
Schlafen 12 Slows TNBC Tumor Growth, Induces Luminal Markers, and Predicts Favorable Survival

 , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Study
2.1.1. Animal Care
2.1.2. Animals
2.1.3. Animal Surgery
2.1.4. RNA and RT-qPCR
2.1.5. Genomics
2.2. Study Population
Xenograft Samples
2.3. Bulk-RNA Data Preprocessing
Human Samples
2.4. Statistical Methods
2.5. Functional and Pathway Analysis
2.6. Single-Cell Analysis
2.7. Computation of Signature (Module) Score for High SLFN12 Expression Biomarker
2.8. Data Availability
3. Results
3.1. Schlafen 12 Overexpression Reduces the Growth of Mammary Xenograft Tumors
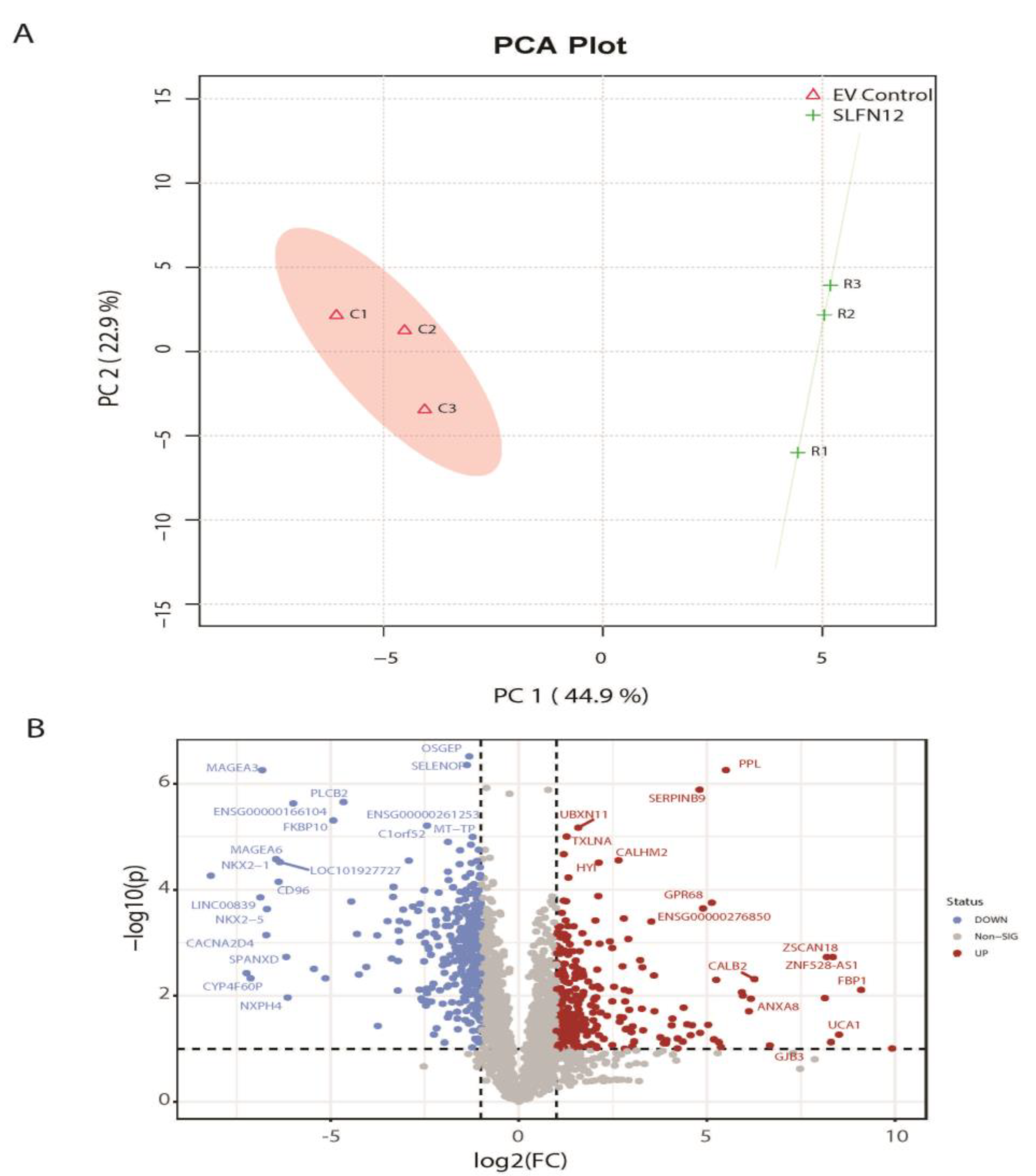
3.2. Global Gene Expression Analysis for Xenografts
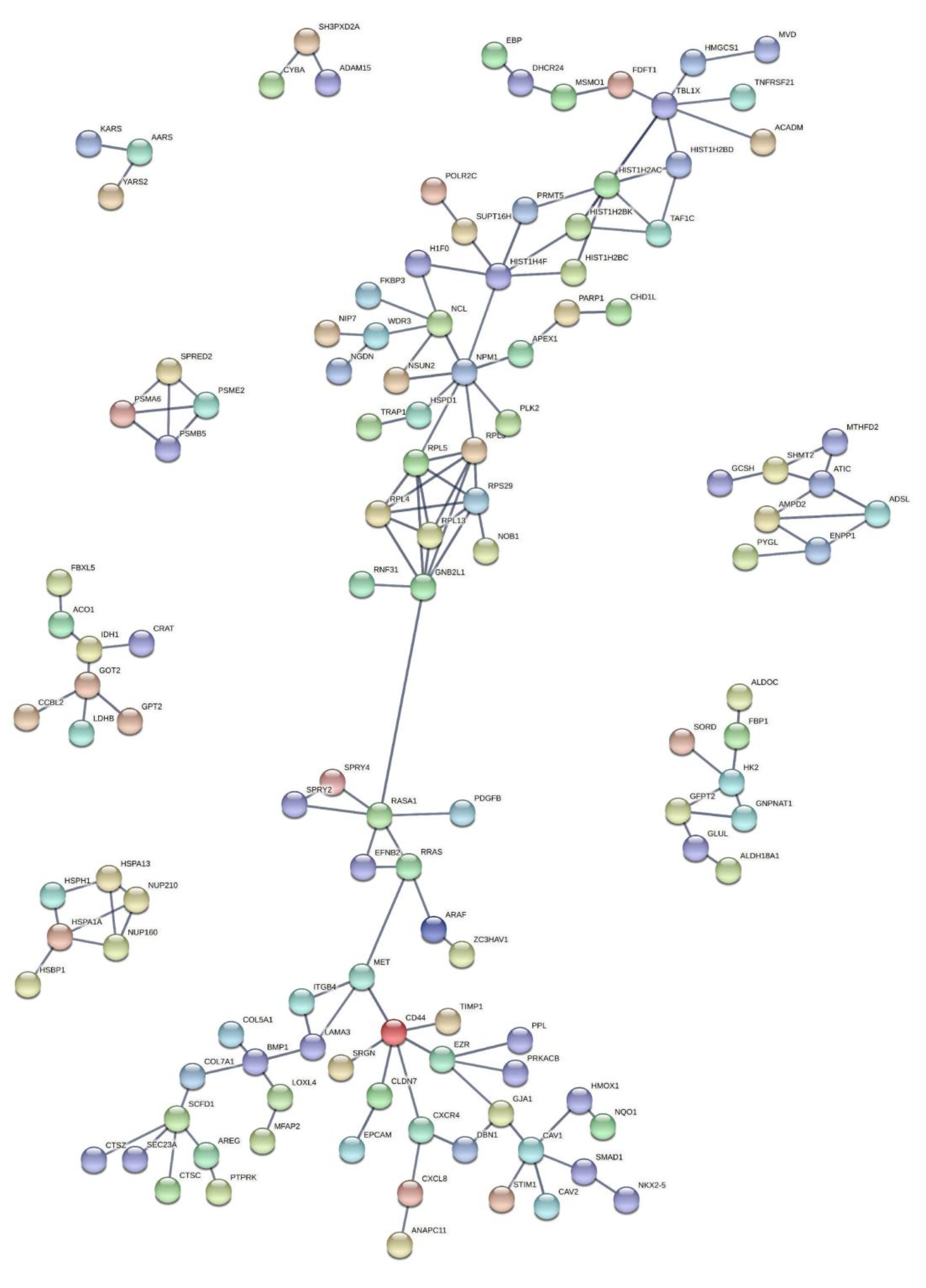
3.3. Functional and Pathway Analysis of Significant Genes
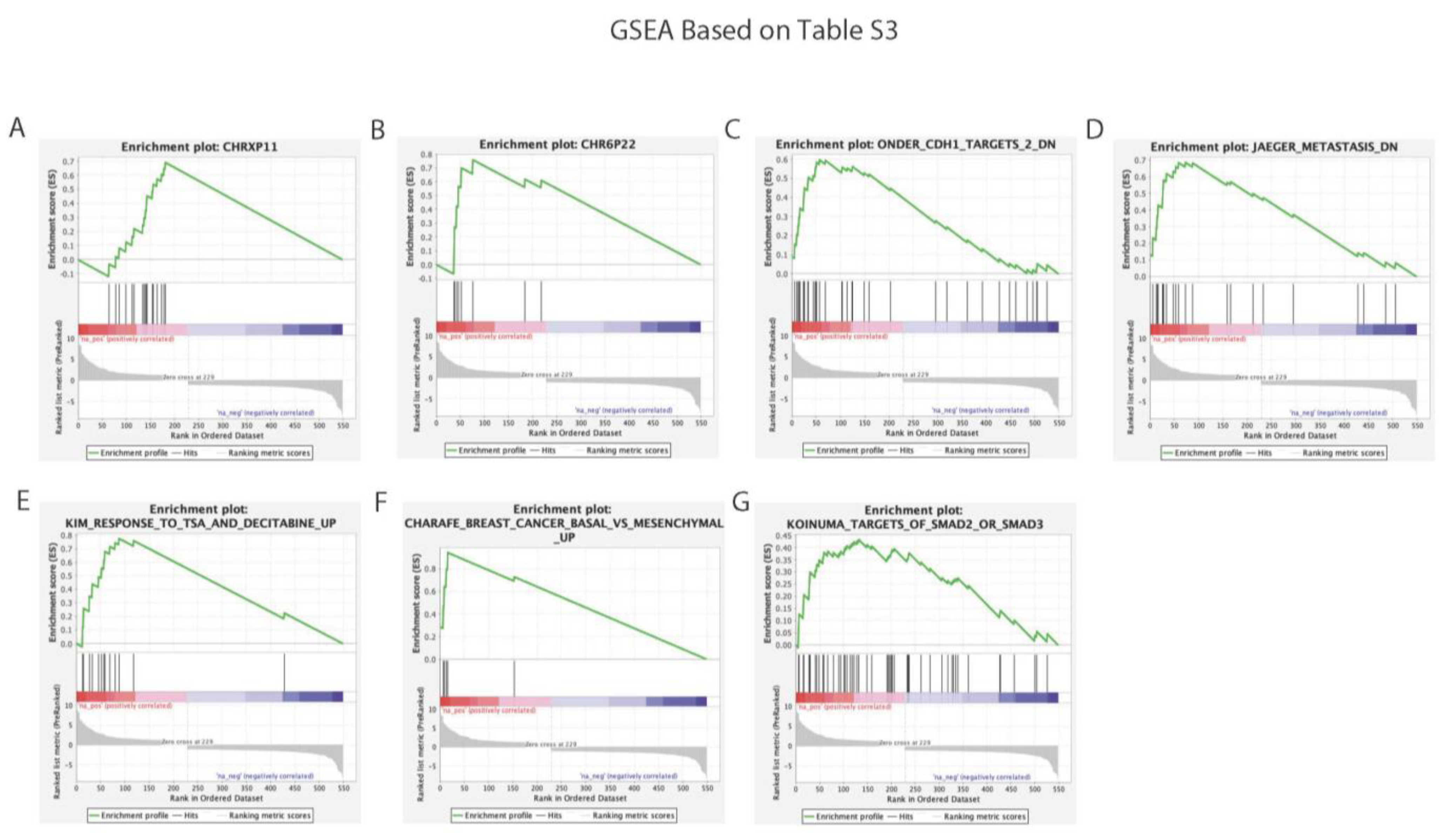
3.3.1. Functional Enrichment Analysis
3.3.2. Gene Set Enrichment Analysis
3.3.3. Ingenuity Pathway Analysis
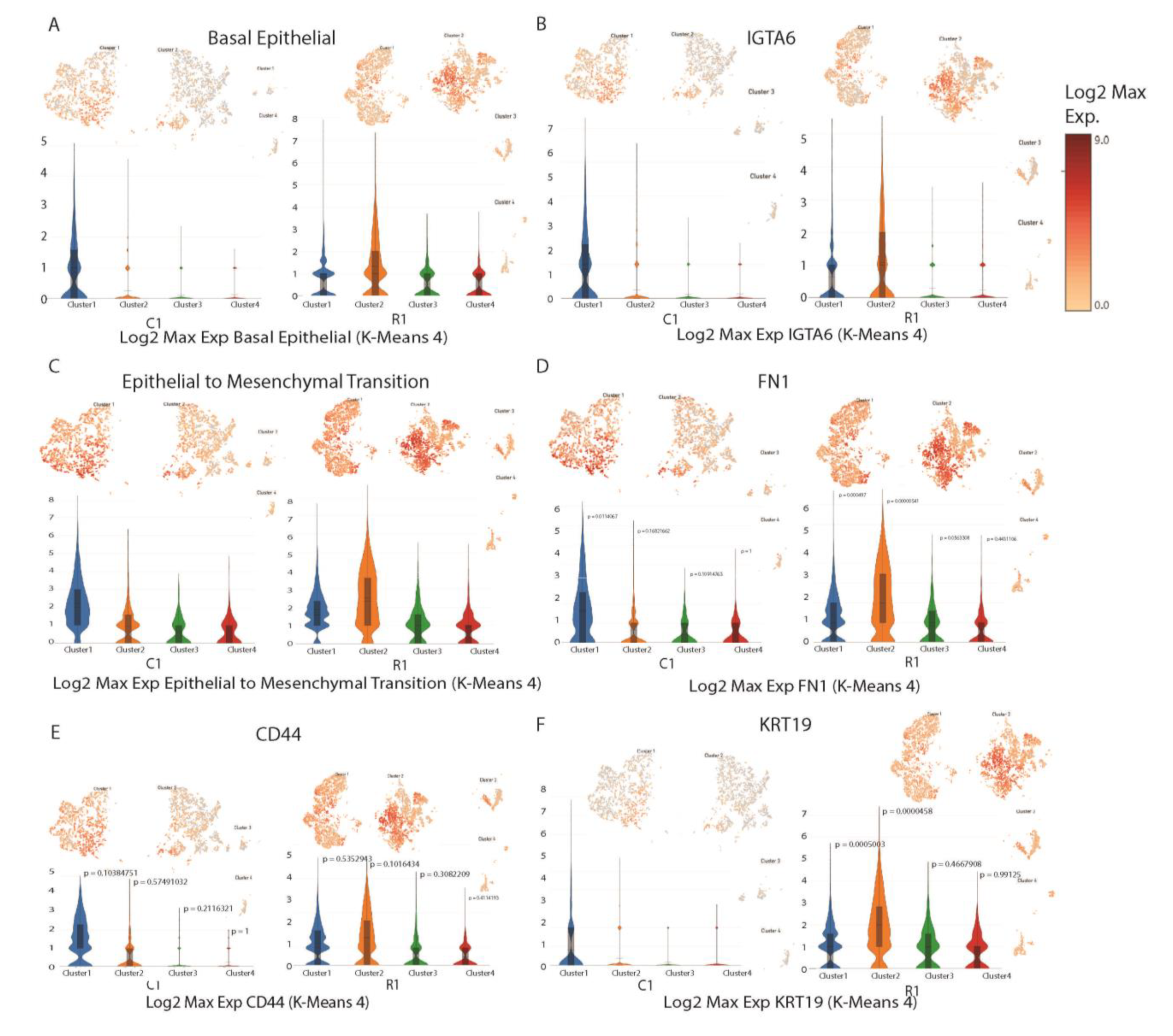
3.4. Single-Cell Analysis for Patient-Derived Xenografts
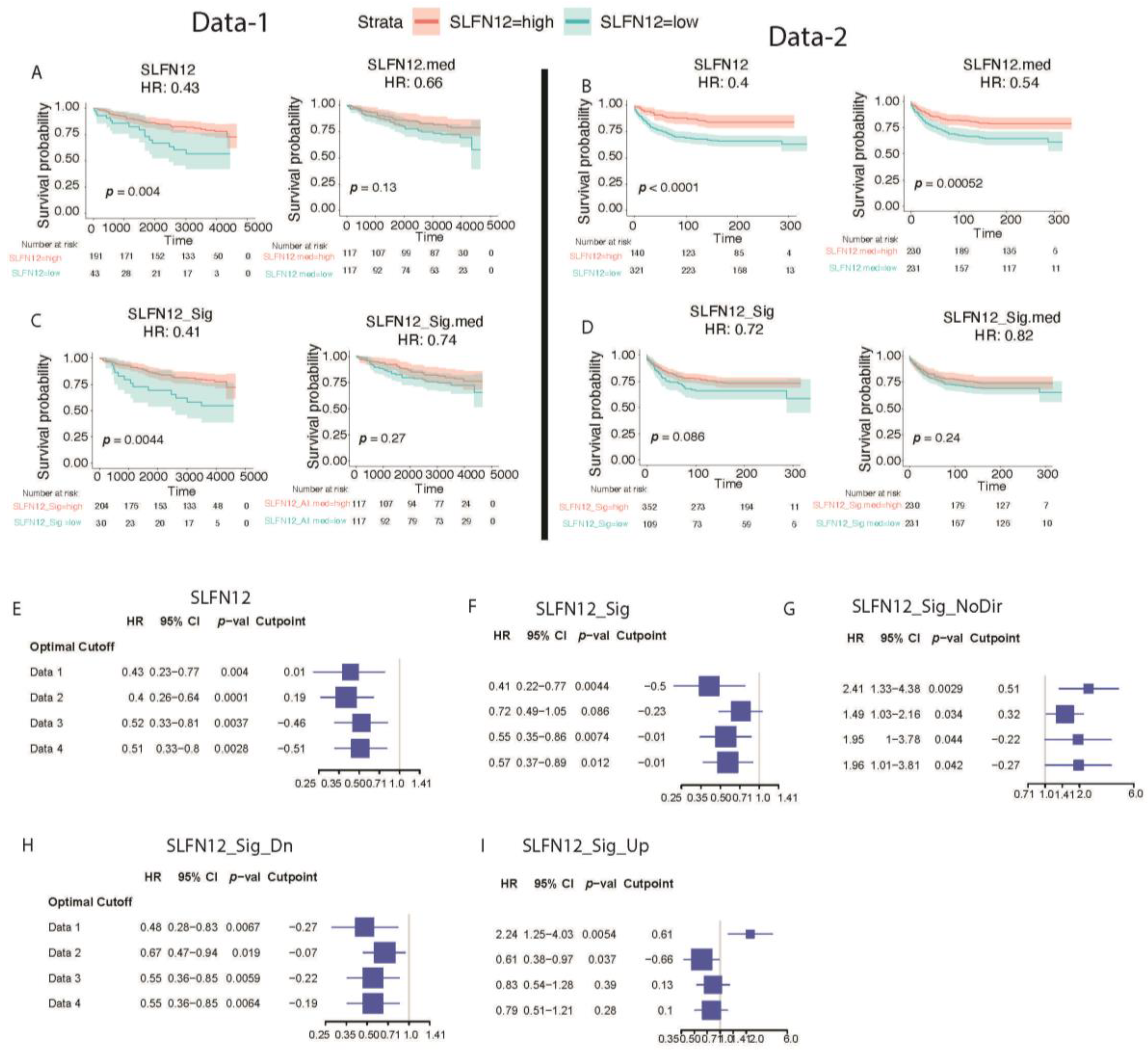
3.5. Favorable Survival with High Expression of SLFN12
Conformation qPCR Analysis of SLFN12 Signature Genes
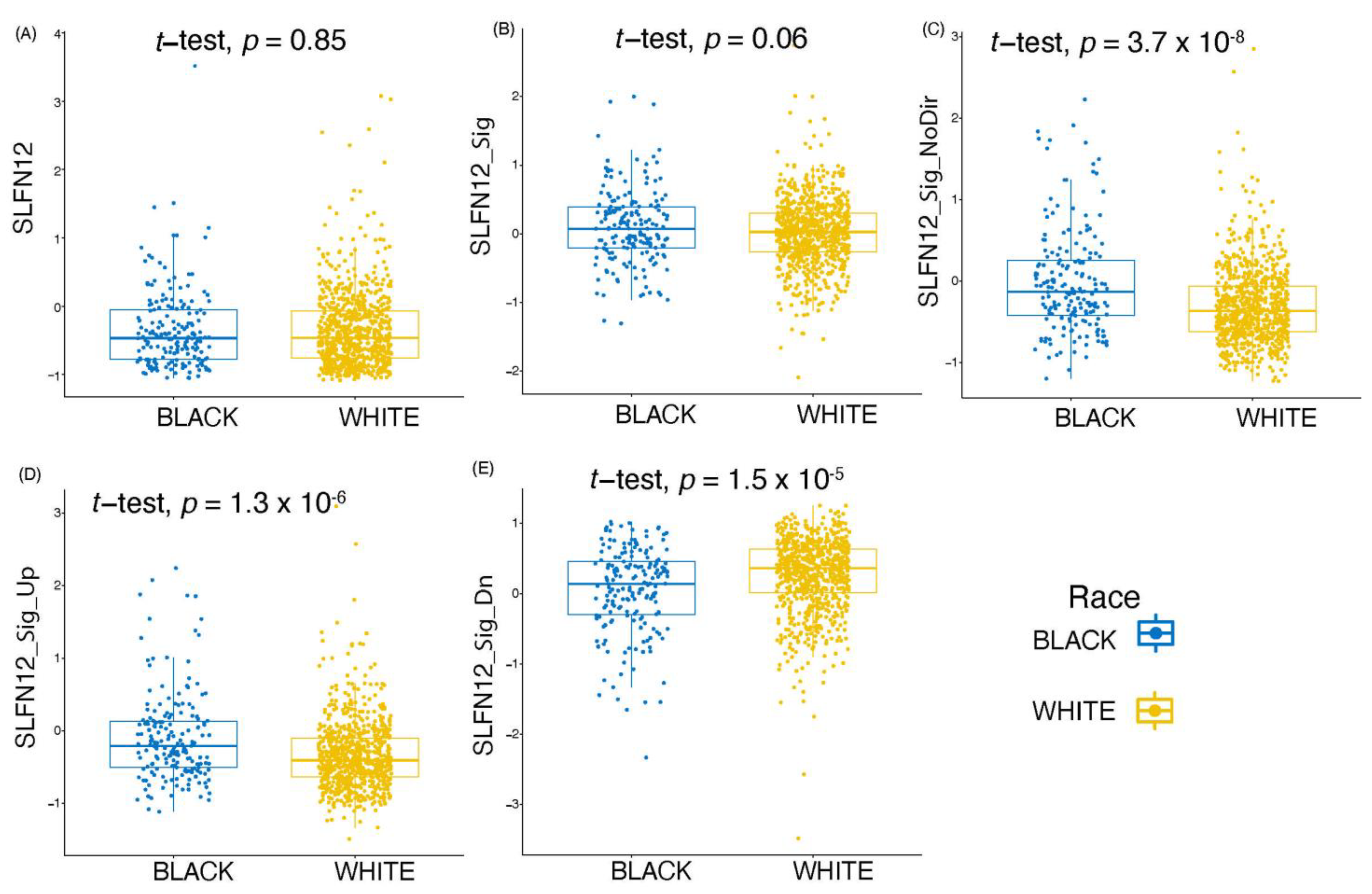
3.6. SLFN12 Expression Behaves Differently amongst Different Races
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Al-Marsoummi, S.; Vomhof-DeKrey, E.; Basson, M.D. Schlafen12 Reduces the Aggressiveness of Triple Negative Breast Cancer through Post-Transcriptional Regulation of ZEB1 That Drives Stem Cell Differentiation. Cell. Physiol. Biochem. 2019, 53, 999–1014. [Google Scholar]
- Al-Marsoummi, S.; Pacella, J.; Dockter, K.; Soderberg, M.; Singhal, S.K.; Vomhof-DeKrey, E.E.; Basson, M.D. Schlafen 12 Is Prognostically Favorable and Reduces C-Myc and Proliferation in Lung Adenocarcinoma but Not in Lung Squamous Cell Carcinoma. Cancers 2020, 12, 2738. [Google Scholar] [CrossRef] [PubMed]
- Basson Marc, D.; Wang, Q.; Chaturvedi Lakshmi, S.; More, S.; Vomhof-DeKrey Emilie, E.; Al-Marsoummi, S.; Sun, K.; Kuhn Leslie, A.; Kovalenko, P.; Kiupel, M. Schlafen 12 Interaction with SerpinB12 and Deubiquitylases Drives Human Enterocyte Differentiation. Cell. Physiol. Biochem. 2018, 48, 1274–1290. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, L.; Sun, K.; Walsh, M.F.; Kuhn, L.A.; Basson, M.D. The P-loop region of Schlafen 3 acts within the cytosol to induce differentiation of human Caco-2 intestinal epithelial cells. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 3029–3037. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.R.; Yi, M.; Cho, S.A.; Kim, W.Y.; Min, J.; Shin, J.G.; Lee, S.J. Identification and functional study of genetic polymorphisms in cyclic nucleotide phosphodiesterase 3A (PDE3A). Ann. Hum. Genet. 2021, 85, 80–91. [Google Scholar] [CrossRef]
- Rhead, B.; Brorson, I.S.; Berge, T.; Adams, C.; Quach, H.; Moen, S.M.; Berg-Hansen, P.; Celius, E.G.; Sangurdekar, D.P.; Bronson, P.G.; et al. Increased DNA methylation of SLFN12 in CD4+ and CD8+ T cells from multiple sclerosis patients. PLoS ONE 2018, 13, e0206511. [Google Scholar] [CrossRef] [Green Version]
- Kovalenko PLDVMP, Basson MDMDPMBA: Schlafen 12 expression modulates prostate cancer cell differentiation. J. Surg. Res. 2014, 190, 177–184. [CrossRef] [Green Version]
- Raafat Elsayed, A.A.; Al-Marsoummi, S.; Vomhof-Dekrey, E.E.; Basson, M.D. SLFN12 Over-expression Sensitizes Triple Negative Breast Cancer Cells to Chemotherapy Drugs and Radiotherapy. Cancer Genom. Proteom. 2022, 19, 328–338. [Google Scholar] [CrossRef]
- Wu, X.; Schnitzler, G.R.; Gao, G.F.; Diamond, B.; Baker, A.R.; Kaplan, B.; Williamson, K.; Westlake, L.; Lorrey, S.; Lewis, T.A.; et al. Mechanistic insights into cancer cell killing through interaction of phosphodiesterase 3A and schlafen family member 12. J. Biol. Chem. 2020, 295, 3431–3446. [Google Scholar] [CrossRef]
- Garvie, C.W.; Wu, X.; Papanastasiou, M.; Lee, S.; Fuller, J.; Schnitzler, G.R.; Horner, S.W.; Baker, A.; Zhang, T.; Mullahoo, J.P.; et al. Structure of PDE3A-SLFN12 complex reveals requirements for activation of SLFN12 RNase. Nat. Commun. 2021, 12, 4375. [Google Scholar] [CrossRef]
- Li, D.; Chen, J.; Ai, Y.; Gu, X.; Li, L.; Che, D.; Jiang, Z.; Li, L.; Chen, S.; Huang, H.; et al. Estrogen-Related Hormones Induce Apoptosis by Stabilizing Schlafen-12 Protein Turnover. Mol. Cell 2019, 75, 1103–1116.e1109. [Google Scholar] [CrossRef]
- Cheng, R.Y.S.; Patel, N.L.; Back, T.; Basudhar, D.; Somasundaram, V.; Kalen, J.D.; Wink, D.A.; Ridnour, L.A. Studying Triple Negative Breast Cancer Using Orthotopic Breast Cancer Model. J. Vis. Exp. 2020, 157, e60316. [Google Scholar] [CrossRef] [Green Version]
- Faustino-Rocha, A.; Oliveira, P.A.; Pinho-Oliveira, J.; Teixeira-Guedes, C.; Soares-Maia, R.; da Costa, R.G.; Colaço, B.; Pires, M.J.; Colaço, J.; Ferreira, R.; et al. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab Anim. 2013, 42, 217–224. [Google Scholar] [CrossRef]
- Euhus, D.M.; Hudd, C.; Laregina, M.C.; Johnson, F.E. Tumor measurement in the nude mouse. J. Surg. Oncol. 1986, 31, 229–234. [Google Scholar] [CrossRef]
- Du, Y.; Huang, Q.; Arisdakessian, C.; Garmire, L.X. Evaluation of STAR and Kallisto on Single Cell RNA-Seq Data Alignment. G3 Genes Genomes Genet. 2020, 10, 1775–1783. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland Jane, E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef]
- Fanidis, D.; Moulos, P. Integrative, normalization-insusceptible statistical analysis of RNA-Seq data, with improved differential expression and unbiased downstream functional analysis. Brief. Bioinform. 2021, 22, bbaa156. [Google Scholar] [CrossRef]
- Miller, L.D.; Smeds, J.; George, J.; Vega, V.B.; Vergara, L.; Ploner, A.; Pawitan, Y.; Hall, P.; Klaar, S.; Liu, E.T.; et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc. Natl. Acad. Sci. USA 2005, 102, 13550–13555. [Google Scholar] [CrossRef] [Green Version]
- Heimes, A.S.; Härtner, F.; Almstedt, K.; Krajnak, S.; Lebrecht, A.; Battista, M.J.; Edlund, K.; Brenner, W.; Hasenburg, A.; Sahin, U.; et al. Prognostic Significance of Interferon-γ and Its Signaling Pathway in Early Breast Cancer Depends on the Molecular Subtypes. Int. J. Mol. Sci. 2020, 21, 7178. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, R.; Finetti, P.; Cervera, N.; Lambaudie, E.; Esterni, B.; Mamessier, E.; Tallet, A.; Chabannon, C.; Extra, J.M.; Jacquemier, J.; et al. A gene expression signature identifies two prognostic subgroups of basal breast cancer. Breast Cancer Res. Treat. 2011, 126, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatier, R.; Finetti, P.; Adelaide, J.; Guille, A.; Borg, J.P.; Chaffanet, M.; Lane, L.; Birnbaum, D.; Bertucci, F. Down-regulation of ECRG4, a candidate tumor suppressor gene, in human breast cancer. PLoS ONE 2011, 6, e27656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovici, V.; Chen, W.; Gallas, B.G.; Hatzis, C.; Shi, W.; Samuelson, F.W.; Nikolsky, Y.; Tsyganova, M.; Ishkin, A.; Nikolskaya, T.; et al. Effect of training-sample size and classification difficulty on the accuracy of genomic predictors. Breast Cancer Res. 2010, 12, R5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J.J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Gambardella, G.; Viscido, G.; Tumaini, B.; Isacchi, A.; Bosotti, R.; di Bernardo, D. A single-cell analysis of breast cancer cell lines to study tumour heterogeneity and drug response. Nat. Commun. 2022, 13, 1714. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Singhal, S.K.; Desmedt, C.; Haibe-Kains, B.; Criscitiello, C.; Andre, F.; Loi, S.; Piccart, M.; Michiels, S.; Sotiriou, C. Gene modules and response to neoadjuvant chemotherapy in breast cancer subtypes: A pooled analysis. J. Clin. Oncol. 2012, 30, 1996–2004. [Google Scholar] [CrossRef]
- Hothorn, T.; Lausen, B. On the exact distribution of maximally selected rank statistics. Comput. Stat. Data Anal. 2003, 43, 121–137. [Google Scholar] [CrossRef]
- Kim, M.-H.; Kim, H.-B.; Yoon, S.P.; Lim, S.-C.; Cha, M.J.; Jeon, Y.J.; Park, S.G.; Chang, I.-Y.; You, H.J. Colon cancer progression is driven by APEX1-mediated upregulation of Jagged. J. Clin. Investig. 2013, 123, 3211–3230. [Google Scholar] [CrossRef] [Green Version]
- Qin, G.; Wang, X.; Ye, S.; Li, Y.; Chen, M.; Wang, S.; Qin, T.; Zhang, C.; Li, Y.; Long, Q.; et al. NPM1 upregulates the transcription of PD-L1 and suppresses T cell activity in triple-negative breast cancer. Nat. Commun. 2020, 11, 1669. [Google Scholar] [CrossRef] [Green Version]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD Metabolism in Cancer Therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Song, B.-L.; Xu, C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; He, J.; Lu, H.; Kong, Q.; Lin, S. KRT8 and KRT19, associated with EMT, are hypomethylated and overexpressed in lung adenocarcinoma and link to unfavorable prognosis. Biosci. Rep. 2020, 40, BSR20193468. [Google Scholar] [CrossRef]
- Tan, H.-S.; Jiang, W.-H.; He, Y.; Wang, D.-S.; Wu, Z.-J.; Wu, D.-S.; Gao, L.; Bao, Y.; Shi, J.-Z.; Liu, B.; et al. KRT8 upregulation promotes tumor metastasis and is predictive of a poor prognosis in clear cell renal cell carcinoma. Oncotarget 2017, 8, 76189–76203. [Google Scholar] [CrossRef] [Green Version]
- Khoshbakht, S.; Azimzadeh Jamalkandi, S.; Masudi-Nejad, A. Involvement of immune system and Epithelial-Mesenchymal-Transition in increased invasiveness of clustered circulatory tumor cells in breast cancer. BMC Med. Genom. 2021, 14, 273. [Google Scholar] [CrossRef]
- Saha, S.K.; Choi, H.Y.; Kim, B.W.; Dayem, A.A.; Yang, G.M.; Kim, K.S.; Yin, Y.F.; Cho, S.G. KRT19 directly interacts with β-catenin/RAC1 complex to regulate NUMB-dependent NOTCH signaling pathway and breast cancer properties. Oncogene 2017, 36, 332–349. [Google Scholar] [CrossRef] [Green Version]
- Rakha, E.A.; Boyce, R.W.G.; Abd El-Rehim, D.; Kurien, T.; Green, A.R.; Paish, E.C.; Robertson, J.F.R.; Ellis, I.O. Expression of mucins (MUC1, MUC2, MUC3, MUC4, MUC5AC and MUC6) and their prognostic significance in human breast cancer. Mod. Pathol. 2005, 18, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Jia, R.; Li, Z.; Liang, W.; Ji, Y.; Weng, Y.; Liang, Y.; Ning, P. Identification of key genes unique to the luminal a and basal-like breast cancer subtypes via bioinformatic analysis. World J. Surg. Oncol. 2020, 18, 268. [Google Scholar] [CrossRef] [PubMed]
- Hennigs, A.; Riedel, F.; Gondos, A.; Sinn, P.; Schirmacher, P.; Marmé, F.; Jäger, D.; Kauczor, H.-U.; Stieber, A.; Lindel, K.; et al. Prognosis of breast cancer molecular subtypes in routine clinical care: A large prospective cohort study. BMC Cancer 2016, 16, 734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capici, S.; Ammoni, L.C.; Meli, N.; Cogliati, V.; Pepe, F.F.; Piazza, F.; Cazzaniga, M.E. Personalised Therapies for Metastatic Triple-Negative Breast Cancer: When Target Is Not Everything. Cancers 2022, 14, 3729. [Google Scholar] [CrossRef] [PubMed]
- Hoeferlin, L.A.; Chalfant, E.C.; Park, M.A. Challenges in the Treatment of Triple Negative and HER2-Overexpressing Breast Cancer. J Surg. Sci. 2013, 1, 3–7. [Google Scholar] [PubMed]
- Targeted Drug Therapy|Breast Cancer Treatment. Available online: https://www.cancer.org/cancer/breast-cancer/treatment/targeted-therapy-for-breast-cancer.html (accessed on 15 August 2022).
- Ueno, N.T.; Zhang, D. Targeting EGFR in Triple Negative Breast Cancer. J. Cancer 2011, 2, 324–328. [Google Scholar] [CrossRef]
- Nakai, K.; Hung, M.C.; Yamaguchi, H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar]
- Amjad, S.; Nisar, S.; Bhat, A.A.; Shah, A.R.; Frenneaux, M.P.; Fakhro, K.; Haris, M.; Reddy, R.; Patay, Z.; Baur, J.; et al. Role of NAD+ in regulating cellular and metabolic signaling pathways. Mol. Metab. 2021, 49, 101195. [Google Scholar] [CrossRef]
- Poljšak, B. NAD + in Cancer Prevention and Treatment: Pros and Cons. J. Clin. Exp. Oncol. 2016, 5, 1000165. [Google Scholar] [CrossRef]
- González-Ortiz, A.; Galindo-Hernández, O.; Hernández-Acevedo, G.N.; Hurtado-Ureta, G.; García-González, V. Impact of cholesterol-pathways on breast cancer development, a metabolic landscape. J. Cancer 2021, 12, 4307–4321. [Google Scholar] [CrossRef]
- McCarthy, A.M.; Friebel-Klingner, T.; Ehsan, S.; He, W.; Welch, M.; Chen, J.; Kontos, D.; Domchek, S.M.; Conant, E.F.; Semine, A.; et al. Relationship of established risk factors with breast cancer subtypes. Cancer Med. 2021, 10, 6456–6467. [Google Scholar] [CrossRef]
- Chinn, J.J.; Martin, I.K.; Redmond, N. Health Equity among Black Women in the United States. J. Women Health 2021, 30, 212–219. [Google Scholar] [CrossRef]
- Ansari-Pour, N.; Zheng, Y.; Yoshimatsu, T.F.; Sanni, A.; Ajani, M.; Reynier, J.-B.; Tapinos, A.; Pitt, J.J.; Dentro, S.; Woodard, A.; et al. Whole-genome analysis of Nigerian patients with breast cancer reveals ethnic-driven somatic evolution and distinct genomic subtypes. Nat. Commun. 2021, 12, 6946. [Google Scholar] [CrossRef]
- Pollock, N.C.; Ramroop, J.R.; Hampel, H.; Troester, M.A.; Conway, K.; Hu, J.J.; Freudenheim, J.L.; Olopade, O.I.; Huo, D.; Ziv, E.; et al. Differences in somatic TP53 mutation type in breast tumors by race and receptor status. Breast Cancer Res. Treat. 2022, 192, 639–648. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C1 | |||||
|---|---|---|---|---|---|
| Cluster 1 (236) | Cluster 2 (271) | Cluster 3 (91) | Cluster 4 (0) | ||
| R1 | Cluster 1 (200) | 87 | 112 | 41 | 0 |
| Cluster 2 (225) | 99 | 126 | 40 | 0 | |
| Cluster 3 (15) | 4 | 4 | 20 | 0 | |
| Cluster 4 (25) | 6 | 19 | 19 | 0 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singhal, S.K.; Al-Marsoummi, S.; Vomhof-DeKrey, E.E.; Lauckner, B.; Beyer, T.; Basson, M.D. Schlafen 12 Slows TNBC Tumor Growth, Induces Luminal Markers, and Predicts Favorable Survival. Cancers 2023, 15, 402. https://doi.org/10.3390/cancers15020402
Singhal SK, Al-Marsoummi S, Vomhof-DeKrey EE, Lauckner B, Beyer T, Basson MD. Schlafen 12 Slows TNBC Tumor Growth, Induces Luminal Markers, and Predicts Favorable Survival. Cancers. 2023; 15(2):402. https://doi.org/10.3390/cancers15020402
Chicago/Turabian StyleSinghal, Sandeep K., Sarmad Al-Marsoummi, Emilie E. Vomhof-DeKrey, Bo Lauckner, Trysten Beyer, and Marc D. Basson. 2023. "Schlafen 12 Slows TNBC Tumor Growth, Induces Luminal Markers, and Predicts Favorable Survival" Cancers 15, no. 2: 402. https://doi.org/10.3390/cancers15020402
APA StyleSinghal, S. K., Al-Marsoummi, S., Vomhof-DeKrey, E. E., Lauckner, B., Beyer, T., & Basson, M. D. (2023). Schlafen 12 Slows TNBC Tumor Growth, Induces Luminal Markers, and Predicts Favorable Survival. Cancers, 15(2), 402. https://doi.org/10.3390/cancers15020402