
Application Prospects of Triphenylphosphine-Based Mitochondria-Targeted Cancer Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Mitochondria-Targeted Agents
3. Advantages of TPP with Targeted Tumor Cell Delivery
3.1. TPP-Based Mitochondrial Targeting System Preferentially Delivers Drugs into Tumor Cell Mitochondria
3.2. TPP Can Carry Drug Molecules across the Membrane Barrier
3.3. Safety of TPP Compounds
4. TPP-Based Mitocans
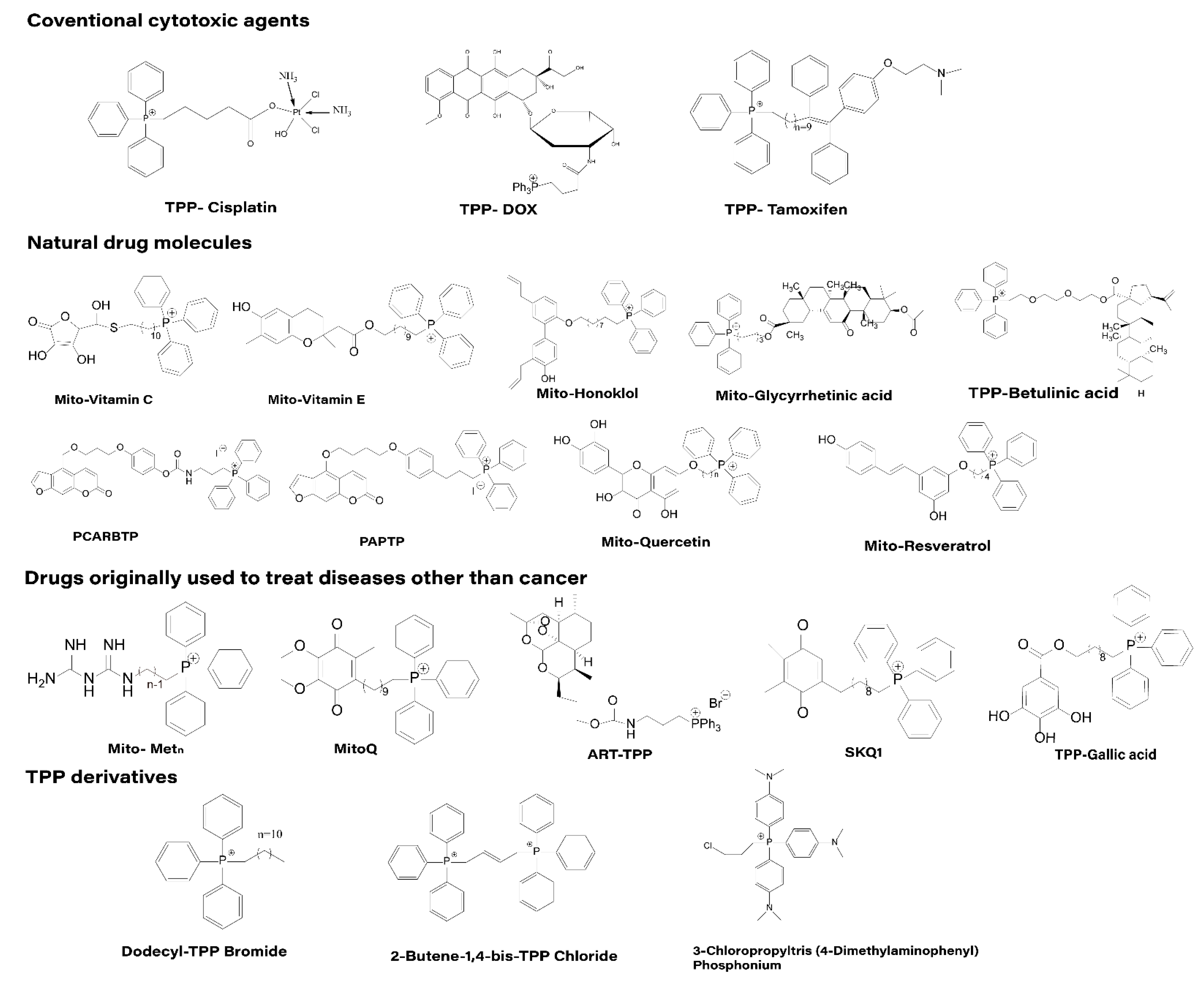
4.1. Direct Affixation of TPP to Drug Molecules
4.1.1. Traditional Cytotoxic Drugs
4.1.2. Natural Drug Molecules
4.1.3. Drugs Initially Used Outside of Cancer Treatment
4.1.4. TPP Derivatives
4.2. TPP-Modified Targeted Mitochondrial Nanosystems
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA-Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- de la Puente, P.; Azab, A.K. Nanoparticle delivery systems, general approaches, and their implementation in multiple myeloma. Eur. J. Haematol. 2017, 98, 529–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shtil, A.A.; Mandlekar, S.; Yu, R.; Walter, R.J.; Hagen, K.; Tan, T.H.; Roninson, I.B.; Kong, A.N. Differential regulation of mitogen-activated protein kinases by microtubule-binding agents in human breast cancer cells. Oncogene 1999, 18, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Filigheddu, N.; Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Schmiedel, J.; Jackson, S.; Schafer, J.; Reichmann, H. Mitochondrial cytopathies. J. Neurol. 2003, 250, 267–277. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Izyumov, D.S.; Avetisyan, A.V.; Pletjushkina, O.Y.; Sakharov, D.V.; Wirtz, K.W.; Chernyak, B.V.; Skulachev, V.P. "Wages of fear": Transient threefold decrease in intracellular ATP level imposes apoptosis. Biochim. Biophys. Acta 2004, 1658, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zou, P.; Cohen, A.E. Voltage imaging with genetically encoded indicators. Curr. Opin. Chem. Biol. 2017, 39, 1–10. [Google Scholar] [CrossRef]
- Klier, P.E.Z.; Gest, A.M.M.; Martin, J.G.; Roo, R.; Navarro, M.X.; Lesiak, L.; Deal, P.E.; Dadina, N.; Tyson, J.; Schepartz, A.; et al. Bioorthogonal, Fluorogenic Targeting of Voltage-Sensitive Fluorophores for Visualizing Membrane Potential Dynamics in Cellular Organelles. J. Am. Chem. Soc. 2022, 144, 12138–12146. [Google Scholar] [CrossRef]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuevo-Tapioles, C.; Santacatterina, F.; Stamatakis, K.; Nunez de Arenas, C.; Gomez de Cedron, M.; Formentini, L.; Cuezva, J.M. Coordinate beta-adrenergic inhibition of mitochondrial activity and angiogenesis arrest tumor growth. Nat. Commun. 2020, 11, 3606. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.-L.; Qiao, G.; Wang, L.-L.; Cheng, L.; Lin, X.-K. Protopine inhibits the growth of hepatocellular carcinoma through a mitochondrially mediated apoptosis pathway. Yaoxue Xuebao 2021, 56, 2223–2229. [Google Scholar] [CrossRef]
- Ralph, S.J.; Low, P.; Dong, L.; Lawen, A.; Neuzil, J. Mitocans: Mitochondrial targeted anti-cancer drugs as improved therapies and related patent documents. Recent Pat. Anti-Cancer Drug Discov. 2006, 1, 327–346. [Google Scholar] [CrossRef] [PubMed]
- Dakubo, G.D. Mitochondrial Genetics and Cancer; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2010; pp. 1–356. [Google Scholar]
- Bonekamp, N.A.; Peter, B.; Hillen, H.S.; Felser, A.; Bergbrede, T.; Choidas, A.; Horn, M.; Unger, A.; Di Lucrezia, R.; Atanassov, I.; et al. Small-molecule inhibitors of human mitochondrial DNA transcription. Nature 2020, 588, 712–716. [Google Scholar] [CrossRef]
- Ilmi, R.; Tseriotou, E.; Stylianou, P.; Christou, Y.A.; Ttofi, I.; Dietis, N.; Pitris, C.; Odysseos, A.D.; Georgiades, S.N. A Novel Conjugate of Bis[((4-bromophenyl)amino)quinazoline], a EGFR-TK Ligand, with a Fluorescent Ru(II)-Bipyridine Complex Exhibits Specific Subcellular Localization in Mitochondria. Mol. Pharm. 2019, 16, 4260–4273. [Google Scholar] [CrossRef]
- Weissig, V.; Torchilin, V.P. Towards mitochondrial gene therapy: DQAsomes as a strategy. J. Drug Target. 2001, 9, 1–13. [Google Scholar] [CrossRef]
- Li, Q.; Huang, Y. Mitochondrial targeted strategies and their application for cancer and other diseases treatment. J. Pharm. Investig. 2020, 50, 271–293. [Google Scholar] [CrossRef]
- Wu, J.; Li, J.; Wang, H.; Liu, C.-B. Mitochondrial-targeted penetrating peptide delivery for cancer therapy. Expert Opin. Drug Deliv. 2018, 15, 951–964. [Google Scholar] [CrossRef]
- Liberman, E.A.; Topaly, V.P.; Tsofina, L.M.; Iasaitis, A.A.; Skulachev, V.P. Ion transport and electrical potential of mitochondrial membranes. Biokhimiia 1969, 34, 1083–1087. [Google Scholar] [PubMed]
- Liberman, E.A.; Topaly, V.P.; Tsofina, L.M.; Jasaitis, A.A.; Skulachev, V.P. Mechanism of coupling of oxidative phosphorylation and the membrane potential of mitochondria. Nature 1969, 222, 1076–1078. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol. 1997, 15, 326–330. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A.J. Drug delivery to mitochondria: The key to mitochondrial medicine. Adv. Drug Deliv. Rev. 2000, 41, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 2021, 22, 54–70. [Google Scholar] [CrossRef]
- Neupert, W.; Herrmann, J.M. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Borgeld, H.J.; Zhang, J.; Muramatsu, S.; Gong, J.S.; Yoneda, M.; Shamoto, M.; Fuku, N.; Kurata, M.; Yamada, Y.; et al. Gene therapy for mitochondrial disease by delivering restriction endonuclease Smal into mitochondria. J. Biomed. Sci. 2002, 9, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P.; Rammohan, R.; Weissig, V.; Levchenko, T.S. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 8786–8791. [Google Scholar] [CrossRef] [Green Version]
- Shokolenko, I.N.; Alexeyev, M.F.; LeDoux, S.P.; Wilson, G.L. TAT-mediated protein transduction and targeted delivery of fusion proteins into mitochondria of breast cancer cells. DNA Repair 2005, 4, 511–518. [Google Scholar] [CrossRef]
- Johnson, L.V.; Walsh, M.L.; Bockus, B.J.; Chen, L.B. Monitoring of relative mitochondrial membrane potential in living cells by fluorescence microscopy. J. Cell Biol. 1981, 88, 526–535. [Google Scholar] [CrossRef]
- Johnson, L.V.; Walsh, M.L.; Chen, L.B. Localization of mitochondria in living cells with rhodamine 123. Proc. Natl. Acad. Sci. USA 1980, 77, 990–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonenko, Y.N.; Avetisyan, A.V.; Bakeeva, L.E.; Chernyak, B.V.; Chertkov, V.A.; Domnina, L.V.; Ivanova, O.Y.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: Synthesis and in vitro studies. Biochemistry 2008, 73, 1273–1287. [Google Scholar] [CrossRef] [PubMed]
- El Baraka, M.; Deumié, M.; Viallet, P.; Lampidis, T.J. Fluorescence properties and partitioning behaviour of esterified and unesterified rhodamines. J. Photochem. Photobiol. A Chem. 1991, 62, 195–216. [Google Scholar] [CrossRef]
- Qi, X.J. Advacnaces in mitochondrial targeting molecules in anticancer applications. J. Chin. Pharm. Sci. 2015, 50, 741–744. [Google Scholar]
- Weiss, M.J.; Wong, J.R.; Ha, C.S.; Bleday, R.; Salem, R.R.; Steele, G.D., Jr.; Chen, L.B. Dequalinium, a topical antimicrobial agent, displays anticarcinoma activity based on selective mitochondrial accumulation. Proc. Natl. Acad. Sci. USA 1987, 84, 5444–5448. [Google Scholar] [CrossRef] [Green Version]
- Christman, J.E.; Miller, D.S.; Coward, P.; Smith, L.H.; Teng, N.N. Study of the selective cytotoxic properties of cationic, lipophilic mitochondrial-specific compounds in gynecologic malignancies. Gynecol. Oncol. 1990, 39, 72–79. [Google Scholar] [CrossRef]
- Pathak, R.K.; Kolishetti, N.; Dhar, S. Targeted nanoparticles in mitochondrial medicine. Wiley Interdiscip. Rev.-Nanomed. Nanobiotechnol. 2015, 7, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.M.; Pabba, S.; Torchilin, V.P.; Fowle, W.; Kimpfler, A.; Schubert, R.; Weissig, V. Towards mitochondria-specific delivery of apoptosis-inducing agents: DQAsomal incorporated paclitaxel. J. Drug Deliv. Sci. Technol. 2005, 15, 81–86. [Google Scholar] [CrossRef]
- Song, Y.-f.; Liu, D.-z.; Cheng, Y.; Liu, M.; Ye, W.-l.; Zhang, B.-l.; Liu, X.-y.; Zhou, S.-y. Dual subcellular compartment delivery of doxorubicin to overcome drug resistant and enhance antitumor activity. Sci. Rep. 2015, 5, 16125. [Google Scholar] [CrossRef] [Green Version]
- Fantin, V.R.; Berardi, M.J.; Scorrano, L.; Korsmeyer, S.J.; Leder, P. A novel mitochondriotoxic small molecule that selectively inhibits tumor cell growth. Cancer Cell 2002, 2, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Fantin, V.R.; Leder, P. F16, a mitochondriotoxic compound, triggers apoptosis or necrosis depending on the genetic background of the target carcinoma cell. Cancer Res. 2004, 64, 329–336. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Li, D.-W.; Yang, L.-Y.; Fu, L.; Zhu, X.-J.; Wong, W.-K.; Jiang, F.-L.; Liu, Y. A novel bifunctional mitochondria-targeted anticancer agent with high selectivity for cancer cells. Sci. Rep. 2015, 5, 13543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.A.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.J.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells—Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [Green Version]
- Apostolova, N.; Victor, V.M. Molecular Strategies for Targeting Antioxidants to Mitochondria: Therapeutic Implications. Antioxid. Redox Signal. 2015, 22, 686–729. [Google Scholar] [CrossRef] [Green Version]
- Khairul, I.; Wang, Q.Q.; Jiang, Y.H.; Wang, C.; Naranmandura, H. Metabolism, toxicity and anticancer activities of arsenic compounds. Oncotarget 2017, 8, 23905–23926. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.-Y.; Liu, Y.-J.; Cai, Y.-M.; Wang, A.-D.; Xia, Y.-Z.; Hu, Y.-J.; Jiang, F.-L.; Liu, Y. A mitochondria-targeted organic arsenical accelerates mitochondrial metabolic disorder and function injury. Bioorganic Med. Chem. 2019, 27, 760–768. [Google Scholar] [CrossRef]
- Lohlamoh, W.; Soontornworajit, B.; Rotkrua, P. Anti-Proliferative Effect of Doxorubicin-Loaded AS1411 Aptamer on Colorectal Cancer Cell. Asian Pac. J. Cancer Prev. 2021, 22, 2209–2219. [Google Scholar] [CrossRef]
- Pan, Z.Z.; Wang, H.Y.; Zhang, M.; Lin, T.T.; Zhang, W.Y.; Zhao, P.F.; Tang, Y.S.; Xiong, Y.; Zeng, Y.E.; Huang, Y.Z. Nuclear-targeting TAT-PEG-Asp8-doxorubicin polymeric nanoassembly to overcome drug-resistant colon cancer. Acta Pharmacol. Sin. 2016, 37, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.B.; Lavasanifar, A. Traceable multifunctional micellar nanocarriers for cancer-targeted co-delivery of MDR-1 siRNA and doxorubicin. ACS Nano 2011, 5, 5202–5213. [Google Scholar] [CrossRef]
- Han, M.; Vakili, M.R.; Abyaneh, H.S.; Molavi, O.; Lai, R.; Lavasanifar, A. Mitochondrial Delivery of Doxorubicin via Triphenylphosphine Modification for Overcoming Drug Resistance in MDA-MB-435/DOX Cells. Mol. Pharm. 2014, 11, 2640–2649. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Cho, H.; Chen, H.H.; Panagia, M.; Sosnovik, D.E.; Josephson, L. Fluorescent and radiolabeled triphenylphosphonium probes for imaging mitochondria. Chem. Commun. 2013, 49, 10361–10363. [Google Scholar] [CrossRef] [PubMed]
- Safin, D.A.; Mitoraj, M.P.; Robeyns, K.; Filinchuk, Y.; Vande Velde, C.M.L. Luminescent mononuclear mixed ligand complexes of copper(I) with 5-phenyl-2,2 '-bipyridine and triphenylphosphine. Dalton Trans. 2015, 44, 16824–16832. [Google Scholar] [CrossRef] [PubMed]
- Caplan, S.R.; Essig, A. Oxidative phosphorylation: Thermodynamic criteria for the chemical and chemiosmotic hypotheses. Proc. Natl. Acad. Sci. USA 1969, 64, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef] [PubMed]
- Indig, G.L.; Anderson, G.S.; Nichols, M.G.; Bartlett, J.A.; Mellon, W.S.; Sieber, F. Effect of molecular structure on the performance of triarylmethane dyes as therapeutic agents for photochemical purging of autologous bone marrow grafts from residual tumor cells. J. Pharm. Sci. 2000, 89, 88–99. [Google Scholar] [CrossRef]
- Chen, L.B. Mitochondrial membrane potential in living cells. Annu. Rev. Cell Biol. 1988, 4, 155–181. [Google Scholar] [CrossRef]
- Rottenberg, H. Membrane potential and surface potential in mitochondria: Uptake and binding of lipophilic cations. J. Membr. Biol. 1984, 81, 127–138. [Google Scholar] [CrossRef]
- Ono, A.; Miyauchi, S.; Demura, M.; Asakura, T.; Kamo, N. Activation energy for permeation of phosphonium cations through phospholipid bilayer membrane. Biochemistry 1994, 33, 4312–4318. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Xiao, Y.; Fu, B.; Qin, Z. TPP-based mitocans: A potent strategy for anticancer drug design. RSC Med. Chem. 2020, 11, 858–875. [Google Scholar] [CrossRef]
- Ross, M.F.; Kelso, G.F.; Blaikie, F.H.; James, A.M.; Cocheme, H.M.; Filipovska, A.; Da Ros, T.; Hurd, T.R.; Smith, R.A.J.; Murphy, M.P. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry 2005, 70, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Aprille, J.R. Basis for the selective cytotoxicity of rhodamine 123. Cancer Res. 1987, 47, 4361–4365. [Google Scholar] [PubMed]
- Modica-Napolitano, J.S.; Weiss, M.J.; Chen, L.B.; Aprille, J.R. Rhodamine 123 inhibits bioenergetic function in isolated rat liver mitochondria. Biochem. Biophys. Res. Commun. 1984, 118, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Propper, D.J.; Braybrooke, J.P.; Taylor, D.J.; Lodi, R.; Styles, P.; Cramer, J.A.; Collins, W.C.; Levitt, N.C.; Talbot, D.C.; Ganesan, T.S.; et al. Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumours. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1999, 10, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Li, Q.; Wang, Y.; Ge, C.; Feng, C.; Xie, S.; He, H.; Xu, X.; Wang, C. Design, Synthesis, and Biological Evaluation of Mitochondria Targeted Flavone-Naphthalimide- Polyamine Conjugates with Antimetastatic Activity. J. Med. Chem. 2017, 60, 2071–2083. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O'Sullivan, J.D.; Fung, V.; Smith, R.A.J.; Murphy, M.P.; Taylor, K.M.; Protect Study, G. A Double-Blind, Placebo-Controlled Study to Assess the Mitochondria-Targeted Antioxidant MitoQ as a Disease-Modifying Therapy in Parkinson's Disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.; Weilert, F.; Orr, D.W.; Keogh, G.F.; Gibson, M.; Lockhart, M.M.; Frampton, C.M.; Taylor, K.M.; Smith, R.A.J.; Murphy, M.P. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010, 30, 1019–1026. [Google Scholar] [CrossRef]
- Fuller, N.; Rand, R.P. The influence of lysolipids on the spontaneous curvature and bending elasticity of phospholipid membranes. Biophys. J. 2001, 81, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi, R.; Urig, S.; Hartmann, M.; Helmke, B.M.; Koncarevic, S.; Allenberger, B.; Kienhoefer, C.; Neher, M.; Steiner, H.H.; Unterberg, A.; et al. Antiglioma activity of 2,2′: 6′, 2′′-terpyridineplatinum(II) complexes in a rat model—Effects on cellular redox metabolism. Free. Radic. Biol. Med. 2006, 40, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Marrache, S.; Pathak, R.K.; Dhar, S. Detouring of cisplatin to access mitochondrial genome for overcoming resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 10444–10449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Wang, Z.; Zhang, C.; Wang, Y.; Zhang, H.; Gan, Z.; Guo, Z.; Wang, X. Mitochondrion-targeted platinum complexes suppressing lung cancer through multiple pathways involving energy metabolism. Chem. Sci. 2019, 10, 3089–3095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, D.; Maare, C.; Skovsgaard, T. Cellular resistance to anthracyclines. Gen. Pharmacol. 1996, 27, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Tavallaie, M.S.; Voshtani, R.; Deng, X.; Qiao, Y.; Jiang, F.; Collman, J.P.; Fu, L. Moderation of mitochondrial respiration mitigates metabolic syndrome of aging. Proc. Natl. Acad. Sci. USA 2020, 117, 9840–9850. [Google Scholar] [CrossRef]
- Kik, K.; Studzian, K.; Wasowska-Lukawska, M.; Oszczapwicz, I.; Szmigiero, L. Cytotoxicity and inhibitory properties against topoisomerase II of doxorubicin and its formamidine derivatives. Acta Biochim. Pol. 2009, 56, 135–142. [Google Scholar] [CrossRef]
- Liu, H.N.; Guo, N.N.; Guo, W.W.; Huang-Fu, M.Y.; Vakili, M.R.; Chen, J.J.; Xu, W.H.; Wei, Q.C.; Han, M.; Lavasanifar, A.; et al. Delivery of mitochondriotropic doxorubicin derivatives using self-assembling hyaluronic acid nanocarriers in doxorubicin-resistant breast cancer. Acta Pharmacol. Sin. 2018, 39, 1681–1692. [Google Scholar] [CrossRef] [Green Version]
- Theodossiou, T.A.; Sideratou, Z.; Katsarou, M.E.; Tsiourvas, D. Mitochondrial Delivery of Doxorubicin by Triphenylphosphonium-Functionalized Hyperbranched Nanocarriers Results in Rapid and Severe Cytotoxicity. Pharm. Res. 2013, 30, 2832–2842. [Google Scholar] [CrossRef]
- Shagufta; Ahmad, I. Tamoxifen a pioneering drug: An update on the therapeutic potential of tamoxifen derivatives. Eur. J. Med. Chem. 2018, 143, 515–531. [Google Scholar] [CrossRef]
- Rohlenova, K.; Sachaphibulkij, K.; Stursa, J.; Bezawork-Geleta, A.; Blecha, J.; Endaya, B.; Werner, L.; Cerny, J.; Zobalova, R.; Goodwin, J.; et al. Selective Disruption of Respiratory Supercomplexes as a New Strategy to Suppress Her2(high) Breast Cancer. Antioxid. Redox Signal. 2017, 26, 84–103. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Garrett, W.S.; Chan, A.T. Nutrients, foods, and colorectal cancer prevention. Gastroenterology 2015, 148, 1244–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Zhang, Q.; Liu, Q.; Komas, S.M.; Kalyanaraman, B.; Lubet, R.A.; Wang, Y.; You, M. Honokiol Inhibits Lung Tumorigenesis through Inhibition of Mitochondrial Function. Cancer Prev. Res. 2014, 7, 1149–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Lee, Y.; Cheng, G.; Zielonka, J.; Zhang, Q.; Bajzikova, M.; Xiong, D.; Tsaih, S.-W.; Hardy, M.; Flister, M.; et al. Mitochondria-Targeted Honokiol Confers a Striking Inhibitory Effect on Lung Cancer via Inhibiting Complex I Activity. Iscience 2018, 3, 192–207. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Dai, L.; Ji, M.; Wang, H. Mitochondria-targeted triphenylphosphonium conjugated glycyrrhetinic acid derivatives as potent anticancer drugs. Bioorganic Chem. 2019, 85, 179–190. [Google Scholar] [CrossRef]
- Mattarei, A.; Romio, M.; Manago, A.; Zoratti, M.; Paradisi, C.; Szabo, I.; Leanza, L.; Biasutto, L. Novel Mitochondria-Targeted Furocoumarin Derivatives as Possible Anti-Cancer Agents. Front. Oncol. 2018, 8, 122. [Google Scholar] [CrossRef] [Green Version]
- Leanza, L.; Romio, M.; Becker, K.A.; Azzolini, M.; Trentin, L.; Manago, A.; Venturini, E.; Zaccagnino, A.; Mattarei, A.; Carraretto, L.; et al. Direct Pharmacological Targeting of a Mitochondrial Ion Channel Selectively Kills Tumor Cells In Vivo. Cancer Cell 2017, 31, 516–531. [Google Scholar] [CrossRef] [Green Version]
- Shankar, S.; Singh, G.; Srivastava, R.K. Chemoprevention by resveratrol: Molecular mechanisms and therapeutic potential. Front. Biosci.-Landmark 2007, 12, 4839–4854. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.A.; Shukitt-Hale, B.; Lau, F.C. Fruit polyphenols and their effects on neuronal signaling and behavior in senescence. In Biogerontology: Mechanisms and Interventions; Rattan, S.I.S., Akman, S., Eds.; Blackwell Publisher: Hoboken, NJ, USA, 2007; Volume 1100, pp. 470–485. [Google Scholar]
- Valenzano, D.R.; Terzibasi, E.; Genade, T.; Cattaneo, A.; Domenici, L.; Cellerino, A. Resveratrol prolongs lifespan and retards the onset of age-related markers in a short-lived vertebrate. Curr. Biol. 2006, 16, 296–300. [Google Scholar] [CrossRef] [Green Version]
- Mattarei, A.; Biasutto, L.; Marotta, E.; De Marchi, U.; Sassi, N.; Garbisa, S.; Zoratti, M.; Paradisi, C. A Mitochondriotropic Derivative of Quercetin: A Strategy to Increase the Effectiveness of Polyphenols. Chembiochem 2008, 9, 2633–2642. [Google Scholar] [CrossRef]
- Sassi, N.; Mattarei, A.; Azzolini, M.; Bernardi, P.; Szabo, I.; Paradisi, C.; Zoratti, M.; Biasutto, L. Mitochondria-targeted Resveratrol Derivatives Act as Cytotoxic Pro-oxidants. Curr. Pharm. Des. 2014, 20, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Betulinic acid for cancer treatment and prevention. Int. J. Mol. Sci. 2008, 9, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsepaeva, O.V.; Nemtarev, A.V.; Salikhova, T.I.; Abdullin, T.I.; Grigor'eva, L.R.; Khozyainova, S.A.; Mironov, V.F. Synthesis, Anticancer, and Antibacterial Activity of Betulinic and Betulonic Acid C-28-Triphenylphosphonium Conjugates with Variable Alkyl Linker Length. Anti-Cancer Agents Med. Chem. 2020, 20, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.Y.; Nedopekina, D.A.; Shakurova, E.R.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Dzhemilev, U.M.; Bel'skii, Y.P.; Bel'skaya, N.V.; Stankevich, S.A.; et al. Synthesis of lupane triterpenoids with triphenylphosphonium substituents and studies of their antitumor activity. Russ. Chem. Bull. 2013, 62, 188–198. [Google Scholar] [CrossRef]
- Chopra, B.; Dhingra, A.K. Natural products: A lead for drug discovery and development. Phytother. Res. 2021, 35, 4660–4702. [Google Scholar] [CrossRef]
- Lee, M.-S.; Hsu, C.-C.; Wahlqvist, M.L.; Tsai, H.-N.; Chang, Y.-H.; Huang, Y.-C. Type 2 diabetes increases and metformin reduces total, colorectal, liver and pancreatic cancer incidences in Taiwanese: A representative population prospective cohort study of 800,000 individuals. BMC Cancer 2011, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Investig. 2007, 117, 1422–1431. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Zielonka, J.; Ouari, O.; Lopez, M.; McAllister, D.; Boyle, K.; Barrios, C.S.; Weber, J.J.; Johnson, B.D.; Hardy, M.; et al. Mitochondria-Targeted Analogues of Metformin Exhibit Enhanced Antiproliferative and Radiosensitizing Effects in Pancreatic Cancer Cells. Cancer Res. 2016, 76, 3904–3915. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Hebrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [Green Version]
- Nagel, R.; Stigter-van Walsum, M.; Buijze, M.; van den Berg, J.; van der Meulen, I.H.; Hodzic, J.; Piersma, S.R.; Pham, T.V.; Jimenez, C.R.; van Beusechem, V.W.; et al. Genome-wide siRNA Screen Identifies the Radiosensitizing Effect of Downregulation of MASTL and FOXM1 in NSCLC. Mol. Cancer Ther. 2015, 14, 1434–1444. [Google Scholar] [CrossRef] [Green Version]
- Rao, V.A.; Klein, S.R.; Bonar, S.J.; Zielonka, J.; Mizuno, N.; Dickey, J.S.; Keller, P.W.; Joseph, J.; Kalyanaraman, B.; Shacter, E. The Antioxidant Transcription Factor Nrf2 Negatively Regulates Autophagy and Growth Arrest Induced by the Anticancer Redox Agent Mitoquinone. J. Biol. Chem. 2010, 285, 34447–34459. [Google Scholar] [CrossRef] [PubMed]
- Agapova, L.S.; Chernyak, B.V.; Domnina, L.V.; Dugina, V.B.; Efimenko, A.Y.; Fetisova, E.K.; Ivanova, O.Y.; Kalinina, N.I.; Khromova, N.V.; Kopnin, B.P.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 3. Inhibitory effect of SkQ1 on tumor development from p53-deficient cells. Biochemistry 2008, 73, 1300–1316. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Zhang, Z.; Liao, F.; Jiang, T.; Tu, Y. The birth of artemisinin. Pharmacol. Ther. 2020, 216, 107658. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.N.; Ma, N.; Meng, Y.; Zhang, X.; Wong, Y.K.; Xu, C.; Liao, F.; Jiang, T.; Tu, Y.; Wang, J. Study towards improving artemisinin-based combination therapies. Nat. Prod. Rep. 2021, 38, 1243–1250. [Google Scholar] [CrossRef]
- Ho, W.E.; Peh, H.Y.; Chan, T.K.; Wong, W.S.F. Artemisinins: Pharmacological actions beyond anti-malarial. Pharmacol. Ther. 2014, 142, 126–139. [Google Scholar] [CrossRef]
- Zhang, C.-J.; Wang, J.; Zhang, J.; Lee, Y.M.; Feng, G.; Lim, T.K.; Shen, H.-M.; Lin, Q.; Liu, B. Mechanism-Guided Design and Synthesis of a Mitochondria-Targeting Artemisinin Analogue with Enhanced Anticancer Activity. Angew. Chem.-Int. Ed. 2016, 55, 13770–13774. [Google Scholar] [CrossRef]
- Fiuza, S.M.; Gomes, C.; Teixeira, L.J.; da Cruz, M.T.G.; Cordeiro, M.; Milhazes, N.; Borges, F.; Marques, M.P.M. Phenolic acid derivatives with potential anticancer properties—A structure-activity relationship study. Part 1: Methyl, propyl and octyl esters of caffeic and gallic acids. Bioorg. Med. Chem. 2004, 12, 3581–3589. [Google Scholar] [CrossRef] [Green Version]
- Calcabrini, A.; Garcia-Martinez, J.M.; Gonzalez, L.; Julian Tendero, M.; Agullo Ortuno, M.T.; Crateri, P.; Lopez-Rivas, A.; Arancia, G.; Gonzalez-Porque, P.; Martin-Perez, J. Inhibition of proliferation and induction of apoptosis in human breast cancer cells by lauryl gallate. Carcinogenesis 2006, 27, 1699–1712. [Google Scholar] [CrossRef]
- Jara, J.A.; Castro-Castillo, V.; Saavedra-Olavarria, J.; Peredo, L.; Pavanni, M.; Jana, F.; Eugenia Letelier, M.; Parra, E.; Ines Becker, M.; Morello, A.; et al. Antiproliferative and Uncoupling Effects of Delocalized, Lipophilic, Cationic Gallic Acid Derivatives on Cancer Cell Lines. Validation in Vivo in Singenic Mice. J. Med. Chem. 2014, 57, 2440–2454. [Google Scholar] [CrossRef]
- Jara, J.A.; Rojas, D.; Castro-Castillo, V.; Fuentes-Retamal, S.; Sandoval-Acuna, C.; Parra, E.; Pavani, M.; Maya, J.D.; Ferreira, J.; Catalan, M. Novel benzoate-lipophilic cations selectively induce cell death in human colorectal cancer cell lines. Toxicol. Vitr. 2020, 65, 104814. [Google Scholar] [CrossRef]
- Rideout, D.; Bustamante, A.; Patel, J. Mechanism of inhibition of FaDu hypopharyngeal carcinoma cell growth by tetraphenylphosphonium chloride. Int. J. Cancer 1994, 57, 247–253. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Ozsvari, B.; Sotgia, F.; Lisanti, M.P. Dodecyl-TPP Targets Mitochondria and Potently Eradicates Cancer Stem Cells (CSCs): Synergy With FDA-Approved Drugs and Natural Compounds (Vitamin C and Berberine). Front. Oncol. 2019, 9, 615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rideout, D.C.; Calogeropoulou, T.; Jaworski, J.S.; Dagnino, R., Jr.; McCarthy, M.R. Phosphonium salts exhibiting selective anti-carcinoma activity in vitro. Anti-Cancer Drug Des. 1989, 4, 265–280. [Google Scholar]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Battogtokh, G.; Gotov, O.; Kang, J.H.; Cho, J.; Jeong, T.H.; Chimed, G.; Ko, Y.T. Triphenylphosphine-docetaxel conjugate-incorporated albumin nanoparticles for cancer treatment. Nanomedicine 2018, 13, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Tamam, H.; Park, J.; Gadalla, H.H.; Masters, A.R.; Abdel-Aleem, J.A.; Abdelrahman, S.I.; Abdelrahman, A.A.; Lyle, L.T.; Yeo, Y. Development of Liposomal Gemcitabine with High Drug Loading Capacity. Mol. Pharm. 2019, 16, 2858–2871. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Khan, A.R.; Yang, X.; Dong, B.; Ji, J.; Zhai, G. The reversal of chemotherapy-induced multidrug resistance by nanomedicine for cancer therapy. J. Control. Release 2021, 335, 1–20. [Google Scholar] [CrossRef]
- Han, L.; Lv, H.; Wang, D.; Wang, J.; Tang, M. Mitochondrial targeting function of norcantharidin TPP-PEG-PCL nanomicelles promotes apoptosis of liver tumor cells. Chin. Tradit. Herb. Drugs 2020, 51, 4943–4953. [Google Scholar]
- Biswas, S.; Dodwadkar, N.S.; Piroyan, A.; Torchilin, V.P. Surface conjugation of triphenylphosphonium to target poly(amidoamine) dendrimers to mitochondria. Biomaterials 2012, 33, 4773–4782. [Google Scholar] [CrossRef] [Green Version]
- Slika, L.; Patra, D. A short review on chemical properties, stability and nano-technological advances for curcumin delivery. Expert Opin. Drug Deliv. 2020, 17, 61–75. [Google Scholar] [CrossRef]
- Liang, X.; Xu, S.; Zhang, J.; Li, J.; Sheng, Q. Cascade Amplifiers of Intracellular Reactive Oxygen Species Based on Mitochondria-Targeted Core-Shell ZnO-TPP@D/H Nanorods for Breast Cancer Therapy. ACS Appl. Mater. Interfaces 2018, 10, 38749–38759. [Google Scholar] [CrossRef] [PubMed]
- Vlodaysky, I.; Singh, P.; Boyango, I.; Gutter-Kapon, L.; Elkin, M.; Sanderson, R.D.; Lian, N. Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resist. Updates 2016, 29, 54–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Lee, H.; Bae, K.H.; Park, T.G. Heparin immobilized gold nanoparticles for targeted detection and apoptotic death of metastatic cancer cells. Biomaterials 2010, 31, 6530–6536. [Google Scholar] [CrossRef] [PubMed]
- Bilia, A.R.; Piazzini, V.; Guccione, C.; Risaliti, L.; Asprea, M.; Capecchi, G.; Bergonzi, M.C. Improving on Nature: The Role of Nanomedicine in the Development of Clinical Natural Drugs. Planta Med. 2017, 83, 366–381. [Google Scholar] [CrossRef] [Green Version]
- Yoon, N.G.; Lee, H.; Kim, S.Y.; Hu, S.; Kim, D.; Yang, S.; Hong, K.B.; Lee, J.H.; Kang, S.; Kim, B.G.; et al. Mitoquinone Inactivates Mitochondrial Chaperone TRAP1 by Blocking the Client Binding Site. J. Am. Chem. Soc. 2021, 143, 19684–19696. [Google Scholar] [CrossRef]
- Dairkee, S.H.; Hackett, A.J. Differential retention of rhodamine 123 by breast carcinoma and normal human mammary tissue. Breast Cancer Res. Treat. 1991, 18, 57–61. [Google Scholar] [CrossRef]
- Summerhayes, I.C.; Lampidis, T.J.; Bernal, S.D.; Nadakavukaren, J.J.; Nadakavukaren, K.K.; Shepherd, E.L.; Chen, L.B. Unusual retention of rhodamine 123 by mitochondria in muscle and carcinoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 5292–5296. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, X.; Feng, D.; Lv, J.; Cui, X.; Wang, Y.; Wang, Q.; Zhang, L. Application Prospects of Triphenylphosphine-Based Mitochondria-Targeted Cancer Therapy. Cancers 2023, 15, 666. https://doi.org/10.3390/cancers15030666
Cheng X, Feng D, Lv J, Cui X, Wang Y, Wang Q, Zhang L. Application Prospects of Triphenylphosphine-Based Mitochondria-Targeted Cancer Therapy. Cancers. 2023; 15(3):666. https://doi.org/10.3390/cancers15030666
Chicago/Turabian StyleCheng, Xiaoxia, Dong Feng, Junyu Lv, Xiaoman Cui, Yichen Wang, Qun Wang, and Lei Zhang. 2023. "Application Prospects of Triphenylphosphine-Based Mitochondria-Targeted Cancer Therapy" Cancers 15, no. 3: 666. https://doi.org/10.3390/cancers15030666
APA StyleCheng, X., Feng, D., Lv, J., Cui, X., Wang, Y., Wang, Q., & Zhang, L. (2023). Application Prospects of Triphenylphosphine-Based Mitochondria-Targeted Cancer Therapy. Cancers, 15(3), 666. https://doi.org/10.3390/cancers15030666