
Sphingosine-1-Phosphate Recruits Macrophages and Microglia and Induces a Pro-Tumorigenic Phenotype That Favors Glioma Progression
 , , , , , , and
, , , , , , and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. PBMC Isolation and Monocyte Enrichment
2.3. Animal Models
2.4. Microglia Isolation
2.5. In Vitro Microglia-Glioma co-Culture
2.6. Chemoattraction Assays
2.7. Plasmid Construction
2.8. Lentivirus Production
2.9. Bioluminescence Imaging
2.10. Flow Cytometry and Cell Sorting
2.11. Trizol-Based RNA Isolation
2.12. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.13. Active Rac1/RhoA Pull-Down Assay
2.14. Immunoblotting
2.15. Enzyme-Linked Immunosorbent Assay (ELISA)
2.16. Griess Reagent Assay
2.17. S1P Determination by UPLC-ESI-(QqQ)-MS2
2.18. Human Glioma Datasets
2.19. Survival Analysis
2.20. Statistical Analysis
3. Results
3.1. High SPHK1 Expression Correlates with Poor Survival and High Microglia Recruitment in GB
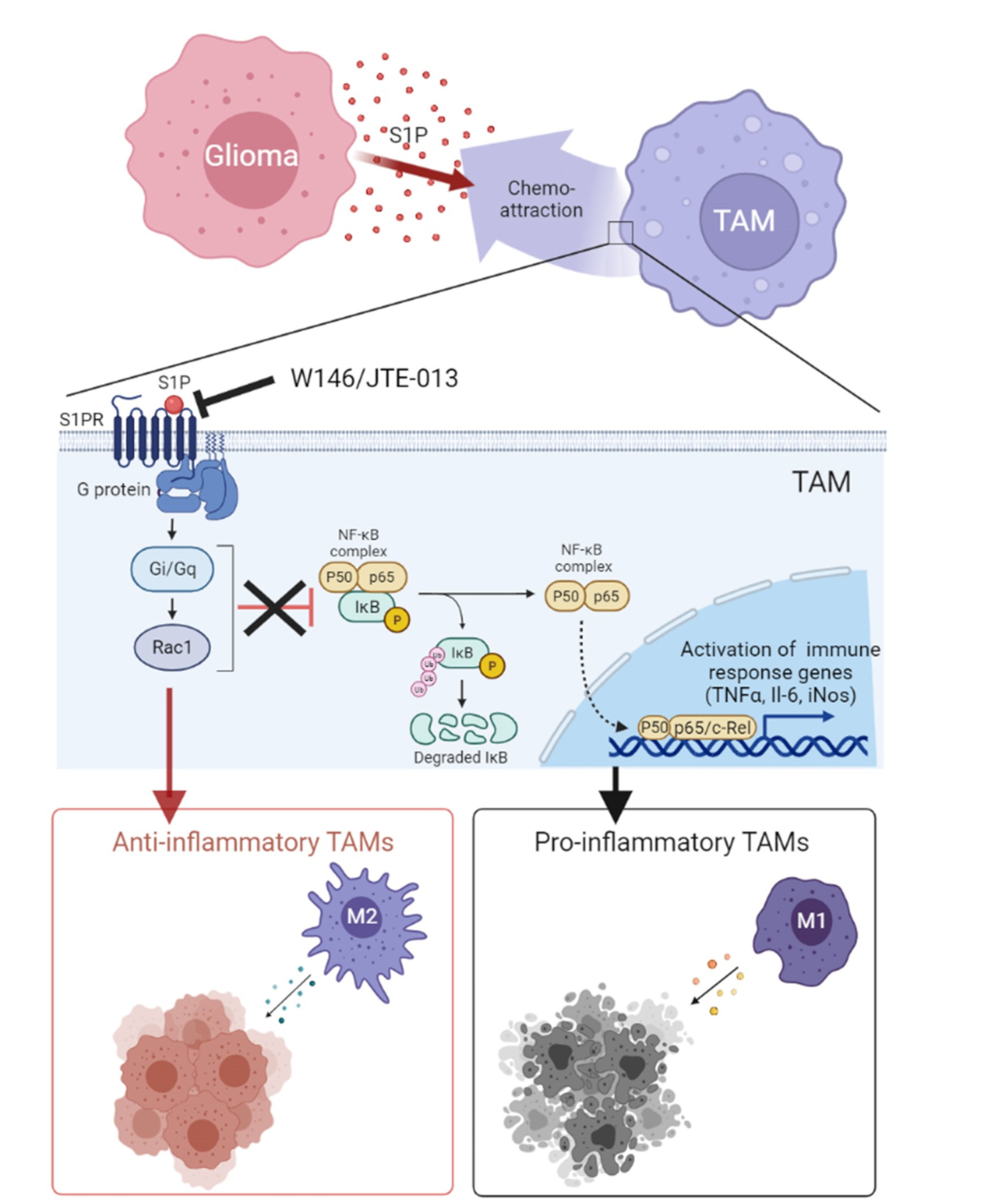
3.2. S1P Acts as a Chemoattractant for Murine Macrophages via the Modulation of Rac1/RhoA
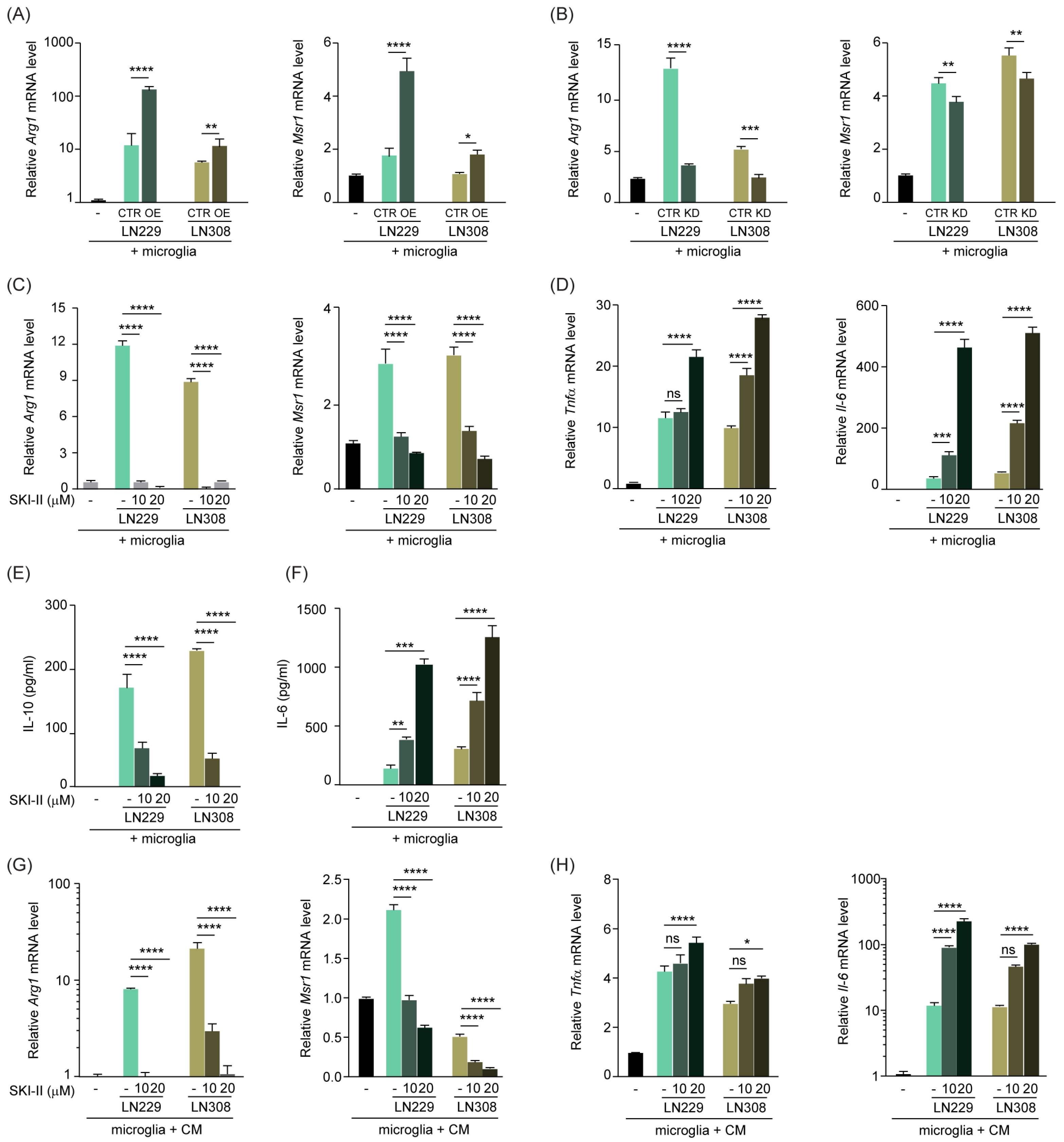
3.3. S1P Shifts TAMs to an Anti-Inflammatory Phenotype, Which Is Rescued by SPHK1 Inhibition
3.4. S1P Reduces NFkB-Mediated Inflammation via S1PR1- and S1PR2-Dependent Signaling
3.5. S1P Exerts a Supporting Role in Orthotopic Brain Tumors and Correlates with Worse Outcome in GB Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Buonfiglioli, A.; Hambardzumyan, D. Macrophages and microglia: The cerberus of glioblastoma. Acta Neuropathol. Commun. 2021, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Mildenberger, W.; Stifter, S.A.; Greter, M. Diversity and function of brain-associated macrophages. Curr. Opin. Immunol. 2022, 76, 102181. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A. The many alternative faces of macrophage activation. Front. Immunol. 2015, 6, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Pyne, S.; Pyne, N.J. Translational aspects of sphingosine 1-phosphate biology. Trends Mol. Med. 2011, 17, 463–472. [Google Scholar] [CrossRef]
- Blaho, V.A.; Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 2014, 55, 1596–1608. [Google Scholar] [CrossRef] [Green Version]
- Pyne, S.; Adams, D.R.; Pyne, N.J. Sphingosine 1-phosphate and sphingosine kinases in health and disease: Recent advances. Prog. Lipid Res. 2016, 62, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Spiegel, S. Sphingosine-1-phosphate signaling: A novel target for simultaneous adjuvant treatment of triple negative breast cancer and chemotherapy-induced neuropathic pain. Adv. Biol. Regul. 2020, 75, 100670. [Google Scholar] [CrossRef]
- Hernandez-Coronado, C.G.; Guzman, A.; Castillo-Juarez, H.; Zamora-Gutierrez, D.; Rosales-Torres, A.M. Sphingosine-1-phosphate (S1P) in ovarian physiology and disease. Ann. Endocrinol. 2019, 80, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Sukocheva, O.A.; Furuya, H.; Ng, M.L.; Friedemann, M.; Menschikowski, M.; Tarasov, V.V.; Chubarev, V.N.; Klochkov, S.G.; Neganova, M.E.; Mangoni, A.A.; et al. Sphingosine kinase and sphingosine-1-phosphate receptor signaling pathway in inflammatory gastrointestinal disease and cancers: A novel therapeutic target. Pharmacol. Therapeut. 2020, 207, 107464. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Rohrbach, T.; Milstien, S.; Spiegel, S. Role of Sphingosine Kinase 1 and Sphingosine-1-Phosphate Axis in Hepatocellular Carcinoma. Handb. Exp. Pharmacol. 2020, 259, 3–17. [Google Scholar]
- Tea, M.N.; Poonnoose, S.I.; Pitson, S.M. Targeting the Sphingolipid System as a Therapeutic Direction for Glioblastoma. Cancers 2020, 12, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.K. Understanding the Role of Sphingosine 1-Phosphate in Regulating the Microglia and Glioma Interactions. Master’s Thesis, Universitätsbibliothek Heidelberg, Heidelberg, Germany, 2019. [Google Scholar]
- Osada, M.; Yatomi, Y.; Ohmori, T.; Ikeda, H.; Ozaki, Y. Enhancement of sphingosine 1-phosphate-induced migration of vascular endothelial cells and smooth muscle cells by an EDG-5 antagonist. Biochem. Biophys. Res. Commun. 2002, 299, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Bien-Möller, S.; Lange, S.; Holm, T.; Böhm, A.; Paland, H.; Küpper, J.; Herzog, S.; Weitmann, K.; Havemann, C.; Vogelgesang, S.; et al. Expression of S1P metabolizing enzymes and receptors correlate with survival time and regulate cell migration in glioblastoma multiforme. Oncotarget 2016, 7, 13031–13046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadik, A.; Patterson, L.F.S.; Öztürk, S.; Mohapatra, S.R.; Panitz, V.; Secker, P.F.; Pfänder, P.; Loth, S.; Salem, H.; Prentzell, M.T.; et al. IL4I1 Is a Metabolic Immune Checkpoint that Activates the AHR and Promotes Tumor Progression. Cell 2020, 182, 1252–1270.e34. [Google Scholar] [CrossRef]
- Yang, Y.; Torta, F.; Arai, K.; Wenk, M.R.; Herr, D.R.; Wong, P.T.; Lai, M.K. Sphingosine kinase inhibition ameliorates chronic hypoperfusion-induced white matter lesions. Neurochem. Int. 2016, 94, 90–97. [Google Scholar] [CrossRef]
- Saura, J.; Tusell, J.M.; Serratosa, J. High-yield isolation of murine microglia by mild trypsinization. Glia 2003, 44, 183–189. [Google Scholar] [CrossRef]
- Wirthschaft, P.; Bode, J.; Soni, H.; Dietrich, F.; Krüwel, T.; Fischer, B.; Knobbe-Thomsen, C.B.; Rossetti, G.; Hentschel, A.; Mack, N.; et al. RhoA regulates translation of the Nogo-A decoy SPARC in white matter-invading glioblastomas. Acta Neuropathol. 2019, 138, 275–293. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Smyth, G.K. Camera: A competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012, 40, e133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Hothorn, T.; Lausen, B. On the exact distribution of maximally selected rank statistics. Comput. Stat. Data Anal. 2003, 43, 121–137. [Google Scholar] [CrossRef]
- Hothorn, T.; Zeileis, A. Generalized Maximally Selected Statistics. Biometrics 2008, 64, 1263–1269. [Google Scholar] [CrossRef] [Green Version]
- Lausen, B.; Schumacher, M. Maximally Selected Rank Statistics. Biometrics 1992, 48, 73–85. [Google Scholar] [CrossRef]
- Guillermet-Guibert, J.; Davenne, L.; Pchejetski, D.; Saint-Laurent, N.; Brizuela, L.; Guilbeau-Frugier, C.; Delisle, M.B.; Cuvillier, O.; Susini, C.; Bousquet, C. Targeting the sphingolipid metabolism to defeat pancreatic cancer cell resistance to the chemotherapeutic gemcitabine drug. Mol. Cancer Ther. 2009, 8, 809–820. [Google Scholar] [CrossRef] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Elmore, M.R.P.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-Stimulating Factor 1 Receptor Signaling Is Necessary for Microglia Viability, Unmasking a Microglia Progenitor Cell in the Adult Brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [Green Version]
- Payne, J.; Maher, F.; Simpson, I.; Mattice, L.; Davies, P. Glucose transporter Glut 5 expression in microglial cells. Glia 1997, 21, 327–331. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, B.A.; Beamer, M.; Ahmed, S. Fractalkine/CX3CL1: A Potential New Target for Inflammatory Diseases. Mol. Interv. 2010, 10, 263–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.-B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Bryan, A.M.; Del Poeta, M. Sphingosine-1-phosphate receptors and innate immunity. Cell Microbiol. 2018, 20, e12836. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhao, J.; Lee, J.F.; Gartung, A.; Jawadi, H.; Zhang, W.; Lominadze, D.; Lee, M.-J. 3-amino-4-(3-hexylphenylamino)-4-oxobutyl phosphonic acid (W146), a Selective Antagonist of Sphingosine-1-phospahte Receptor Subtype 1, Enhances AMD3100-stimulated Mobilization of Hematopoietic Stem Progenitor Cells in Animals. J. Biochem. Pharmacol. Res. 2013, 1, 197–203. [Google Scholar]
- Gonzalez-Cabrera, P.J.; Jo, E.; Sanna, M.G.; Brown, S.; Leaf, N.; Marsolais, D.; Schaeffer, M.-T.; Chapman, J.; Cameron, M.; Guerrero, M.; et al. Full pharmacological efficacy of a novel S1P1 agonist that does not require S1P-like headgroup interactions. Mol. Pharmacol. 2008, 74, 1308–1318. [Google Scholar] [CrossRef] [Green Version]
- French, K.J.; Schrecengost, R.S.; Lee, B.D.; Zhuang, Y.; Smith, S.N.; Eberly, J.L.; Yun, J.; Smith, C.D. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003, 63, 5962–5969. [Google Scholar]
- Xu, C.-Y.; Liu, S.-Q.; Qin, M.-B.; Zhuge, C.-F.; Qin, L.; Qin, N.; Lai, M.-Y.; Huang, J.-A. SphK1 modulates cell migration and EMT-related marker expression by regulating the expression of p-FAK in colorectal cancer cells. Int. J. Mol. Med. 2017, 39, 1277–1284. [Google Scholar] [CrossRef] [Green Version]
- Weissenberger, J.; Priester, M.; Bernreuther, C.; Rakel, S.; Glatzel, M.; Seifert, V.; Kögel, D. Dietary Curcumin Attenuates Glioma Growth in a Syngeneic Mouse Model by Inhibition of the JAK1,2/STAT3 Signaling Pathway. Clin. Cancer Res. 2010, 16, 5781–5795. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Lee, K.P.; Kang, S.; Lee, J.; Sato, K.; Chung, H.Y.; Okajima, F.; Im, D.-S. Sphingosine 1-phosphate induced anti-atherogenic and atheroprotective M2 macrophage polarization through IL-4. Cell. Signal. 2014, 26, 2249–2258. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.E.; Srinivasan, S.; Lynch, K.R.; Proia, R.L.; Ferdek, P.; Hedrick, C.C. Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circ. Res. 2008, 102, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.J.; Lynch, C.L.; Neway, W.; Mills, S.G.; Hajdu, R.; Keohane, C.A.; Rosenbach, M.J.; Milligan, J.A.; Shei, G.-J.; Parent, S.A.; et al. A rational utilization of high-throughput screening affords selective, orally bioavailable 1-benzyl-3-carboxyazetidine sphingosine-1-phosphate-1 receptor agonists. J. Med. Chem. 2004, 47, 6662–6665. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [Green Version]
- Engler, J.R.; Robinson, A.E.; Smirnov, I.; Hodgson, J.G.; Berger, M.S.; Gupta, N.; James, C.D.; Molinaro, A.; Phillips, J.J. Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS ONE 2012, 7, e43339. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.H.; Hu, B.L.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [Green Version]
- Anton, K.; Baehring, J.M.; Mayer, T. Glioblastoma multiforme: Overview of current treatment and future perspectives. Hematol./Oncol. Clin. North Am. 2012, 26, 825–853. [Google Scholar] [CrossRef]
- Paugh, B.S.; Bryan, L.; Paugh, S.; Wilczynska, K.M.; Alvarez, S.M.; Singh, S.K.; Kapitonov, D.; Rokita, H.; Wright, S.; Griswold-Prenner, I.; et al. Interleukin-1 regulates the expression of sphingosine kinase 1 in glioblastoma cells. J. Biol. Chem. 2009, 284, 3408–3417. [Google Scholar] [CrossRef] [Green Version]
- Young, N.; Pearl, D.K.; Van Brocklynl, J.R. Sphingosine-1-Phosphate Regulates Glioblastoma Cell Invasiveness through the Urokinase Plasminogen Activator System and CCN1/Cyr61. Mol. Cancer Res. 2009, 7, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Shida, D.; Takabe, K.; Kapitonov, D.; Milstien, S.; Spiegel, S. Targeting SphK1 as a new strategy against cancer. Curr. Drug Targets 2008, 9, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Murph, M.; Mills, G.B. Targeting the lipids LPA and S1P and their signalling pathways to inhibit tumour progression. Expert Rev. Mol. Med. 2007, 9, 1–18. [Google Scholar] [CrossRef]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2011, 59, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Van Brocklyn, J.R.; Jackson, C.A.; Pearl, D.K.; Kotur, M.S.; Snyder, P.J.; Prior, T.W. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: Roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J. Neuropath. Exp. Neur. 2005, 64, 695–705. [Google Scholar] [CrossRef]
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.; Daniels, B.; Laaksonen, M.A.; Teo, C.; et al. A Metabolic Shift Favoring Sphingosine 1-Phosphate at the Expense of Ceramide Controls Glioblastoma Angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef] [Green Version]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, B.; Bai, Y.; Liu, Y.H.; Zhang, B.Y.; Jin, J. Upregulation of sphingosine kinase 1 is associated with recurrence and poor prognosis in papillary thyroid carcinoma. Oncol. Lett. 2019, 18, 5374–5382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gachechiladze, M.; Tichy, T.; Kolek, V.; Grygarkova, I.; Klein, J.; Mgebrishvili, G.; Kharaishvili, G.; Janíková, M.; Smičková, P.; Cierna, L.; et al. Sphingosine kinase-1 predicts overall survival outcomes in non-small cell lung cancer patients treated with carboplatin and navelbine. Oncol. Lett. 2019, 18, 1259–1266. [Google Scholar] [CrossRef] [Green Version]
- Acharya, S.; Yao, J.; Li, P.; Zhang, C.Y.; Lowery, F.J.; Zhang, Q.L.; Guo, H.; Qu, J.; Yang, F.; Wistuba., I.I.; et al. Sphingosine Kinase 1 Signaling Promotes Metastasis of Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 4211–4226. [Google Scholar] [CrossRef] [Green Version]
- Bae, G.E.; Do, S.I.; Kim, K.; Park, J.H.; Cho, S.; Kim, H.S. Increased Sphingosine Kinase 1 Expression Predicts Distant Metastasis and Poor Outcome in Patients with Colorectal Cancer. Anticancer Res. 2019, 39, 663–670. [Google Scholar] [CrossRef]
- Gude, D.R.; Alvarez, S.E.; Paugh, S.W.; Mitra, P.; Yu, J.D.; Griffiths, R.; Barbour, S.E.; Milstien, S.; Spiegel, S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. Faseb J. 2008, 22, 2629–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chumanevich, A.; Wedman, P.; Oskeritzian, C.A. Sphingosine-1-Phosphate/Sphingosine-1-Phosphate Receptor 2 Axis Can Promote Mouse and Human Primary Mast Cell Angiogenic Potential through Upregulation of Vascular Endothelial Growth Factor-A and Matrix Metalloproteinase-2. Mediat. Inflamm. 2016, 2016, 1503206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelbaset-Ismail, A.; Cymer, M.; Borkowska-Rzeszotek, S.; Brzeźniakiewicz-Janus, K.; Rameshwar, P.; Kakar, S.S.; Ratajczak, J.; Ratajczak, M.Z. Bioactive Phospholipids Enhance Migration and Adhesion of Human Leukemic Cells by Inhibiting Heme Oxygenase 1 (HO-1) and Inducible Nitric Oxygenase Synthase (iNOS) in a p38 MAPK-Dependent Manner. Stem Cell Rev. Rep. 2019, 15, 139–154. [Google Scholar] [CrossRef]
- Schneider, G.; Sellers, Z.P.; Bujko, K.; Kakar, S.S.; Kucia, M.; Ratajczak, M.Z. Novel pleiotropic effects of bioactive phospholipids in human lung cancer metastasis. Oncotarget 2017, 8, 58247–58263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malchinkhuu, E.; Sato, K.; Maehama, T.; Mogi, C.; Tomura, H.; Ishiuchi, S.; Yoshimoto, Y.; Kurose, H.; Okajima, F. S1P(2) receptors mediate inhibition of glioma cell migration through Rho signaling pathways independent of PTEN. Biochem. Biophys. Res. Commun. 2008, 366, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Kim, M.-K.; Lee, H.Y.; Kim, S.D.; Lee, S.Y.; Kim, J.M.; Ryu, S.H.; Bae, Y.-S. S1P stimulates chemotactic migration and invasion in OVCAR3 ovarian cancer cells. Biochem. Biophys. Res. Commun. 2007, 356, 239–244. [Google Scholar] [CrossRef]
- Schneider, G.; Bryndza, E.; Abdel-Latif, A.; Ratajczak, J.; Maj, M.; Tarnowski, M.; Klyachkin, Y.M.; Houghton, P.; Morris, A.J.; Vater, A.; et al. Bioactive Lipids S1P and C1P Are Prometastatic Factors in Human Rhabdomyosarcoma, and Their Tissue Levels Increase in Response to Radio/Chemotherapy. Mol. Cancer Res. 2013, 11, 793–807. [Google Scholar] [CrossRef] [Green Version]
- Weigert, A.; Tzieply, N.; Von Knethen, A.; Johann, A.M.; Schmidt, H.; Geisslinger, G.; Brüne, B. Tumor cell apoptosis polarizes macrophages-Role of sphingosine-1-phosphate. Mol. Biol. Cell 2007, 18, 3810–3819. [Google Scholar] [CrossRef] [Green Version]
- Weigert, A.; Johann, A.M.; von Knethen, A.; Schmidt, H.; Geisslinger, G.; Brune, B. Apoptotic cells promote macrophage survival by releasing the antiapoptotic mediator sphingosine-1-phosphate. Blood 2006, 108, 1635–1642. [Google Scholar] [CrossRef] [Green Version]
- Green, J.A.; Suzuki, K.; Cho, B.; Willison, L.D.; Palmer, D.; Allen, C.D.C.; Schmidt, T.H.; Xu, Y.; Proia, R.; Coughlin, S.R.; et al. The sphingosine 1-phosphate receptor S1P(2) maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat. Immunol. 2011, 12, 672–680. [Google Scholar] [CrossRef]
- Sugimoto, N.; Takuwa, N.; Okamoto, H.; Sakurada, S.; Takuwa, Y. Inhibitory and stimulatory regulation of Rac and cell motility by the G12/13-Rho and Gi pathways integrated downstream of a single G protein-coupled sphingosine-1-phosphate receptor isoform. Mol. Cell Biol. 2003, 23, 1534–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brecht, K.; Weigert, A.; Hu, J.; Popp, R.; Fisslthaler, B.; Korff, T.; Fleming, I.; Geisslinger, G.; Brüne, B. Macrophages programmed by apoptotic cells promote angiogenesis via prostaglandin E2. FASEB J. 2011, 25, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Campos, L.S.; Rodriguez, Y.I.; Leopoldino, A.M.; Hait, N.C.; Lopez Bergami, P.; Castro, M.G.; Sanchez, E.S.; Maceyka, M.; Spiegel, S.; Alvarez, S.E. Filamin A Expression Negatively Regulates Sphingosine-1-Phosphate-Induced NF-kappaB Activation in Melanoma Cells by Inhibition of Akt Signaling. Mol. Cell Biol. 2016, 36, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [Green Version]
- Maceyka, M.; Alvarez, S.E.; Milstien, S.; Spiegel, S. Filamin a links sphingosine kinase 1 and sphingosine-1-phosphate receptor 1 at lamellipodia to orchestrate cell migration. Mol. Cell Biol. 2008, 28, 5687–5697. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, A.; Ellinger-Ziegelbauer, H.; Franzoso, G.; Brown, K.; Siebenlist, U. Physical and functional interaction of filamin (actin-binding protein-280) and tumor necrosis factor receptor-associated factor 2. J. Biol. Chem. 2000, 275, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Deng, J.H.; Wang, L.; Lee, H.; Armstrong, B.; Scuto, A.; Kowolik, C.; Weiss, L.M.; Forman, S.; Yu, H. S1PR1 is an effective target to block STAT3 signaling in activated B cell-like diffuse large B-cell lymphoma. Blood 2012, 120, 1458–1465. [Google Scholar] [CrossRef]
- Chia, K.; Mazzolini, J.; Mione, M.; Sieger, D. Tumor initiating cells induce Cxcr4-mediated infiltration of pro-tumoral macrophages into the brain. Elife 2018, 7, e31918. [Google Scholar] [CrossRef]
- Tian, Y.X.; Ke, Y.Q.; Ma, Y.X. High expression of stromal signatures correlated with macrophage infiltration, angiogenesis and poor prognosis in glioma microenvironment. Peerj 2020, 8, e9038. [Google Scholar] [CrossRef]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro-Oncology 2006, 8, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrusiewicz, K.; Ellert-Miklaszewska, A.; Lipko, M.; Sielska, M.; Frankowska, M.; Kaminska, B. Characteristics of the Alternative Phenotype of Microglia/Macrophages and its Modulation in Experimental Gliomas. PLoS ONE 2011, 6, e23902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, J.C.; Gonzalez, G.C.; Zhang, L.; Ibrahim, G.; Kelly, J.J.; Gustafson, M.P.; Lin, Y.; Dietz, A.B.; Forsyth, P.A.; Yong, V.W.; et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro-Oncology 2010, 12, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [Green Version]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef]
- Guo, X.D.; Ji, J.; Xue, T.F.; Sun, Y.Q.; Guo, R.B.; Cheng, H.; Sun, X.L. FTY720 Exerts Anti-Glioma Effects by Regulating the Glioma Microenvironment Through Increased CXCR4 Internalization by Glioma-Associated Microglia. Front. Immunol. 2020, 11, 178. [Google Scholar] [CrossRef] [Green Version]
- Chiba, K.; Adachi, K. Discovery of fingolimod, the sphingosine 1-phosphate receptor modulator and its application for the therapy of multiple sclerosis. Future Med. Chem. 2012, 4, 771–781. [Google Scholar] [CrossRef]
- Hawkins, C.C.; Ali, T.; Ramanadham, S.; Hjelmeland, A.B. Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate. Biomolecules 2020, 10, 1357. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arseni, L.; Sharma, R.; Mack, N.; Nagalla, D.; Ohl, S.; Hielscher, T.; Singhal, M.; Pilz, R.; Augustin, H.; Sandhoff, R.; et al. Sphingosine-1-Phosphate Recruits Macrophages and Microglia and Induces a Pro-Tumorigenic Phenotype That Favors Glioma Progression. Cancers 2023, 15, 479. https://doi.org/10.3390/cancers15020479
Arseni L, Sharma R, Mack N, Nagalla D, Ohl S, Hielscher T, Singhal M, Pilz R, Augustin H, Sandhoff R, et al. Sphingosine-1-Phosphate Recruits Macrophages and Microglia and Induces a Pro-Tumorigenic Phenotype That Favors Glioma Progression. Cancers. 2023; 15(2):479. https://doi.org/10.3390/cancers15020479
Chicago/Turabian StyleArseni, Lavinia, Rakesh Sharma, Norman Mack, Deepthi Nagalla, Sibylle Ohl, Thomas Hielscher, Mahak Singhal, Robert Pilz, Hellmut Augustin, Roger Sandhoff, and et al. 2023. "Sphingosine-1-Phosphate Recruits Macrophages and Microglia and Induces a Pro-Tumorigenic Phenotype That Favors Glioma Progression" Cancers 15, no. 2: 479. https://doi.org/10.3390/cancers15020479
APA StyleArseni, L., Sharma, R., Mack, N., Nagalla, D., Ohl, S., Hielscher, T., Singhal, M., Pilz, R., Augustin, H., Sandhoff, R., Herold-Mende, C., Tews, B., Lichter, P., & Seiffert, M. (2023). Sphingosine-1-Phosphate Recruits Macrophages and Microglia and Induces a Pro-Tumorigenic Phenotype That Favors Glioma Progression. Cancers, 15(2), 479. https://doi.org/10.3390/cancers15020479