
Organ-on-a-Chip and Microfluidic Platforms for Oncology in the UK

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Organ-Chip Technology
Mechanobiology in Organ Chips
3. Cancer Phenomena Modelled Using Organ Chips
3.1. Tumour Growth/Proliferation
3.2. Modelling the Metastatic Cascade
3.3. Cancer-Associated Behaviours
3.4. Modelling Responses to Cancer Treatments
4. Perspective on Organ Chip Research in UK Cancer Research
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Day, C.-P.; Merlino, G.; Van Dyke, T. Preclinical mouse cancer models: A maze of opportunities and challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef] [Green Version]
- United States Senate. S.5002—FDA Modernization Act 2.0. In United States Senate—117th Congress; United States Senate: Washington, DC, USA, 2022. [Google Scholar]
- United States House of Representatives. H.R.2565—FDA Modernization Act of 2021. In United States House of Representatives—117th Congress; United States House of Representatives: Washington, DC, USA, 2022. [Google Scholar]
- Killion, J.J.; Radinsky, R.; Fidler, I.J. Orthotopic models are necessary to predict therapy of transplantable tumors in mice. Cancer Metastasis Rev. 1998, 17, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.E.; Junttila, M.R.; de Sauvage, F.J. Translational value of mouse models in oncology drug development. Nat. Med. 2015, 21, 431–439. [Google Scholar] [CrossRef]
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer 2019, 19, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, C.J.; Fuchs, S.; Hermanns, M.I.; Peters, K.; Unger, R.E. Cell culture models of higher complexity in tissue engineering and regenerative medicine. Biomaterials 2007, 28, 5193–5198. [Google Scholar] [CrossRef]
- Zhang, S. Beyond the Petri dish. Nat. Biotechnol. 2004, 22, 151–152. [Google Scholar] [CrossRef]
- Wilkinson, J.M. Need for alternative testing methods and opportunities for organ-on-a-chip systems. In Organ-on-a-Chip; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–11. [Google Scholar]
- Justus, C.R.; Leffler, N.; Ruiz-Echevarria, M.; Yang, L.V. In vitro cell migration and invasion assays. JoVE (J. Vis. Exp.) 2014, 88, e51046. [Google Scholar]
- Mehta, G.; Hsiao, A.Y.; Ingram, M.; Luker, G.D.; Takayama, S. Opportunities and challenges for use of tumor spheroids as models to test drug delivery and efficacy. J. Control. Release 2012, 164, 192–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschhaeuser, F.; Menne, H.; Dittfeld, C.; West, J.; Mueller-Klieser, W.; Kunz-Schughart, L.A. Multicellular tumor spheroids: An underestimated tool is catching up again. J. Biotechnol. 2010, 148, 3–15. [Google Scholar] [CrossRef]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Polacheck, W.J.; Charest, J.L.; Kamm, R.D. Interstitial flow influences direction of tumor cell migration through competing mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 11115–11120. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H.; Rubin, K.; Pietras, K.; Östman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.-P.; Yu, X.; Guo, J.-J.; Wang, Y.; Wang, T.; Li, J.-Y.; Konstantopoulos, K.; Wang, Z.-Y.; Wang, P. By activating matrix metalloproteinase-7, shear stress promotes chondrosarcoma cell motility, invasion and lung colonization. Oncotarget 2015, 6, 9140. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.P.; Kulkarni, S.; Perkins, M.W.; Hieber, K.; Pessu, R.L.; Gambles, K.; Maniar, M.; Kao, T.-C.; Seed, T.M.; Kumar, K.S. Amelioration of radiation-induced hematopoietic and gastrointestinal damage by Ex-RAD® in mice. J. Radiat. Res. 2012, 53, 526–536. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, P.K.; Low, B.C.; Lim, C.T. Mechanobiology of tumor growth. Chem. Rev. 2018, 118, 6499–6515. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Novak, R.; Didier, M.; Calamari, E.; Ng, C.F.; Choe, Y.; Clauson, S.L.; Nestor, B.A.; Puerta, J.; Fleming, R.; Firoozinezhad, S.J. Scalable fabrication of stretchable, dual channel, microfluidic organ chips. JoVE (J. Vis. Exp.) 2018, 140, e58151. [Google Scholar]
- Hughes, D.L.; Hughes, A.; Soonawalla, Z.; Mukherjee, S.; O’Neill, E. Dynamic Physiological Culture of Ex Vivo Human Tissue: A Systematic Review. Cancers 2021, 13, 2870. [Google Scholar] [CrossRef]
- Jain, A.; Barrile, R.; van der Meer, A.D.; Mammoto, A.; Mammoto, T.; De Ceunynck, K.; Aisiku, O.; Otieno, M.A.; Louden, C.S.; Hamilton, G.A.; et al. Primary Human Lung Alveolus-on-a-chip Model of Intravascular Thrombosis for Assessment of Therapeutics. Clin. Pharmacol. Ther. 2018, 103, 332–340. [Google Scholar] [CrossRef]
- Barrile, R.; van der Meer, A.D.; Park, H.; Fraser, J.P.; Simic, D.; Teng, F.; Conegliano, D.; Nguyen, J.; Jain, A.; Zhou, M. Organ-on-chip recapitulates thrombosis induced by an anti-CD154 monoclonal antibody: Translational potential of advanced microengineered systems. Clin. Pharmacol. Ther. 2018, 104, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.; Greenman, J.; Haswell, S.J. Development of microfluidic devices for biomedical and clinical application. J. Chem. Technol. Biotechnol. 2011, 86, 10–17. [Google Scholar] [CrossRef]
- Jaalouk, D.E.; Lammerding, J. Mechanotransduction gone awry. Nat. Rev. Mol. Cell Biol. 2009, 10, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Polacheck, W.J.; Li, R.; Uzel, S.G.M.; Kamm, R.D. Microfluidic platforms for mechanobiology. Lab Chip 2013, 13, 2252–2267. [Google Scholar] [CrossRef] [Green Version]
- Kaarj, K.; Yoon, J.-Y. Methods of delivering mechanical stimuli to organ-on-a-chip. Micromachines 2019, 10, 700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, C.L.; Fu, S.; Heywood, H.K.; Knight, M.M.; Thorpe, S.D. Mechanical stimulation: A crucial element of organ-on-chip models. Front. Bioeng. Biotechnol. 2020, 8, 602646. [Google Scholar] [CrossRef] [PubMed]
- Slay, E.E.; Meldrum, F.C.; Pensabene, V.; Amer, M.H. Embracing Mechanobiology in Next Generation Organ-On-A-Chip Models of Bone Metastasis. Front. Med. Technol. 2021, 3, 722501. [Google Scholar] [CrossRef]
- Ayuso, J.M.; Virumbrales-Munoz, M.; Lacueva, A.; Lanuza, P.M.; Checa-Chavarria, E.; Botella, P.; Fernandez, E.; Doblare, M.; Allison, S.J.; Phillips, R.M.; et al. Development and characterization of a microfluidic model of the tumour microenvironment. Sci. Rep. 2016, 6, 36086. [Google Scholar] [CrossRef] [Green Version]
- Huebner, J.; Raschke, M.; Ruetschle, I.; Graessle, S.; Hasenberg, T.; Schirrmann, K.; Lorenz, A.; Schnurre, S.; Lauster, R.; Maschmeyer, I.; et al. Simultaneous evaluation of anti-EGFR-induced tumour and adverse skin effects in a microfluidic human 3D co-culture model. Sci. Rep. 2018, 8, 15010. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Cheng, L.; Li, J.; Liu, Y.; Yin, S.; Xu, B.; Wang, D.; Lu, H.; Liu, C. A microfluidic platform culturing two cell lines paralleled under in-vivo like fluidic microenvironment for testing the tumor targeting of nanoparticles. Talanta 2020, 208, 120355. [Google Scholar] [CrossRef]
- Singh, D.; Deosarkar, S.P.; Cadogan, E.; Flemington, V.; Bray, A.; Zhang, J.; Reiserer, R.S.; Schaffer, D.K.; Gerken, G.B.; Britt, C.M. A microfluidic system that replicates pharmacokinetic (PK) profiles in vitro improves prediction of in vivo efficacy in preclinical models. PLoS Biol. 2022, 20, e3001624. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Ross, K.N.; Lander, E.S.; Golub, T.R. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2003, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Bertulli, C.; Gerigk, M.; Piano, N.; Liu, Y.; Zhang, D.; Mueller, T.; Knowles, T.J.; Huang, Y.Y.S. Image-Assisted Microvessel-on-a-Chip Platform for Studying Cancer Cell Transendothelial Migration Dynamics. Sci. Rep. 2018, 8, 12480. [Google Scholar] [CrossRef]
- Kühlbach, C.; Da Luz, S.; Baganz, F.; Hass, V.C.; Mueller, M.M. A microfluidic system for the investigation of tumor cell extravasation. Bioengineering 2018, 5, 40. [Google Scholar] [CrossRef] [Green Version]
- Verbruggen, S.W.; Thompson, C.L.; Duffy, M.P.; Lunetto, S.; Nolan, J.; Pearce, O.M.T.; Jacobs, C.R.; Knight, M.M. Mechanical Stimulation Modulates Osteocyte Regulation of Cancer Cell Phenotype. Cancers 2021, 13, 2906. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.; Pyne, E.; Christensen, M.; Iles, A.; Pamme, N.; Pires, I.M. Spheroid-on-chip microfluidic technology for the evaluation of the impact of continuous flow on metastatic potential in cancer models in vitro. Biomicrofluidics 2021, 15, 44103. [Google Scholar] [CrossRef]
- Lee, S.W.L.; Seager, R.J.; Litvak, F.; Spill, F.; Sieow, J.L.; Leong, P.H.; Kumar, D.; Tan, A.S.M.; Wong, S.C.; Adriani, G.; et al. Integrated in silico and 3D in vitro model of macrophage migration in response to physical and chemical factors in the tumor microenvironment. Integr. Biol. 2020, 12, 90–108. [Google Scholar] [CrossRef]
- Gerigk, M.; Bulstrode, H.; Shi, H.H.; Tonisen, F.; Cerutti, C.; Morrison, G.; Rowitch, D.; Huang, Y.Y.S. On-chip perivascular niche supporting stemness of patient-derived glioma cells in a serum-free, flowable culture. Lab Chip 2021, 21, 2343–2358. [Google Scholar] [CrossRef]
- Li, S.-S.; Xu, L.-Z.; Zhou, W.; Yao, S.; Wang, C.-L.; Xia, J.-L.; Wang, H.-F.; Kamran, M.; Xue, X.-Y.; Dong, L.; et al. p62/SQSTM1 interacts with vimentin to enhance breast cancer metastasis. Carcinogenesis 2017, 38, 1092–1103. [Google Scholar] [CrossRef] [Green Version]
- Boussommier-Calleja, A.; Atiyas, Y.; Haase, K.; Headley, M.; Lewis, C.; Kamm, R.D. The effects of monocytes on tumor cell extravasation in a 3D vascularized microfluidic model. Biomaterials 2019, 198, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Cai, P.; Li, Z.; Keneth, E.S.; Wang, L.; Wan, C.; Jiang, Y.; Hu, B.; Wu, Y.-L.; Wang, S.; Lim, C.T.; et al. Differential Homeostasis of Sessile and Pendant Epithelium Reconstituted in a 3D-Printed “GeminiChip” . Adv. Mater. 2019, 31, e1900514. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, A.; Sharples, L.; Corbo, F.; Franchini, C.; Vacca, A.; Catalano, A.; Carocci, A.; Kamm, R.D.; Pavesi, A.; Adriani, G. Phthalimide Derivative Shows Anti-angiogenic Activity in a 3D Microfluidic Model and No Teratogenicity in Zebrafish Embryos. Front. Pharmacol. 2019, 10, 349. [Google Scholar] [CrossRef] [PubMed]
- Algarni, A.; Greenman, J.; Madden, L.A. Procoagulant tumor microvesicles attach to endothelial cells on biochips under microfluidic flow. Biomicrofluidics 2019, 13, 64124. [Google Scholar] [CrossRef]
- Algarni, A.; Greenman, J.; Madden, L.A. Assessment of the procoagulant potential state of tumour-MP in cancer patients. Thromb. Res. 2016, 140, S194. [Google Scholar] [CrossRef] [PubMed]
- Smietana, K.; Siatkowski, M.; Møller, M. Trends in clinical success rates. Nat. Rev. Drug Discov. 2016, 15, 379–380. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [PubMed] [Green Version]
- Virumbrales-Muñoz, M.; Ayuso, J.M.; Olave, M.; Monge, R.; de Miguel, D.; Martínez-Lostao, L.; Le Gac, S.; Doblare, M.; Ochoa, I.; Fernandez, L.J. Multiwell capillarity-based microfluidic device for the study of 3D tumour tissue-2D endothelium interactions and drug screening in co-culture models. Sci. Rep. 2017, 7, 11998. [Google Scholar] [CrossRef] [Green Version]
- Ouattara, D.A.; Prot, J.-M.; Bunescu, A.; Dumas, M.-E.; Elena-Herrmann, B.; Leclerc, E.; Brochot, C. Metabolomics-on-a-chip and metabolic flux analysis for label-free modeling of the internal metabolism of HepG2/C3A cells. Mol. Biosyst. 2012, 8, 1908–1920. [Google Scholar] [CrossRef] [PubMed]
- Choucha Snouber, L.; Bunescu, A.; Naudot, M.; Legallais, C.; Brochot, C.; Dumas, M.E.; Elena-Herrmann, B.; Leclerc, E. Metabolomics-on-a-chip of hepatotoxicity induced by anticancer drug flutamide and its active metabolite hydroxyflutamide using HepG2/C3a microfluidic biochips. Toxicol. Sci. 2013, 132, 8–20. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, D.; Wu, G.; Wu, J.; Lu, S.; Lo, J.; He, Y.; Zhao, C.; Zhao, X.; Zhang, H. Metastasis-on-a-chip mimicking the progression of kidney cancer in the liver for predicting treatment efficacy. Theranostics 2020, 10, 300. [Google Scholar] [CrossRef]
- Azimi, T.; Loizidou, M.; Dwek, M.V. Cancer cells grown in 3D under fluid flow exhibit an aggressive phenotype and reduced responsiveness to the anti-cancer treatment doxorubicin. Sci. Rep. 2020, 10, 12020. [Google Scholar] [CrossRef]
- Petreus, T.; Cadogan, E.; Hughes, G.; Smith, A.; Pilla Reddy, V.; Lau, A.; O’Connor, M.J.; Critchlow, S.; Ashford, M.; Oplustil O’Connor, L. Tumour-on-chip microfluidic platform for assessment of drug pharmacokinetics and treatment response. Commun. Biol. 2021, 4, 1001. [Google Scholar] [CrossRef]
- Riley, A.; Green, V.; Cheah, R.; McKenzie, G.; Karsai, L.; England, J.; Greenman, J. A novel microfluidic device capable of maintaining functional thyroid carcinoma specimens ex vivo provides a new drug screening platform. BMC Cancer 2019, 19, 259. [Google Scholar] [CrossRef] [Green Version]
- Cheah, R.; Srivastava, R.; Stafford, N.D.; Beavis, A.W.; Green, V.; Greenman, J. Measuring the response of human head and neck squamous cell carcinoma to irradiation in a microfluidic model allowing customized therapy. Int. J. Oncol. 2017, 51, 1227–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, S.D.; Green, V.L.; Stafford, N.D.; Greenman, J. Analysis of radiation-induced cell death in head and neck squamous cell carcinoma and rat liver maintained in microfluidic devices. Otolaryngol. Neck Surg. 2014, 150, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.; Kuvshinov, D.; Sdrolia, A.; Kuvshinova, E.; Hilton, K.; Crank, S.; Beavis, A.W.; Green, V.; Greenman, J. A patient tumour-on-a-chip system for personalised investigation of radiotherapy based treatment regimens. Sci. Rep. 2019, 9, 6327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumovska, E.; Aalderink, G.; Valencia, C.W.; Kosim, K.; Nicolas, A.; Brown, S.; Vulto, P.; Erdmann, K.S.; Kurek, D. Direct On-Chip Differentiation of Intestinal Tubules from Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2020, 21, 4964. [Google Scholar] [CrossRef] [PubMed]
- Alepee, N.; Bahinski, A.; Daneshian, M.; De Wever, B.; Fritsche, E.; Goldberg, A.; Hansmann, J.; Hartung, T.; Haycock, J.; Hogberg, H.T. t4 workshop report: State-of-the-art of 3D cultures (organs-on-a-chip) in safety testing and pathophysiology. ALTEX 2014, 31, 441. [Google Scholar] [CrossRef]
- Candarlioglu, P.L.; Dal Negro, G.; Hughes, D.; Balkwill, F.; Harris, K.; Screen, H.; Morgan, H.; David, R.; Beken, S.; Guenat, O. Organ-on-a-chip: Current gaps and future directions. Biochem. Soc. Trans. 2022, 50, 665–673. [Google Scholar] [CrossRef]
- Haddrick, M.; Simpson, P.B. Organ-on-a-chip technology: Turning its potential for clinical benefit into reality. Drug Discov. Today 2019, 24, 1217–1223. [Google Scholar] [CrossRef]
- Singh, B.; Abdelgawad, M.E.; Ali, Z.; Bailey, J.; Budyn, E.; Civita, P.; Clift, M.J.D.; Connelly, J.T.; Constant, S.; Hittinger, M.; et al. Towards More Predictive, Physiological and Animal-free In Vitro Models: Advances in Cell and Tissue Culture 2020 Conference Proceedings. Altern. Lab. Anim. 2021, 49, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Busek, M.; Aizenshtadt, A.; Amirola-Martinez, M.; Delon, L.; Krauss, S. Academic User View: Organ-on-a-Chip Technology. Biosensors 2022, 12, 126. [Google Scholar] [CrossRef] [PubMed]
- Allwardt, V.; Ainscough, A.J.; Viswanathan, P.; Sherrod, S.D.; McLean, J.A.; Haddrick, M.; Pensabene, V. Translational roadmap for the organs-on-a-chip industry toward broad adoption. Bioengineering 2020, 7, 112. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
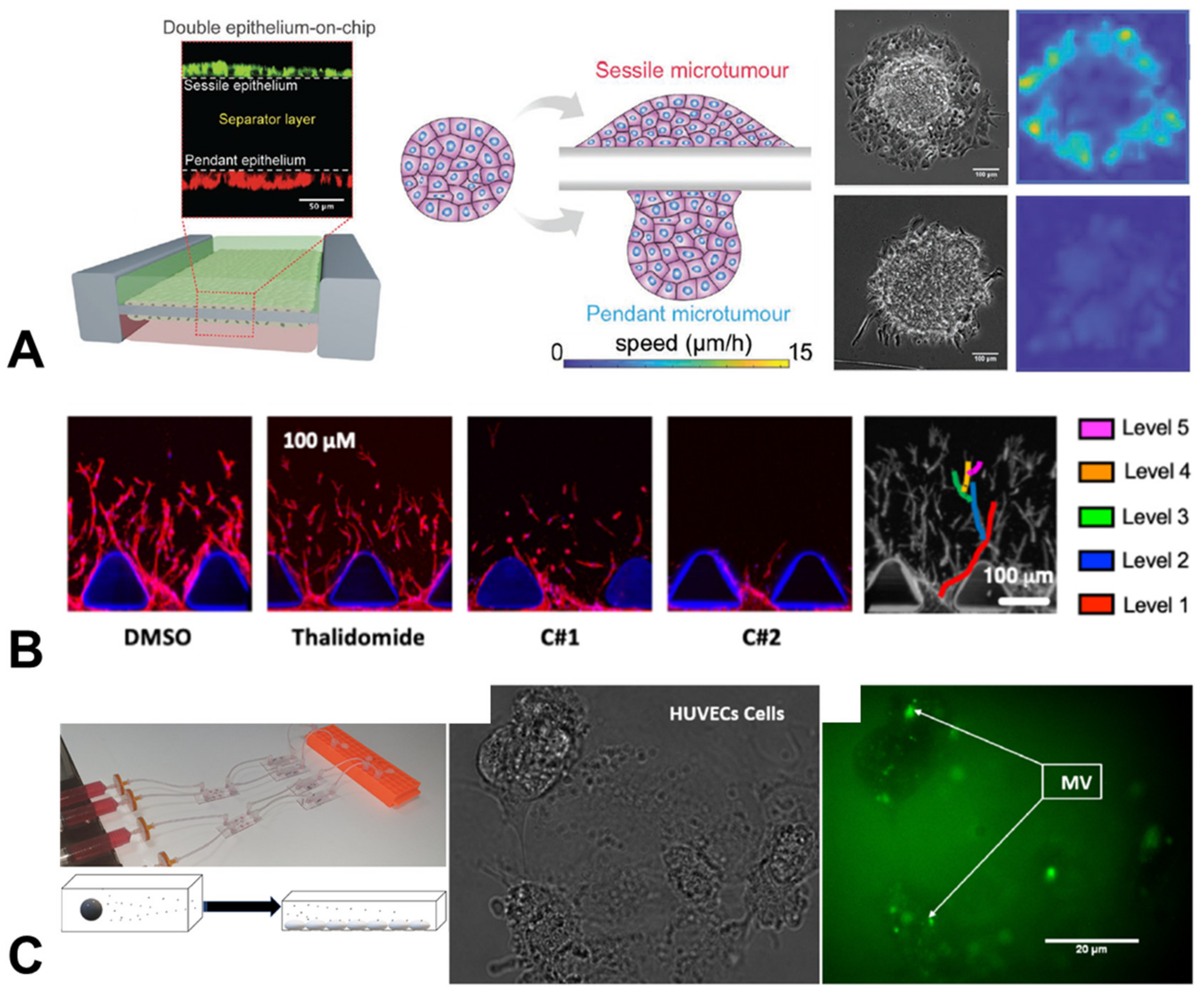{kind=link}
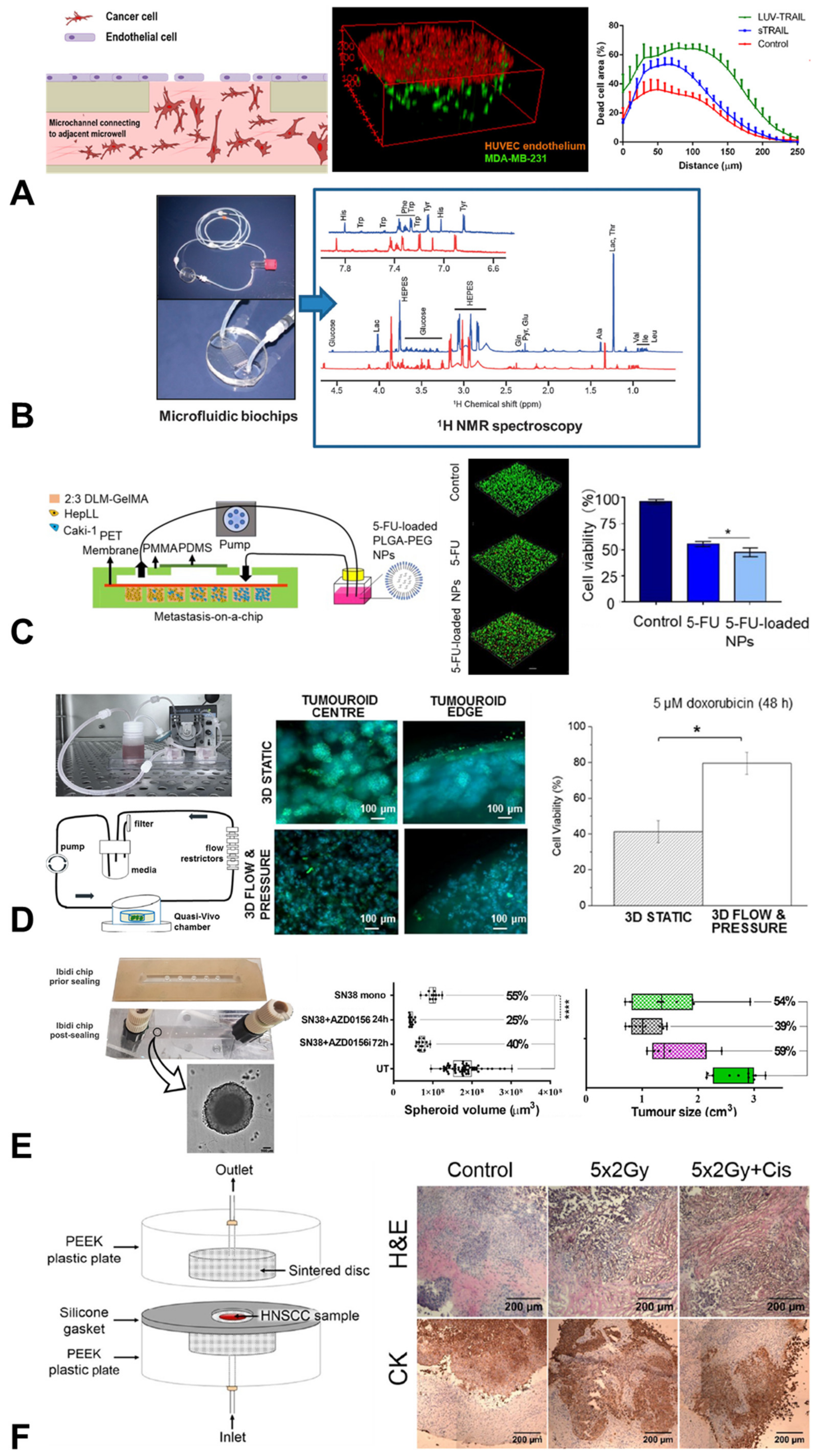{kind=link}
| Study | Chip System | Chip Material * | Organ/Cancer | Cell Lines/Tissue | Drug Treatment | Mechanical Stimulation |
|---|---|---|---|---|---|---|
| Algarni et al., Bio-Microfluidics 2019 [46] | µSlide I Luer, Vena8 | PS, COC | Ovarian, Brain | ES-2, U87, HUVECS | - | Flow |
| Algarni et al., Thromb. Res. 2016 [47] | Custom | PS | Pancreas, Brain | ASPC1, U87 | - | Flow |
| Ayuso et al., Sci. Rep. 2016 [31] | Custom | PS | Colon, Brain | HCT-116, U-251 MG, Jurkat | Doxorubicin | Flow |
| Azimi et al., Sci. Rep. 2020 [54] | Quasi-vivo | PDMS | Breast | MDA-MB-231, SKBR3 | Doxorubicin | Flow |
| Bertulli et al., Sci. Rep. 2018 [36] | Custom | PDMS | Breast | MDA-MB-231, LM2-4175 | - | - |
| Boussommier-Calleja et al., Biomaterials 2019 [43] | Custom | PDMS | Breast | MDA-MB-231, MDA-MB-435, HUVECS, Donor monocytes | - | - |
| Cai et al., Adv. Mat. 2019 [44] | GeminiChip | Glass | Liver epithelial | HepG2 | - | Compression |
| Carr et al., Head Neck Surg. 2014 [58] | Custom | Glass | Head & Neck | Human Tissue | Radiation | - |
| Cheah et al., Int. J. Onc. 2017 [57] | Custom | Glass | Head & Neck | Human Primary Cells | Radiation | - |
| Collins et al., Bio- microfluidics 2021 [39] | Custom | Glass | Brain, Breast | U87 MG, MCF-7 | - | Flow |
| Gerigk et al., Lab Chip 2021 [41] | Custom | PDMS | Brain | U87, Primary, HUVECS | - | - |
| Hübner et al., Sci. Rep. 2018 [32] | MOC | PS | Lung | NCI-H292, Skin tissue | Cetuximab | Flow |
| Kennedy et al., Sci. Rep. 2019 [59] | Custom | PEEK, sintered disk | Head & Neck | Human Tissue | Radiation, Cisplatin | - |
| Kühlbach et al., Bioengineering 2018 [37] | Custom | PET, PDMS | Lung, Skin | H838, SK-Mel 28, HPAEC | - | Flow |
| Lee et al., Int. Biol. 2020 [40] | Custom | PDMS | Pancreas | Panc1, hTERT-HPNE | - | Flow |
| Li et al., Carcin 2017 [42] | Custom | PDMS | Breast | MDA-MB-231, SKBR-3, BT549, MCF-7, SUM149, MCF-10A, HEK293T | - | - |
| Mercurio et al., Front Pharmacol. 2019 [45] | AIM Biotech | PS | Angiogenesis | HUVECS | Thalidomide | - |
| Naumovska et al., Int. J. Mol. Sci. 2020 [60] | MIMETAS | PS | Colon | Caco-2, hiPSC | - | Flow |
| Ouattara et al., Mol. Biosyst. 2012 [51] | Custom | PDMS | Liver epithelial | HepG2/C3a | - | - |
| Petreus et al., Commun. Biol. 2021 [55] | Ibidi | PS | Colorectal | SW620 | Irinotecan (SN38) | - |
| Riley et al., BMC Cancer 2019 [56] | Custom | PEEK, sintered disk | Thyroid | Human Tissue | - | - |
| Singh et al., PLOS Biol. 2022 [34] | Custom | PDMS | Pharyngeal, Lung | FaDu, Calu-6, A549 | - | - |
| Choucha Snouber et al., Toxic Sci. 2013 [52] | Custom | PDMS | Liver epithelial | HepG2/C3a | Flutamide | - |
| Verbruggen et al., Cancers 2021 [38] | Emulate | PDMS | Breast, Prostate, Bone | MDA-MB-231, PC3, MLO-Y4 | - | Flow |
| Virumbrales-Muñoz et al., Sci. Rep. 2017 [50] | Custom | SU-8 | Breast | MDA-MB-231, HUVECS | TRAIL | - |
| Wang et al., Theranostics 2020 [53] | Custom | PDMS | Kidney, Liver | Caki-1, HepLL | 5-Fluorouracil | - |
| Wei et al., Talanta 2021 [33] | Custom | PDMS | Cervical, Lung | HeLa, A549 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nolan, J.; Pearce, O.M.T.; Screen, H.R.C.; Knight, M.M.; Verbruggen, S.W. Organ-on-a-Chip and Microfluidic Platforms for Oncology in the UK. Cancers 2023, 15, 635. https://doi.org/10.3390/cancers15030635
Nolan J, Pearce OMT, Screen HRC, Knight MM, Verbruggen SW. Organ-on-a-Chip and Microfluidic Platforms for Oncology in the UK. Cancers. 2023; 15(3):635. https://doi.org/10.3390/cancers15030635
Chicago/Turabian StyleNolan, Joanne, Oliver M. T. Pearce, Hazel R. C. Screen, Martin M. Knight, and Stefaan W. Verbruggen. 2023. "Organ-on-a-Chip and Microfluidic Platforms for Oncology in the UK" Cancers 15, no. 3: 635. https://doi.org/10.3390/cancers15030635
APA StyleNolan, J., Pearce, O. M. T., Screen, H. R. C., Knight, M. M., & Verbruggen, S. W. (2023). Organ-on-a-Chip and Microfluidic Platforms for Oncology in the UK. Cancers, 15(3), 635. https://doi.org/10.3390/cancers15030635