
Targeting Translation and the Cell Cycle Inversely Affects CTC Metabolism but Not Metastasis

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
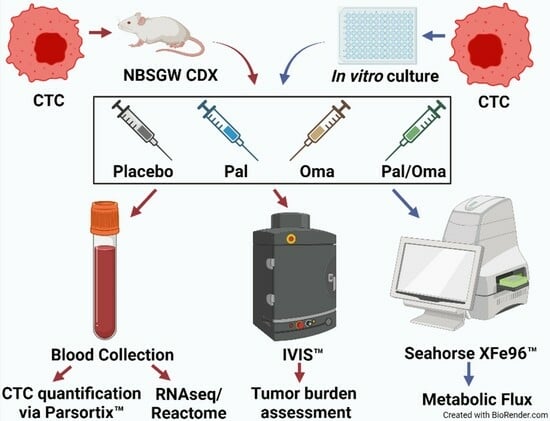
2. Materials and Methods
2.1. Patients’ Blood Collection
2.2. CTC-Derived Xenografts (CDXs)
2.3. CTC/CTC Cluster Capture
2.4. RNA Sequencing
2.5. RNA-Seq Analyses and Bioinformatics
2.6. Cell Culture
2.7. Blood Separation and Lineage Sorting
2.8. Metabolic Flux Analysis on Patient-Derived PBMCs and CTC-Derived Clone
2.9. mRNA Isolation and RT-qPCR
3. Results
3.1. The Translation Machinery and/or Cell Proliferation Are Critical for Successful CTC Micro/Macrometastases
3.2. Inhibition of Translation and/or Cell Proliferation Decreased CTC Numbers and Altered Gene Signatures
3.3. Metabolic Profiling of CTCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- In, G.K.; Poorman, K.; Saul, M.; O’Day, S.; Farma, J.M.; Olszanski, A.J.; Gordon, M.S.; Thomas, J.S.; Eisenberg, B.; Flaherty, L. Molecular profiling of melanoma brain metastases compared to primary cutaneous melanoma and to extracranial metastases. Oncotarget 2020, 11, 3118. [Google Scholar] [CrossRef]
- Eroglu, Z.; Holmen, S.L.; Chen, Q.; Khushalani, N.I.; Amaravadi, R.; Thomas, R.; Ahmed, K.A.; Tawbi, H.; Chandra, S.; Markowitz, J. Melanoma central nervous system metastases: An update to approaches, challenges, and opportunities. Pigment Cell Melanoma Res. 2019, 32, 458–469. [Google Scholar] [CrossRef]
- Cohen, J.V.; Tawbi, H.; Margolin, K.A.; Amravadi, R.; Bosenberg, M.; Brastianos, P.K.; Chiang, V.L.; de Groot, J.; Glitza, I.C.; Herlyn, M.J.P.c. Melanoma central nervous system metastases: Current approaches, challenges, and opportunities. Pigment Cell Melanoma Res. 2016, 29, 627–642. [Google Scholar] [CrossRef]
- Zakrzewski, J.; Geraghty, L.N.; Rose, A.E.; Christos, P.J.; Mazumdar, M.; Polsky, D.; Shapiro, R.; Berman, R.; Darvishian, F.; Hernando, E. Clinical variables and primary tumor characteristics predictive of the development of melanoma brain metastases and post-brain metastases survival. Cancer 2011, 117, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.A.; Liu, P.; McIntyre, S.; Kim, K.B.; Papadopoulos, N.; Hwu, W.J.; Hwu, P.; Bedikian, A. Prognostic factors for survival in melanoma patients with brain metastases. Cancer 2011, 117, 1687–1696. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Carter, J.H.; Friedman, A.H.; Seigler, H.F. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J. Neurosurg. 1998, 88, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef]
- Fife, K.; Colman, M.; Stevens, G.; Firth, I.; Moon, D.; Shannon, K.; Harman, R.; Petersen-Schaefer, K.; Zacest, A.; Besser, M. Determinants of outcome in melanoma patients with cerebral metastases. J. Clin. Oncol. 2004, 22, 1293–1300. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Schur, S.; Füreder, L.M.; Gatterbauer, B.; Dieckmann, K.; Widhalm, G.; Hainfellner, J.; Zielinski, C.C.; Birner, P.; Bartsch, R. Descriptive statistical analysis of a real life cohort of 2419 patients with brain metastases of solid cancers. ESMO Open 2016, 1, e000024. [Google Scholar] [CrossRef]
- Zoga, E.; Wolff, R.; Ackermann, H.; Meissner, M.; Rödel, C.; Tselis, N.; Chatzikonstantinou, G. Factors associated with hemorrhage of melanoma brain metastases after stereotactic radiosurgery in the era of targeted/immune checkpoint inhibitor therapies. Cancers 2022, 14, 2391. [Google Scholar] [CrossRef]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef]
- Giuliano, M.; Shaikh, A.; Lo, H.C.; Arpino, G.; De Placido, S.; Zhang, X.H.; Cristofanilli, M.; Schiff, R.; Trivedi, M.V. Perspective on circulating tumor cell clusters: Why it takes a village to metastasize. Cancer Res. 2018, 78, 845–852. [Google Scholar] [CrossRef]
- Vishnoi, M.; Boral, D.; Marchetti, D. Transcriptomic Analysis of CTCs. In Circulating Tumor Cells: Advances in Liquid Biopsy Technologies; Springer: Berlin/Heidelberg, Germany, 2023; pp. 277–299. [Google Scholar]
- Ildiz, E.S.; Gvozdenovic, A.; Kovacs, W.J.; Aceto, N. Travelling under pressure—Hypoxia and shear stress in the metastatic journey. Clin. Exp. Metastasis 2023, 40, 375–394. [Google Scholar] [CrossRef] [PubMed]
- Godet, I.; Shin, Y.J.; Ju, J.A.; Ye, I.C.; Wang, G.; Gilkes, D.M. Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nat. Commun. 2019, 10, 4862. [Google Scholar] [CrossRef] [PubMed]
- Werner-Klein, M.; Scheitler, S.; Hoffmann, M.; Hodak, I.; Dietz, K.; Lehnert, P.; Naimer, V.; Polzer, B.; Treitschke, S.; Werno, C. Genetic alterations driving metastatic colony formation are acquired outside of the primary tumour in melanoma. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.A. Selection and adaptation during metastatic cancer progression. Nature 2013, 501, 365–372. [Google Scholar] [CrossRef]
- Gomis, R.R.; Gawrzak, S. Tumor cell dormancy. Mol. Oncol. 2017, 11, 62–78. [Google Scholar] [CrossRef]
- Marrella, A.; Fedi, A.; Varani, G.; Vaccari, I.; Fato, M.; Firpo, G.; Guida, P.; Aceto, N.; Scaglione, S. High blood flow shear stress values are associated with circulating tumor cells cluster disaggregation in a multi-channel microfluidic device. PLoS ONE 2021, 16, e0245536. [Google Scholar] [CrossRef]
- Dianat-Moghadam, H.; Azizi, M.; Eslami-S, Z.; Cortés-Hernández, L.E.; Heidarifard, M.; Nouri, M.; Alix-Panabières, C. The role of circulating tumor cells in the metastatic cascade: Biology, technical challenges, and clinical relevance. Cancers 2020, 12, 867. [Google Scholar] [CrossRef]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C.; Riethdorf, S. Cancer micrometastases. Nat. Rev. Clin. Oncol. 2009, 6, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Lucci, A.; Hall, C.S.; Patel, S.P.; Narendran, B.; Bauldry, J.B.; Royal, R.E.; Karhade, M.; Upshaw, J.R.; Wargo, J.A.; Glitza, I.C.J.C.C.R. Circulating tumor cells and early relapse in node-positive melanoma. Clin. Cancer Res. 2020, 26, 1886–1895. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Sprouse, M.L.; Welte, T.; Boral, D.; Liu, H.N.; Yin, W.; Vishnoi, M.; Goswami-Sewell, D.; Li, L.; Pei, G.; Jia, P. PMN-MDSCs enhance CTC metastatic properties through reciprocal interactions via ROS/Notch/Nodal signaling. Int. J. Mol. Sci. 2019, 20, 1916. [Google Scholar] [CrossRef]
- Donato, C.; Kunz, L.; Castro-Giner, F.; Paasinen-Sohns, A.; Strittmatter, K.; Szczerba, B.M.; Scherrer, R.; Di Maggio, N.; Heusermann, W.; Biehlmaier, O. Hypoxia triggers the intravasation of clustered circulating tumor cells. Cell Rep. 2020, 32, 108105. [Google Scholar] [CrossRef]
- Michor, F.; Liphardt, J.; Ferrari, M.; Widom, J. What does physics have to do with cancer? Nat. Rev. Cancer 2011, 11, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Bowley, T.Y.; Lagutina, I.V.; Francis, C.; Sivakumar, S.; Selwyn, R.G.; Taylor, E.; Guo, Y.; Fahy, B.N.; Tawfik, B.; Marchetti, D. The RPL/RPS gene signature of melanoma CTCs associates with brain metastasis. Cancer Res. Commun. 2022, 2, 1436–1448. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Ebright, R.Y.; Haber, D.A.; Maheswaran, S. Translational regulation of cancer metastasis. Cancer Res. 2021, 81, 517–524. [Google Scholar] [CrossRef]
- Ebright, R.Y.; Lee, S.; Wittner, B.S.; Niederhoffer, K.L.; Nicholson, B.T.; Bardia, A.; Truesdell, S.; Wiley, D.F.; Wesley, B.; Li, S. Deregulation of ribosomal protein expression and translation promotes breast cancer metastasis. Science 2020, 367, 1468–1473. [Google Scholar] [CrossRef]
- Elhamamsy, A.R.; Metge, B.J.; Alsheikh, H.A.; Shevde, L.A.; Samant, R.S. Ribosome biogenesis: A central player in cancer metastasis and therapeutic resistance. Cancer Res. 2022, 82, 2344–2353. [Google Scholar] [CrossRef]
- Robichaud, N.; Sonenberg, N.; Ruggero, D.; Schneider, R. Translational control in cancer. Cold Spring Harb. Perspect. Biol. 2019, 11, a032896. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Li, X.; Nian, W.; Wang, J.; Wang, X.; Sun, L.; Zhu, Y.; Tong, Z. Ribosome proteins represented by RPL27A mark the development and metastasis of triple-negative breast cancer in mouse and human. Front. Cell Dev. Biol. 2021, 9, 716730. [Google Scholar] [CrossRef]
- Volarević, S.a.; Stewart, M.J.; Ledermann, B.; Zilberman, F.; Terracciano, L.; Montini, E.; Grompe, M.; Kozma, S.C.; Thomas, G. Proliferation, but not growth, blocked by conditional deletion of 40 S ribosomal protein S6. Science 2000, 288, 2045–2047. [Google Scholar] [CrossRef]
- Gandhi, V.; Plunkett, W.; Cortes, J.E. Omacetaxine: A protein translation inhibitor for treatment of chronic myelogenous leukemia. Clin. Cancer Res. 2014, 20, 1735–1740. [Google Scholar] [CrossRef] [PubMed]
- Wetzler, M.; Segal, D. Omacetaxine as an anticancer therapeutic: What is old is new again. Curr. Pharm. Des. 2011, 17, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Rosshandler, Y.; Shen, A.Q.; Cortes, J.; Khoury, H.J. Omacetaxine mepesuccinate for chronic myeloid leukemia. Expert Rev. Hematol. 2016, 9, 419–424. [Google Scholar] [CrossRef]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K. Palbociclib in hormone-receptor–positive advanced breast cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ze’ev, A.R.; Osterman, A.L.; Smith, J.W. Comparative metabolic flux profiling of melanoma cell lines: Beyond the Warburg effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef]
- Fischer, G.M.; Guerrieri, R.A.; Hu, Q.; Joon, A.Y.; Kumar, S.; Haydu, L.E.; McQuade, J.L.; Vashisht Gopal, Y.; Knighton, B.; Deng, W. Clinical, molecular, metabolic, and immune features associated with oxidative phosphorylation in melanoma brain metastases. Neuro-Oncol. Adv. 2021, 3, vdaa177. [Google Scholar] [CrossRef]
- Bulfoni, M.; Turetta, M.; Del Ben, F.; Di Loreto, C.; Beltrami, A.P.; Cesselli, D. Dissecting the heterogeneity of circulating tumor cells in metastatic breast cancer: Going far beyond the needle in the haystack. Int. J. Mol. Sci. 2016, 17, 1775. [Google Scholar] [CrossRef]
- Alix-Panabieres, C.; Cayrefourcq, L.; Mazard, T.; Maudelonde, T.; Assenat, E.; Assou, S. Molecular portrait of metastasis-competent circulating tumor cells in colon cancer reveals the crucial role of genes regulating energy metabolism and DNA repair. Clin. Chem. 2017, 63, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, F.; Yao, J.; Mao, C.; Zhu, M.; Qian, M.; Hu, J.; Zhong, H.; Zhou, J.; Shi, X.J. Single-cell metabolic fingerprints discover a cluster of circulating tumor cells with distinct metastatic potential. Nat. Commun. 2023, 14, 2485. [Google Scholar] [CrossRef] [PubMed]
- Stocking, K.L.; Jones, J.C.; Everds, N.E.; Buetow, B.S.; Roudier, M.P.; Miller, R.E. Use of Low-Molecular–Weight Heparin to Decrease Mortality in Mice after Intracardiac Injection of Tumor Cells. Comp. Med. 2009, 59, 37–45. [Google Scholar]
- Devlin, J.R.; Hannan, K.M.; Hein, N.; Cullinane, C.; Kusnadi, E.; Ng, P.Y.; George, A.J.; Shortt, J.; Bywater, M.J.; Poortinga, G. Combination therapy targeting ribosome biogenesis and mRNA translation synergistically extends survival in MYC-driven lymphoma. Cancer Discov. 2016, 6, 59–70. [Google Scholar] [CrossRef]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef]
- Frerich, C.A.; Brayer, K.J.; Painter, B.M.; Kang, H.; Mitani, Y.; El-Naggar, A.K.; Ness, S.A. Transcriptomes define distinct subgroups of salivary gland adenoid cystic carcinoma with different driver mutations and outcomes. Oncotarget 2018, 9, 7341. [Google Scholar] [CrossRef]
- Brown, R.B.; Madrid, N.J.; Suzuki, H.; Ness, S.A. Optimized approach for Ion Proton RNA sequencing reveals details of RNA splicing and editing features of the transcriptome. PLoS ONE 2017, 12, e0176675. [Google Scholar] [CrossRef]
- Pauken, C.M.; Kenney, S.R.; Brayer, K.J.; Guo, Y.; Brown-Glaberman, U.A.; Marchetti, D. Heterogeneity of Circulating Tumor Cell Neoplastic Subpopulations Outlined by Single-Cell Transcriptomics. Cancers 2021, 13, 4885. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology. R Package Version 2.28.0. Cranio 2016. Package. Available online: https://bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 5 March 2023).
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B. Reactome: A database of reactions, pathways, and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar] [CrossRef]
- Lill, M.A.; Danielson, M.L. Computer-aided drug design platform using PyMOL. J. Comput.-Aided Mol. Des. 2011, 25, 13–19. [Google Scholar] [CrossRef]
- Khatter, H.; Myasnikov, A.G.; Natchiar, S.K.; Klaholz, B.P. Structure of the human 80S ribosome. Nature 2015, 520, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Vishnoi, M.; Boral, D.; Liu, H.; Sprouse, M.L.; Yin, W.; Goswami-Sewell, D.; Tetzlaff, M.T.; Davies, M.A.; Oliva, I.C.G.; Marchetti, D. Targeting USP7 identifies a metastasis-competent state within bone marrow–resident melanoma CTCs. Cancer Res. 2018, 78, 5349–5362. [Google Scholar] [CrossRef]
- Lee, A.S.; Wu, J.C. Imaging of embryonic stem cell migration in vivo. In Stem Cell Migration; Springer: Berlin/Heidelberg, Germany, 2011; pp. 101–114. [Google Scholar]
- Tessema, B.; Riemer, J.; Sack, U.; König, B. Cellular Stress Assay in Peripheral Blood Mononuclear Cells: Factors Influencing Its Results. Int. J. Mol. Sci. 2022, 23, 13118. [Google Scholar] [CrossRef]
- König, B.; Lahodny, J. Ozone high dose therapy (OHT) improves mitochondrial bioenergetics in peripheral blood mononuclear cells. Transl. Med. Commun. 2022, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- De Vitis, C.; Capalbo, C.; Torsello, A.; Napoli, C.; Salvati, V.; Loffredo, C.; Blandino, G.; Piaggio, G.; Auciello, F.R.; Pelliccia, F. Opposite Effect of Thyroid Hormones on Oxidative Stress and on Mitochondrial Respiration in COVID-19 Patients. Antioxidants 2022, 11, 1998. [Google Scholar] [CrossRef] [PubMed]
- Amintas, S.; Bedel, A.; Moreau-Gaudry, F.; Boutin, J.; Buscail, L.; Merlio, J.-P.; Vendrely, V.; Dabernat, S.; Buscail, E. Circulating tumor cell clusters: United we stand divided we fall. Int. J. Mol. Sci. 2020, 21, 2653. [Google Scholar] [CrossRef]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef]
- Van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef]
- Kang, J.; Brajanovski, N.; Chan, K.T.; Xuan, J.; Pearson, R.B.; Sanij, E. Ribosomal proteins and human diseases: Molecular mechanisms and targeted therapy. Signal Transduct. Target. Ther. 2021, 6, 323. [Google Scholar] [CrossRef] [PubMed]
- Sulima, S.O.; Kampen, K.R.; De Keersmaecker, K. Cancer Biogenesis in Ribosomopathies. Cells 2019, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Filipp, F.V.; Ratnikov, B.; De Ingeniis, J.; Smith, J.W.; Osterman, A.L.; Scott, D.A. Glutamine-fueled mitochondrial metabolism is decoupled from glycolysis in melanoma. Pigment Cell Melanoma Res. 2012, 25, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Genuth, N.R.; Barna, M. The discovery of ribosome heterogeneity and its implications for gene regulation and organismal life. Mol. Cell 2018, 71, 364–374. [Google Scholar] [CrossRef]
- Guimaraes, J.C.; Zavolan, M. Patterns of ribosomal protein expression specify normal and malignant human cells. Genome Biol. 2016, 17, 1–13. [Google Scholar] [CrossRef]
- de Las Heras-Rubio, A.; Perucho, L.; Paciucci, R.; Vilardell, J.; ME, L.L. Ribosomal proteins as novel players in tumorigenesis. Cancer Metastasis Rev. 2014, 33, 115–141. [Google Scholar] [CrossRef]
- Bortoluzzi, S.; d’Alessi, F.; Romualdi, C.; Danieli, G.A. Differential expression of genes coding for ribosomal proteins in different human tissues. Bioinformatics 2001, 17, 1152–1157. [Google Scholar] [CrossRef]
- Henry, J.L.; Coggin, D.L.; King, C.R. High-Level Expression of the Ribosomal Protein L19 in Human Breast Tumors That Overexpress erb B-2. Cancer Res. 1993, 53, 1403–1408. [Google Scholar]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef]
- Pelletier, J.; Thomas, G.; Volarević, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Penzo, M.; Montanaro, L.; Treré, D.; Derenzini, M. The Ribosome Biogenesis-Cancer Connection. Cells 2019, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G. An encore for ribosome biogenesis in the control of cell proliferation. Nat. Cell Biol. 2000, 2, E71–E72. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, J. Ribosome heterogeneity in stem cells and development. J. Cell Biol. 2020, 219, e202001108. [Google Scholar] [CrossRef]
- Zhang, Y.; Wolf, G.W.; Bhat, K.; Jin, A.; Allio, T.; Burkhart, W.A.; Xiong, Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell Biol. 2003, 23, 8902–8912. [Google Scholar] [CrossRef]
- Shi, Z.; Fujii, K.; Kovary, K.M.; Genuth, N.R.; Röst, H.L.; Teruel, M.N.; Barna, M. Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs genome-wide. Mol. Cell 2017, 67, 71–83.e77. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Liu, Y.; Yu, X.Y.; Pan, X.; Zhang, Y.; Tu, J.; Song, Y.H.; Li, Y. Ribosome biogenesis in disease: New players and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 15. [Google Scholar] [CrossRef]
- Wei, J.; Kishton, R.J.; Angel, M.; Conn, C.S.; Dalla-Venezia, N.; Marcel, V.; Vincent, A.; Catez, F.; Ferré, S.; Ayadi, L.; et al. Ribosomal Proteins Regulate MHC Class I Peptide Generation for Immunosurveillance. Mol. Cell 2019, 73, 1162–1173.e1165. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef]
- Ruggero, D. The role of Myc-induced protein synthesis in cancer. Cancer Res. 2009, 69, 8839–8843. [Google Scholar] [CrossRef]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Ban, Y.; Tan, Y.; Xiong, W.; Li, G.; Xiang, B. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 and 4: A pair of valves for fine-tuning of glucose metabolism in human cancer. Mol. Metab. 2019, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, M.; Szczudło, J.; Pietrzyk, A.; Shah, J.; Trojan, S.E.; Ostrowska, B.; Kocemba-Pilarczyk, K.A. The Warburg effect: A score for many instruments in the concert of cancer and cancer niche cells. Pharmacol. Rep. 2023, 75, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Sittewelle, M.; Kappès, V.; Zhou, C.; Lécuyer, D.; Monsoro-Burq, A.H. PFKFB4 interacts with ICMT and activates RAS/AKT signaling-dependent cell migration in melanoma. Life Sci. Alliance 2022, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Kiesel, V.A.; Sheeley, M.P.; Coleman, M.F.; Cotul, E.K.; Donkin, S.S.; Hursting, S.D.; Wendt, M.K.; Teegarden, D. Pyruvate carboxylase and cancer progression. Cancer Metab. 2021, 9, 1–13. [Google Scholar] [CrossRef]
- Lin, Q.; He, Y.; Wang, X.; Zhang, Y.; Hu, M.; Guo, W.; He, Y.; Zhang, T.; Lai, L.; Sun, Z.; et al. Targeting Pyruvate Carboxylase by a Small Molecule Suppresses Breast Cancer Progression. Adv. Sci. 2020, 7, 1903483. [Google Scholar] [CrossRef]
- Rattigan, K.M.; Brabcova, Z.; Sarnello, D.; Zarou, M.M.; Roy, K.; Kwan, R.; de Beauchamp, L.; Dawson, A.; Ianniciello, A.; Khalaf, A.; et al. Pyruvate anaplerosis is a targetable vulnerability in persistent leukaemic stem cells. Nat. Commun. 2023, 14, 4634. [Google Scholar] [CrossRef]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 2013, 5, ra148–ra180. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Challenges in circulating tumour cell research. Nat. Rev. Cancer 2014, 14, 623–631. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTC Clusters | Placebo | Omacetaxine | Palbociclib | Pal/Oma |
|---|---|---|---|---|
| CTCs/mL | ||||
| Single-cell | 60 | 15 | 35 | 10 |
| 2-cell | 20 | 0 | 10 | 0 |
| 3-cell | 10 | 0 | 0 | 0 |
| 4-cell | 10 | 0 | 0 | 0 |
| 5-cell or greater | 5 | 0 | 0 | 0 |
| Placebo | Omacetaxine | Palbociclib | Both Drugs | CTC Clone | |
|---|---|---|---|---|---|
| RPL12 | 2.14 | 1.71 | 4.81 | 7.95 | 11.4 |
| RPL13 | 5.1 | 3.14 | 5.55 | 3.2 | 177.59 |
| RPL18A | 1 | 2.43 | 1.77 | 1.73 | 16.78 |
| RPL19 | 81.1 | 108.64 | 154.07 | 143.33 | 43.91 |
| RPL26 | 3.29 | 1.71 | 3.27 | 4.35 | 6.5 |
| RPL37 | 15.35 | 15.98 | 22.96 | 6.72 | 73.96 |
| RPL38 | 1.91 | 3.85 | 5.6 | 2.39 | 27.73 |
| RPL7 | 3.29 | 2.42 | 1 | 1.73 | 15.61 |
| RPL7A | 8.72 | 5.27 | 1 | 1 | 46.48 |
| RPS18 | 8.96 | 3.85 | 1.75 | 5.82 | 22.42 |
| RPL6 | 3.05 | 1 | 1 | 1.65 | 7.07 |
| RPL35A | 4.62 | 1.71 | 5.57 | 3.78 | 69.09 |
| RPL23 | 13.01 | 2.43 | 4.81 | 7.37 | 35.69 |
| RPS12 | 60.17 | 48.06 | 33.63 | 81.2 | 17.56 |
| RPS15A | 1 | 1 | 1 | 1 | 20.35 |
| RPS24 | 1 | 1 | 1.75 | 4.35 | 24.82 |
| RPS26 | 1 | 1 | 1 | 1 | 3.51 |
| RPS28 | 1 | 1 | 1 | 1 | 12.91 |
| RPS5 | 5.1 | 1 | 2.54 | 9.42 | 15.37 |
| RPS7 | 1.91 | 1.71 | 2.5 | 3.12 | 5.03 |
| RPSA | 12.39 | 4.56 | 7.09 | 20.05 | 28.92 |
| Average | 11.2 | 10.17 | 12.56 | 14.87 | 32.51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bowley, T.Y.; Merkley, S.D.; Lagutina, I.V.; Ortiz, M.C.; Lee, M.; Tawfik, B.; Marchetti, D. Targeting Translation and the Cell Cycle Inversely Affects CTC Metabolism but Not Metastasis. Cancers 2023, 15, 5263. https://doi.org/10.3390/cancers15215263
Bowley TY, Merkley SD, Lagutina IV, Ortiz MC, Lee M, Tawfik B, Marchetti D. Targeting Translation and the Cell Cycle Inversely Affects CTC Metabolism but Not Metastasis. Cancers. 2023; 15(21):5263. https://doi.org/10.3390/cancers15215263
Chicago/Turabian StyleBowley, Tetiana Y., Seth D. Merkley, Irina V. Lagutina, Mireya C. Ortiz, Margaret Lee, Bernard Tawfik, and Dario Marchetti. 2023. "Targeting Translation and the Cell Cycle Inversely Affects CTC Metabolism but Not Metastasis" Cancers 15, no. 21: 5263. https://doi.org/10.3390/cancers15215263
APA StyleBowley, T. Y., Merkley, S. D., Lagutina, I. V., Ortiz, M. C., Lee, M., Tawfik, B., & Marchetti, D. (2023). Targeting Translation and the Cell Cycle Inversely Affects CTC Metabolism but Not Metastasis. Cancers, 15(21), 5263. https://doi.org/10.3390/cancers15215263