.; Huang, Z.; Chen, R.; Yu, Z.; Yu, K.; Zhang, S. PD-L1 Is Expressed and Promotes the Expansion of Regulatory T Cells in Acute Myeloid Leukemia. Front. Immunol. 2020, 11, 1710. [Google Scholar] [CrossRef]
- Zhou, Q.; Munger, M.E.; Highfill, S.L.; Tolar, J.; Weigel, B.J.; Riddle, M.; Sharpe, A.H.; Vallera, D.A.; Azuma, M.; Levine, B.L.; et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood 2010, 116, 2484–2493. [Google Scholar] [CrossRef]
- Yang, X.; Ma, L.; Zhang, X.; Huang, L.; Wei, J. Targeting PD-1/PD-L1 pathway in myelodysplastic syndromes and acute myeloid leukemia. Exp. Hematol. Oncol. 2022, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Chen, X.; Dalton, R.; Calescibetta, A.; So, T.; Gilvary, D.; Ward, G.; Smith, V.; Eckard, S.; Fox, J.A.; et al. Immunodepletion of MDSC by AMV564, a novel bivalent, bispecific CD33/CD3 T cell engager, ex vivo in MDS and melanoma. Mol. Ther. 2022, 30, 2315–2326. [Google Scholar] [CrossRef]
- Cheng, P.; Eksioglu, E.; Chen, X.; Wei, M.; Guenot, J.; Fox, J.; List, A.F.; Wei, S. Immunodepletion of MDSC By AMV564, a Novel Tetravalent Bispecific CD33/CD3 T Cell Engager Restores Immune Homeostasis in MDS in Vitro. Blood 2017, 130, 51. [Google Scholar] [CrossRef]
- Jitschin, R.; Saul, D.; Braun, M.; Tohumeken, S.; Völkl, S.; Kischel, R.; Lutteropp, M.; Dos Santos, C.; Mackensen, A.; Mougiakakos, D. CD33/CD3-bispecific T-cell engaging (BiTE®) antibody construct targets monocytic AML myeloid-derived suppressor cells. J. Immunother. Cancer 2018, 6, 116. [Google Scholar] [CrossRef]
- Cosenza, M.; Sacchi, S.; Pozzi, S. Cytokine Release Syndrome Associated with T-Cell-Based Therapies for Hematological Malignancies: Pathophysiology, Clinical Presentation, and Treatment. Int. J. Mol. Sci. 2021, 22, 7652. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, G.; Servera, L.A.; Danilin, S.; Challier, J.; Steinhoff, N.; Bossen, C.; Odermatt, A.; Nicolini, V.; Umaña, P.; Klein, C.; et al. Dissecting the mechanism of cytokine release induced by T-cell engagers highlights the contribution of neutrophils. Oncoimmunology 2022, 11, 2039432. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef]
- Sharma, S.; Wang, D.; Lon, H.-K.; Soltantabar, P.; Viqueira, A.; Czibere, A.; Hickman, A.; White, J.L.; Elmeliegy, M. A Systematic Meta-Analysis of Cytokine Release Syndrome Incidence in B-Cell Maturation Antigen-Targeting Chimeric Antigen Receptor T-Cell Therapy and Bispecific Antibodies for Patients with Relapsed and/or Refractory Multiple Myeloma. Blood 2022, 140, 10036–10038. [Google Scholar] [CrossRef]
- Li, J.; Piskol, R.; Ybarra, R.; Chen, Y.J.; Li, J.; Slaga, D.; Hristopoulos, M.; Clark, R.; Modrusan, Z.; Totpal, K.; et al. CD3 bispecific antibody-induced cytokine release is dispensable for cytotoxic T cell activity. Sci. Transl. Med. 2019, 11, eaax8861. [Google Scholar] [CrossRef]
- Ball, K.; Dovedi, S.J.; Vajjah, P.; Phipps, A. Strategies for clinical dose optimization of T cell-engaging therapies in oncology. MAbs 2023, 15, 2181016. [Google Scholar] [CrossRef]
- Trudel, S.; Bahlis, N.J.; Spencer, A.; Kaedbey, R.; Rodriguez Otero, P.; Harrison, S.J.; Wong, C.; Goodman, G.R.; Nakamura, R.; Choeurng, V.; et al. Pretreatment with Tocilizumab Prior to the CD3 Bispecific Cevostamab in Patients with Relapsed/Refractory Multiple Myeloma (RRMM) Showed a Marked Reduction in Cytokine Release Syndrome Incidence and Severity. Blood 2022, 140, 1363–1365. [Google Scholar] [CrossRef]
- Mohan, M.; Nagavally, S.; Dhakal, B.; Radhakrishnan, S.V.; Chhabra, S.; D’Souza, A.; Hari, P. Risk of infections with B-cell maturation antigen-directed immunotherapy in multiple myeloma. Blood Adv. 2022, 6, 2466–2470. [Google Scholar] [CrossRef] [PubMed]
- Mazahreh, F.; Mazahreh, L.; Schinke, C.; Thanendrarajan, S.; Zangari, M.; Shaughnessy, J.D.; Zhan, F.; van Rhee, F.; Al Hadidi, S. Risk of infections associated with the use of bispecific antibodies in multiple myeloma: A pooled analysis. Blood Adv. 2023, 7, 3069–3074. [Google Scholar] [CrossRef] [PubMed]
- Lancman, G.; Parsa, K.; Rodriguez, C.; Richter, J.; Cho, H.J.; Parekh, S.; Richard, S.; Rossi, A.; Sanchez, L.; Thibaud, S.; et al. Infections and Severe Hypogammaglobulinemia in Multiple Myeloma Patients Treated with Anti-BCMA Bispecific Antibodies. Blood 2022, 140, 10073–10074. [Google Scholar] [CrossRef]
- Hammons, L.R.; Szabo, A.; Janardan, A.; Dhakal, B.; Chhabra, S.; D’Souza, A.; Mohan, M. Kinetics of Humoral Immunodeficiency with Bispecific Antibody Therapy in Relapsed Refractory Multiple Myeloma. JAMA Netw. Open 2022, 5, e2238961. [Google Scholar] [CrossRef]
- Moreau, P.; Touzeau, C. T-cell-redirecting bispecific antibodies in multiple myeloma: A revolution? Blood 2022, 139, 3681–3687. [Google Scholar] [CrossRef]
- Juluri, K.R.; Wu, Q.V.; Voutsinas, J.; Hou, J.; Hirayama, A.V.; Mullane, E.; Miles, N.; Maloney, D.G.; Turtle, C.J.; Bar, M.; et al. Severe cytokine release syndrome is associated with hematologic toxicity following CD19 CAR T-cell therapy. Blood Adv. 2022, 6, 2055–2068. [Google Scholar] [CrossRef]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]




| BiAB, Trial | Targets | BiAB Structure | N | Design | ORR, CR (%) | CRS (All Grade, ≥Grade 3) % | ICANS (%) | Infections (%) |
|---|---|---|---|---|---|---|---|---|
| Teclistamab (Ph1-2, NCT04557098) [32] | BCMAxCD3 | Humanized IgG Fc | 165 | SQ, weekly injection at dose of 1.5 mg/kg. Step-up doses of 0.06 mg and 0.3 mg per kilogram. | 63.0, 39.4 | 72.1, 0.6 | 3.0 | 76.4 |
| Elranatamab (Ph2, NCT04649359) [33] | BCMAxCD3 | Humanized IgG2a | 123 | SQ, weekly injection at a dose of 76 mg for a 28-day cycle. Two step-up doses at 12 mg and 32 mg. | 61.0, 27.6 | 56.3, 0.0 | 3.4 | 61.8 |
| Linvoseltamab (Ph2, NCT03761108) [34] | BCMAxCD3 | Fc Fab arms | 252 | Two cohorts received doses of 50 mg and 200 mg, respectively. IV, with two step-up doses. A protocol amendment allowed pts who progressed at 50 mg to dose escalate to 200 mg. | 50 mg cohort: 50.0, 20.2 200 mg cohort: 64.0, 24.1 | 50 mg cohort: 53.0, 1.0 200 mg cohort: 37.0, 2.0 | Grade 3 or 4 50 mg cohort: 1.0 200 mg cohort: 2.0 | 50 mg cohort: 59.0 200 mg cohort: 43.0 |
| Abbv-383 (Ph1, NCT03933735) [35] | BCMAxCD3 | IgG4 Fc | 124 | IV, once every 3 weeks. Doses of 40 mg and 60 mg for escalation and expansion cohorts. | 57.0, 29.0 | 40 mg cohort: 83.0, 0.0 60 mg cohort: 72.0, 2.0 | NR | 40 mg cohort: 50.0 60 mg cohort: 43.0 |
| Talquetamab (Ph1, NCT03399799) [36] | GPRC5DxCD3 | Humanized IgG4 | 232 | 102 patients IV weekly or every other week at doses from 0.5 to 180 μg per kilogram of body weight. 130 patients SQ weekly, every other week, or monthly at doses from 5 to 1600 μg per kilogram. | At SQ doses of 405 μg/kg: 70.0, 23.0 and 800 μg/kg: 64.0, 23.0 | At SQ doses of 405 μg/kg: 77.0, 3.0 and 800 μg/kg: 80.0, 0.0 At IV doses: 49.0, 5.0 | NR | NR |
| Cevostamab (Ph1, NCT03275103) [41] | FcRH5xCD3 | Humanized IgG1 | 160 | IV administration in 21-day cycles. Two step-up doses. | At 160 mg dose: 54% At 90 mg dose: 36.7 | 80.0, 1.3 | NR | 42.5, 18.8 |
| First Author, Year | Phase | N | Study Design and Patient Population | Outcomes | Adverse Events |
|---|---|---|---|---|---|
| Kantarjian et al. 2017 [60] | 3 | 405 | Heavily pretreated relapsed/refractory Philadelphia-negative B-ALL. Randomized 2:1 comparison between blinatumomab and standard-of-care chemotherapy. | Median overall survival in blinatumomab group 7.7 months vs. 4.0 months in standard-of-care group. Complete hematologic remission in 34% in the blinatumomab group vs. 16% in the standard-of-care group. | Infection in 34.1% of the blinatumomab group vs. 52.3% in the standard-of-care group. Neurotoxicity in 9.4% in the blinatumomab group vs. 8.3% in the standard-of-care group. |
| Gökbuget et al. 2018 [67] | 2 | 116 | Open-label, single-arm study, adults with B-cell precursor ALL in hematologic complete remission with MRD (≥10−3). | MRD clearance in 78% of patients. Relapse-free survival at 18 months 54%. Median overall survival of 36.5 months. | Cytokine release syndrome in 3%. Neurotoxicity grade 3 in 10%, grade 4 in 3%. |
| Brown et al. 2021 [69] | 3 | 208 | Ages 1–30 years with first relapse B-ALL. Randomized between 2 cycles of blinatumomab and 2 cycles multi-agent chemotherapy. | 2-year disease free survival 54.4% in the blinatumomab group vs. 39.0% in the chemotherapy group. 2-year overall survival 71.3% in the blinatumomab group vs. 58.4% in the chemotherapy group. | Infection in 15.0% of the blinatumomab group vs. 65.0% in the chemotherapy group. |
| Litzow et al. 2022 [73] | 3 | 224 | Patients with negative MRD (<0.01%) post-induction therapy were randomized to either receive conventional consolidation chemotherapy or blinatumomab in addition to conventional consolidation. | Upper boundary for efficacy analysis was crossed in favor of blinatumomab, with a significant improvement in overall survival in favor of blinatumomab arm. Median overall survival not reached vs. 71.4 months, hazard ratio 0.42, p = 0.003. | NR |
| Salek et al. 2022 [74] | 2 | 29 | Single cycle of blinatumomab followed by high-dose chemotherapy in induction therapy for Philadelphia-negative adult ALL. | 93% of patients achieved complete hematological remission after induction, of which 52% were complete molecular remissions. | Febrile neutropenia in 15%, and hepatotoxicity in 11%. No neurotoxicity observed. |
| Foà et al. 2020 [15] | 2 | 63 | Philadelphia-positive ALL patients. Single-arm trial in which Dasatinib plus glucocorticoids were administered, followed by two cycles of blinatumomab. | Complete remission achieved in 98%. At median follow-up of 18 months, overall survival was 95% with disease-free survival of 88%. | Grade ≥ 3 adverse events included cytomegalovirus reactivation in 6 patients, neutropenia in 4 patients, and neurotoxicity in one patient. |
| Trial ID | Antibody Name | Targets | Patient Population | Phase | Primary Outcomes | Status |
|---|---|---|---|---|---|---|
| NCT02520427 | AMG 330 | CD33xCD3 | Relapsed/refractory AML, MDS | 1 | Safety | Terminated |
| NCT03224819 | AMG 673 | CD33xCD3 | Relapsed/refractory AML | 1 | Safety | Terminated |
| NCT03516760 | GEM333 | CD33xCD3 | Relapsed/refractory AML | 1 | Safety | Terminated |
| NCT03915379 | JNJ-67571244 | CD33xCD3 | Relapsed/refractory AML, MDS | 1 | Safety and efficacy | Completed |
| NCT03144245 | AMV564 | CD33xCD3 | Relapsed/refractory AML | 1 | Safety and efficacy | Completed |
| NCT04582864 | Flotetuzumab | CD123xCD3 | Relapsed/refractory AML | 2 | Efficacy | Recruiting |
| NCT05285813 | XmAb14045 | CD123xCD3 | Relapsed/refractory AML, MDS | 2 | Efficacy | Recruiting |
| NCT03647800 | APVO436 | CD123xCD3 | Relapsed/refractory AML, MDS | 1 | Safety | Recruiting |
| NCT05362773 | MGD024 | CD123xCD3 | Relapsed/refractory AML, MDS, Hodgkin’s lymphoma, B-cell leukemia, hairy cell leukemia, CML, systemic mastocytosis | 1 | Safety | Recruiting |
| NCT02715011 | JNJ-63709178 | CD123xCD3 | Relapsed/refractory AML | 1 | Safety | Completed |
| NCT03038230 | MCLA-117 | CLL-1xCD3 | Relapsed/refractory AML | 1 | Safety | Halted |
| NCT05143996 | CLN-049 | FLT-3xCD3 | Relapsed/refractory AML, MDS | 1 | Safety | Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omer, M.H.; Shafqat, A.; Ahmad, O.; Alkattan, K.; Yaqinuddin, A.; Damlaj, M. Bispecific Antibodies in Hematological Malignancies: A Scoping Review. Cancers 2023, 15, 4550. https://doi.org/10.3390/cancers15184550
Omer MH, Shafqat A, Ahmad O, Alkattan K, Yaqinuddin A, Damlaj M. Bispecific Antibodies in Hematological Malignancies: A Scoping Review. Cancers. 2023; 15(18):4550. https://doi.org/10.3390/cancers15184550
Chicago/Turabian StyleOmer, Mohamed H., Areez Shafqat, Omar Ahmad, Khaled Alkattan, Ahmed Yaqinuddin, and Moussab Damlaj. 2023. "Bispecific Antibodies in Hematological Malignancies: A Scoping Review" Cancers 15, no. 18: 4550. https://doi.org/10.3390/cancers15184550
APA StyleOmer, M. H., Shafqat, A., Ahmad, O., Alkattan, K., Yaqinuddin, A., & Damlaj, M. (2023). Bispecific Antibodies in Hematological Malignancies: A Scoping Review. Cancers, 15(18), 4550. https://doi.org/10.3390/cancers15184550