

Metabolic Health, Mitochondrial Fitness, Physical Activity, and Cancer

 ,
,  ,
,  ,
,  , ,
, ,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Metabolism and Cancer

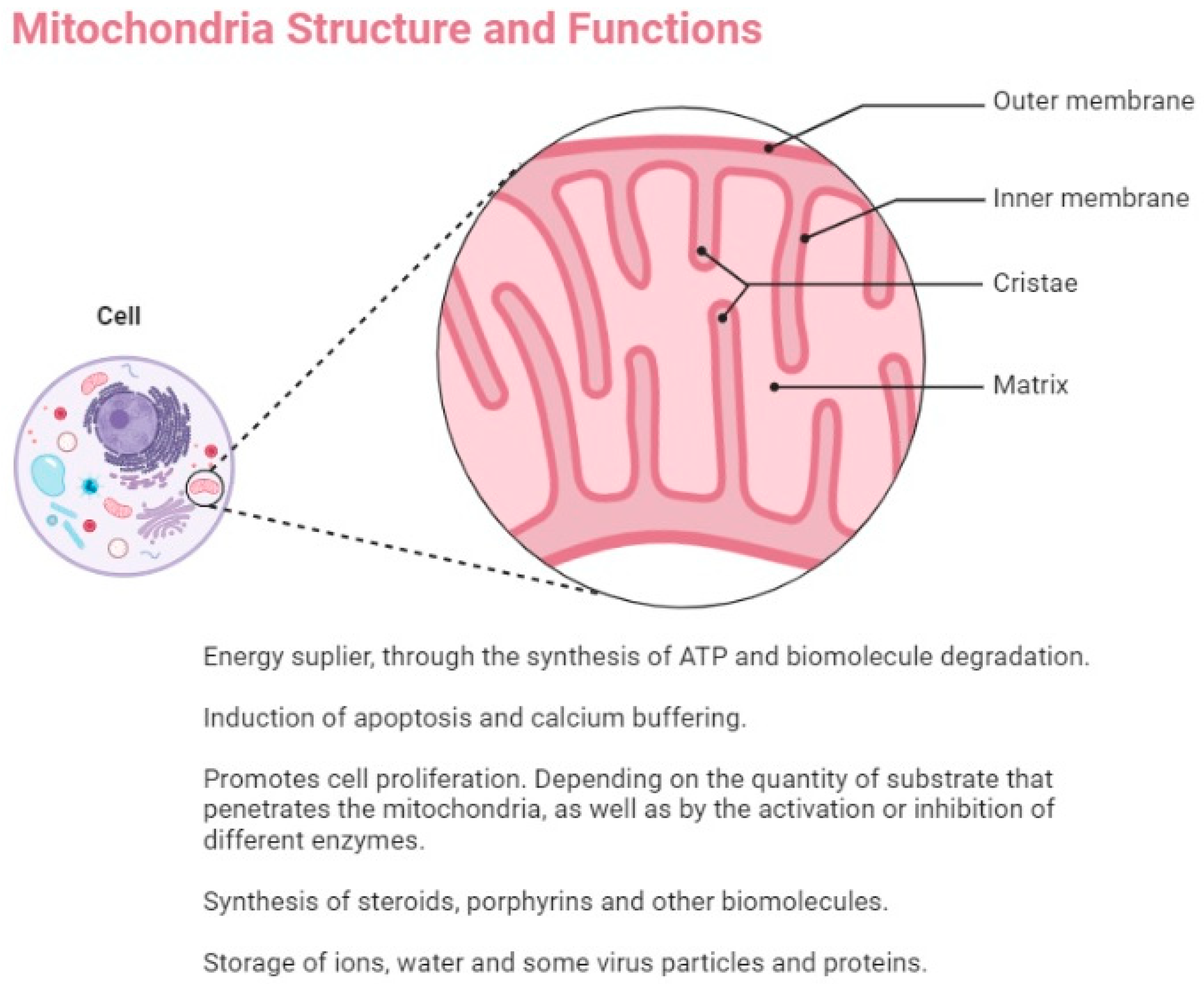{kind=link}
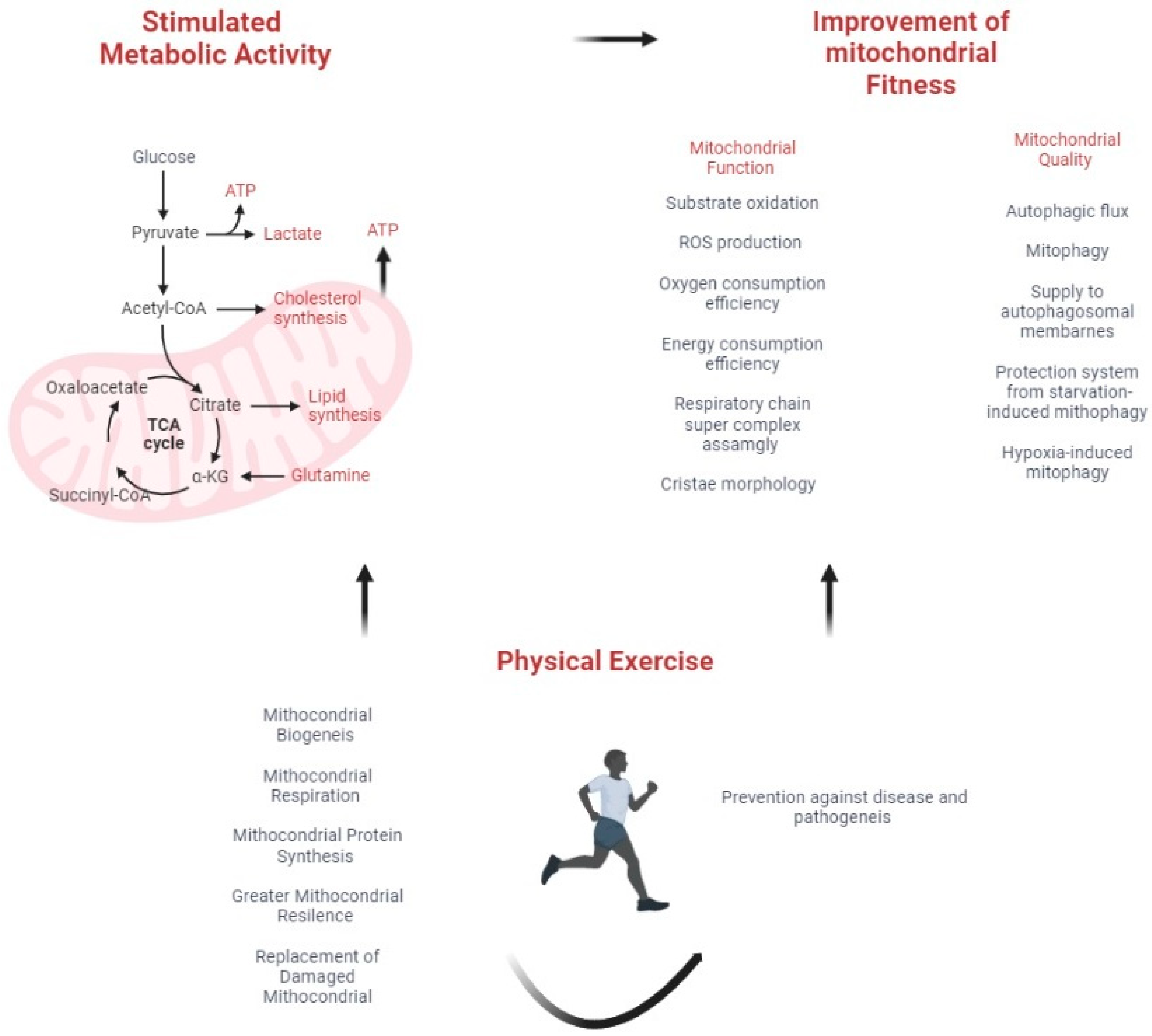{kind=link}
| Protein | Expression in Cancer | Effects on the Metabolism |
|---|---|---|
| Pyruvate kinase isoform M2 (Isoform embryonic) | Increased expression in tumors and cancer cell lines | Increased glycolysis |
| Monocarboxylate transporters (MCTs) | Overexpressed in ovarian, prostate, gastric, and cervical cancers | Increased glycolysis |
| Glutaminase and glutamate oxaloacetate transaminases | Increased expression in cancer | Use of glutamine as a source of ATP and generation of TCA intermediaries |
| Via PI3K/Akt | Dysregulated in cancer | Increased glycolysis |
| HIF-1α | Overexpressed in cancer | Increased glycolysis |
| Myc | Dysregulated in cancer | Promotes the use of glutamine, glycolysis augmented |
| p53 | Mutated in cancer (inactivated) | Active p53 inhibits glycolysis and promotes oxidative phosphorylation. Loss of p53, increased glycolysis |
| Phosphofruct kinase/fructose-2,6-biphosphatase gene B3 (six isoforms) | Augmented expression | Increased glycolytic flux |
| Hexokinase II | Increased expression in hepatoma, cervical cancer | Increased glycolysis |
| Phosphofruct kinase 1 | Hyperactivated in cancer | Increased glycolysis |
| Pyruvate dehydrogenase kinase (PDK, four isoforms) | Increased in cancer | Increased glycolysis |
| Snail (E-cadherin repressor) | Increases epithelial–mesenchymal transition | Suppresses oxidative metabolism in mitochondria |
| Kisspeptin | Cancer metastasis suppressor of thyroid, ovary, bladder, gastric, esophageal, pancreatic, lung, pituitary, and melanoma | Promotes oxidative metabolism, inhibiting glycolysis |
3. Mitochondrial Fitness and Cancer
4. Oxidative Stress
4.1. ROS, Initiation, Promotion, and Progression of Cancer
4.2. Tumor Death by ROS Control
5. Apoptosis
6. Inflammation Response
7. Physical Activity, Cancer, and Stress
8. Effect of Aerobic and Resistance Exercise Interventions on the Cancer Treatment Continuum
9. Active Life Behaviors
9.1. Colon Cancer
9.2. Endometrial Cancer
9.3. Esophageal and Stomach Cancers
9.4. Renal Cancer
9.5. Lung Cancer
9.6. Other Cancers
9.7. Systemic Factors and Cancer
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasso, D.; Zampieri, L.X.; Capelôa, T.; Van de Velde, J.A.; Sonveaux, P. Mitochondria in Cancer. Cell Stress 2020, 4, 114–146. [Google Scholar] [CrossRef] [PubMed]
- Urra, F.A.; Weiss-López, B.; Araya-Maturana, R. Determinants of Anti-Cancer Effect of Mitochondrial Electron Transport Chain Inhibitors: Bioenergetic Profile and Metabolic Flexibility of Cancer Cells. Curr. Pharm. Des. 2016, 22, 5998–6008. [Google Scholar] [CrossRef] [PubMed]
- Bardella, C.; Pollard, P.J.; Tomlinson, I. SDH Mutations in Cancer. Biochim. Biophys. Acta 2011, 1807, 1432–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheriyath, V.; Kaur, J.; Davenport, A.; Khalel, A.; Chowdhury, N.; Gaddipati, L. G1P3 (IFI6), a Mitochondrial Localised Antiapoptotic Protein, Promotes Metastatic Potential of Breast Cancer Cells through MtROS. Br. J. Cancer 2018, 119, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA Damage Response in Cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Alam, M.; Ali, S.; Mohammad, T.; Hasan, G.M.; Yadav, D.K.; Hassan, M.I. B Cell Lymphoma 2: A Potential Therapeutic Target for Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 10442. [Google Scholar] [CrossRef]
- Copeland, W.C.; Wachsman, J.T.; Johnson, F.M.; Penta, J.S. Mitochondrial DNA Alterations in Cancer. Cancer Investig. 2002, 20, 557–569. [Google Scholar] [CrossRef]
- Van Gisbergen, M.W.; Voets, A.M.; Starmans, M.H.W.; de Coo, I.F.M.; Yadak, R.; Hoffmann, R.F.; Boutros, P.C.; Smeets, H.J.M.; Dubois, L.; Lambin, P. How Do Changes in the MtDNA and Mitochondrial Dysfunction Influence Cancer and Cancer Therapy? Challenges, Opportunities and Models. Mutat. Res. Rev. Mutat. Res. 2015, 764, 16–30. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in Cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Grouse, L. Post Hoc Ergo Propter Hoc. J. Thorac. Dis. 2016, 8, E511–E512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsudomi, T.; Steinberg, S.M.; Nau, M.M.; Carbone, D.; D’Amico, D.; Bodner, S.; Oie, H.K.; Linnoila, R.I.; Mulshine, J.L.; Minna, J.D. P53 Gene Mutations in Non-Small-Cell Lung Cancer Cell Lines and Their Correlation with the Presence of Ras Mutations and Clinical Features. Oncogene 1992, 7, 171–180. [Google Scholar] [PubMed]
- McGee, S.L.; Hargreaves, M. Epigenetics and Exercise. Trends Endocrinol. Metab. 2019, 30, 636–645. [Google Scholar] [CrossRef]
- Park, J.H.; Yoo, Y.; Park, Y.J. Epigenetics: Linking Nutrition to Molecular Mechanisms in Aging. Prev. Nutr. Food Sci. 2017, 22, 81–89. [Google Scholar] [CrossRef]
- Matilainen, O.; Quirós, P.M.; Auwerx, J. Mitochondria and Epigenetics—Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef]
- Kossenkov, A.V.; Milcarek, A.; Notta, F.; Jang, G.-H.; Wilson, J.M.; Gallinger, S.; Zhou, D.C.; Ding, L.; Ghosh, J.C.; Perego, M.; et al. Mitochondrial Fitness and Cancer Risk. PLoS ONE 2022, 17, e0273520. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Suárez, V.J.; Beltrán-Velasco, A.I.; Ramos-Campo, D.J.; Mielgo-Ayuso, J.; Nikolaidis, P.A.; Belando, N.; Tornero-Aguilera, J.F. Physical Activity and COVID-19. The Basis for an Efficient Intervention in Times of COVID-19 Pandemic. Physiol. Behav. 2022, 244, 113667. [Google Scholar] [CrossRef]
- Shaw, R.J. Glucose Metabolism and Cancer. Curr. Opin. Cell Biol. 2006, 18, 598–608. [Google Scholar] [CrossRef]
- Locasale, J.W.; Cantley, L.C. Altered Metabolism in Cancer. BMC Biol. 2010, 8, 88. [Google Scholar] [CrossRef] [Green Version]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V.J. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, F.; Chandel, N.S. Mitochondrial Metabolism and Cancer. Ann. N. Y. Acad. Sci. 2009, 1177, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s Contributions to Current Concepts of Cancer Metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.Y.; Soloviev, D.; Brindle, K.M. Imaging Tumor Metabolism Using Positron Emission Tomography. Cancer J. 2015, 21, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The Emerging Role and Targetability of the TCA Cycle in Cancer Metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Feron, O. Cancer Cell Metabolism and Mitochondria: Nutrient Plasticity for TCA Cycle Fueling. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 7–15. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond Aerobic Glycolysis: Transformed Cells Can Engage in Glutamine Metabolism That Exceeds the Requirement for Protein and Nucleotide Synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Valle Mendiola, A.; Soto Cruz, I. Metabolismo Energético y Cáncer. Vertientes Rev. Espec. Cienc. Salud 2014, 17, 108–113. [Google Scholar]
- Marbaniang, C.; Kma, L. Dysregulation of Glucose Metabolism by Oncogenes and Tumor Suppressors in Cancer Cells. Asian Pac. J. Cancer Prev. 2018, 19, 2377–2390. [Google Scholar] [CrossRef]
- Bergers, G.; Fendt, S.-M. The Metabolism of Cancer Cells during Metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef]
- Herrera-González, N.E.; Martínez-García, F.; Mejía-Jiménez, E. El Efecto Warburg: La Mano Derecha En El Desarrollo Del Cáncer. Rev. Espec. Méd. Quir. 2015, 20, 171–177. [Google Scholar]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burtscher, J.; Burtscher, M.; Millet, G.P. The Central Role of Mitochondrial Fitness on Antiviral Defenses: An Advocacy for Physical Activity during the COVID-19 Pandemic. Redox Biol. 2021, 43, 101976. [Google Scholar] [CrossRef]
- Burtscher, J.; Millet, G.P.; Burtscher, M. Low Cardiorespiratory and Mitochondrial Fitness as Risk Factors in Viral Infections: Implications for COVID-19. Br. J. Sport. Med. 2021, 55, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Cuezva, J.M.; Krajewska, M.; de Heredia, M.L.; Krajewski, S.; Santamaría, G.; Kim, H.; Zapata, J.M.; Marusawa, H.; Chamorro, M.; Reed, J.C. The Bioenergetic Signature of Cancer: A Marker of Tumor Progression. Cancer Res. 2002, 62, 6674–6681. [Google Scholar] [PubMed]
- Sánchez-Aragó, M.; Chamorro, M.; Cuezva, J.M. Selection of Cancer Cells with Repressed Mitochondria Triggers Colon Cancer Progression. Carcinogenesis 2010, 31, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Klein, K.; He, K.; Younes, A.I.; Barsoumian, H.B.; Chen, D.; Ozgen, T.; Mosaffa, S.; Patel, R.R.; Gu, M.; Novaes, J.; et al. Role of Mitochondria in Cancer Immune Evasion and Potential Therapeutic Approaches. Front. Immunol. 2020, 11, 573326. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.S.Y. Apoptosis in Cancer: From Pathogenesis to Treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Avery-Kiejda, K.A.; Bowden, N.A.; Croft, A.J.; Scurr, L.L.; Kairupan, C.F.; Ashton, K.A.; Talseth-Palmer, B.A.; Rizos, H.; Zhang, X.D.; Scott, R.J.; et al. P53 in Human Melanoma Fails to Regulate Target Genes Associated with Apoptosis and the Cell Cycle and May Contribute to Proliferation. BMC Cancer 2011, 11, 203. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic Functions of the Tumor Suppressor P53: Implications in Normal Physiology, Metabolic Disorders, and Cancer. Mol. Metab. 2020, 33, 2–22. [Google Scholar] [CrossRef]
- Martincorena, I.; Campbell, P.J. Somatic Mutation in Cancer and Normal Cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Raffo, A.J.; Perlman, H.; Chen, M.W.; Day, M.L.; Streitman, J.S.; Buttyan, R. Overexpression of Bcl-2 Protects Prostate Cancer Cells from Apoptosis in Vitro and Confers Resistance to Androgen Depletion in Vivo. Cancer Res. 1995, 55, 4438–4445. [Google Scholar]
- Mortenson, M.M.; Schlieman, M.G.; Virudalchalam, S.; Bold, R.J. Overexpression of BCL-2 Results in Activation of the AKT/NF-KB Cell Survival Pathway. J. Surg. Res. 2003, 114, 302. [Google Scholar] [CrossRef]
- Izzo, V.; Bravo-San Pedro, J.M.; Sica, V.; Kroemer, G.; Galluzzi, L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016, 26, 655–667. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial Metabolism and Cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in Cancer: Initiators, Amplifiers or an Achilles’ Heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Gaude, E.; Frezza, C. Defects in Mitochondrial Metabolism and Cancer. Cancer Metab. 2014, 2, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Tan, M.; Cai, Q. The Warburg Effect in Tumor Progression: Mitochondrial Oxidative Metabolism as an Anti-Metastasis Mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondria and Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Chen, X.; Sun, X.; Wang, L.; Chen, S. The Glycolytic Switch in Tumors: How Many Players Are Involved? J. Cancer 2017, 8, 3430–3440. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.P.; Sabatini, D.M. Cancer Cell Metabolism: Warburg and Beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial Mutations in Cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar] [CrossRef] [Green Version]
- Pereira, L.; Soares, P.; Máximo, V.; Samuels, D.C. Somatic Mitochondrial DNA Mutations in Cancer Escape Purifying Selection and High Pathogenicity Mutations Lead to the Oncocytic Phenotype: Pathogenicity Analysis of Reported Somatic MtDNA Mutations in Tumors. BMC Cancer 2012, 12, 53. [Google Scholar] [CrossRef]
- Kopinski, P.K.; Singh, L.N.; Zhang, S.; Lott, M.T.; Wallace, D.C. Mitochondrial DNA Variation and Cancer. Nat. Rev. Cancer 2021, 21, 431–445. [Google Scholar] [CrossRef]
- Chang, T.-C.; Lee, H.-T.; Pan, S.-C.; Cho, S.-H.; Cheng, C.; Ou, L.-H.; Lin, C.-I.; Lin, C.-S.; Wei, Y.-H. Metabolic Reprogramming in Response to Alterations of Mitochondrial DNA and Mitochondrial Dysfunction in Gastric Adenocarcinoma. Int. J. Mol. Sci. 2022, 23, 1857. [Google Scholar] [CrossRef]
- Oshima, N.; Ishida, R.; Kishimoto, S.; Beebe, K.; Brender, J.R.; Yamamoto, K.; Urban, D.; Rai, G.; Johnson, M.S.; Benavides, G.; et al. Dynamic Imaging of LDH Inhibition in Tumors Reveals Rapid In Vivo Metabolic Rewiring and Vulnerability to Combination Therapy. Cell Rep. 2020, 30, 1798–1810.e4. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.C.; Lee, I.-M.; Weiderpass, E.; Campbell, P.T.; Sampson, J.N.; Kitahara, C.M.; Keadle, S.K.; Arem, H.; Berrington de Gonzalez, A.; Hartge, P.; et al. Association of Leisure-Time Physical Activity with Risk of 26 Types of Cancer in 1.44 Million Adults. JAMA Intern. Med. 2016, 176, 816–825. [Google Scholar] [CrossRef]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; LLeonart, M.E. Oxidative Stress and Cancer: An Overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Simic, M.G.; Bergtold, D.S.; Karam, L.R. Generation of Oxy Radicals in Biosystems. Mutat. Res. 1989, 214, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [Green Version]
- Veskoukis, A.S.; Tsatsakis, A.M.; Kouretas, D. Dietary Oxidative Stress and Antioxidant Defense with an Emphasis on Plant Extract Administration. Cell Stress Chaperones 2012, 17, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial Dysfunction and Oxidative Stress in Aging and Cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelic, M.D.; Mandic, A.D.; Maricic, S.M.; Srdjenovic, B.U. Oxidative Stress and Its Role in Cancer. J. Cancer Res. Ther. 2021, 17, 22–28. [Google Scholar] [CrossRef]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and Cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive Oxygen Species in Cell Signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, C. Targeted Therapy for Lung Cancer: Present and Future. Ann. Palliat. Med. 2014, 3, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. The Effect of Reactive Oxygen Species on the Synthesis of Prostanoids from Arachidonic Acid. J. Physiol. Pharmacol. Off. J. Polish Physiol. Soc. 2013, 64, 409–421. [Google Scholar]
- Wu, H.; Goel, V.; Haluska, F.G. PTEN Signaling Pathways in Melanoma. Oncogene 2003, 22, 3113–3122. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Wang, Y.; Guo, W.; Zhou, Y.; Lv, C.; Chen, X.; Liu, K. The Significance of the Alteration of 8-OHdG in Serous Ovarian Carcinoma. J. Ovarian Res. 2013, 6, 74. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Liu, N.; Zhu, X.; Wang, L.; Zhao, Y.; Liu, Q.; Gao, C.; Li, J. Subanesthetic Isoflurane Abates ROS-Activated MAPK/NF-ΚB Signaling to Repress Ischemia-Induced Microglia Inflammation and Brain Injury. Aging 2020, 12, 26121–26139. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Ferro, F.; Servais, S.; Besson, P.; Roger, S.; Dumas, J.-F.; Brisson, L. Autophagy and Mitophagy in Cancer Metabolic Remodelling. Semin. Cell Dev. Biol. 2020, 98, 129–138. [Google Scholar] [CrossRef]
- Wang, L.; Azad, N.; Kongkaneramit, L.; Chen, F.; Lu, Y.; Jiang, B.-H.; Rojanasakul, Y. The Fas Death Signaling Pathway Connecting Reactive Oxygen Species Generation and FLICE Inhibitory Protein Down-Regulation. J. Immunol. 2008, 180, 3072–3080. [Google Scholar] [CrossRef] [Green Version]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free Radicals, Antioxidants in Disease and Health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in Cancer Therapy: The Bright Side of the Moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Wang, S.; Konorev, E.A.; Kotamraju, S.; Joseph, J.; Kalivendi, S.; Kalyanaraman, B. Doxorubicin Induces Apoptosis in Normal and Tumor Cells via Distinctly Different Mechanisms. Intermediacy of H(2)O(2)- and P53-Dependent Pathways. J. Biol. Chem. 2004, 279, 25535–25543. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Bermejo, R.; Romo-González, M.; Pérez-Fernández, A.; Ijurko, C.; Hernández-Hernández, Á. Reactive Oxygen Species in Haematopoiesis: Leukaemic Cells Take a Walk on the Wild Side. J. Exp. Clin. Cancer Res. 2018, 37, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mileo, A.M.; Miccadei, S. Polyphenols as Modulator of Oxidative Stress in Cancer Disease: New Therapeutic Strategies. Oxid. Med. Cell. Longev. 2016, 2016, 6475624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubatka, P.; Kapinová, A.; Kello, M.; Kruzliak, P.; Kajo, K.; Výbohová, D.; Mahmood, S.; Murin, R.; Viera, T.; Mojžiš, J.; et al. Fruit Peel Polyphenols Demonstrate Substantial Anti-Tumour Effects in the Model of Breast Cancer. Eur. J. Nutr. 2016, 55, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Raudenská, M.; Balvan, J.; Masařík, M. Cell Death in Head and Neck Cancer Pathogenesis and Treatment. Cell Death Dis. 2021, 12, 192. [Google Scholar] [CrossRef] [PubMed]
- Pchejetski, D.; Golzio, M.; Bonhoure, E.; Calvet, C.; Doumerc, N.; Garcia, V.; Mazerolles, C.; Rischmann, P.; Teissié, J.; Malavaud, B.; et al. Sphingosine Kinase-1 as a Chemotherapy Sensor in Prostate Adenocarcinoma Cell and Mouse Models. Cancer Res. 2005, 65, 11667–11675. [Google Scholar] [CrossRef] [Green Version]
- Pilco-Ferreto, N.; Calaf, G.M. Influence of Doxorubicin on Apoptosis and Oxidative Stress in Breast Cancer Cell Lines. Int. J. Oncol. 2016, 49, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Chance, B.; Williams, G.R. Respiratory Enzymes in Oxidative Phosphorylation. I. Kinetics of Oxygen Utilization. J. Biol. Chem. 1955, 217, 383–393. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative Stress, Mitochondrial Damage and Neurodegenerative Diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS Generation and Its Regulation: Mechanisms Involved in H2O2 Signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and Molecular Targeting Therapy in Cancer. Biomed. Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Zheng, J.; Nussinov, R.; Ma, B. Release of Cytochrome C from Bax Pores at the Mitochondrial Membrane. Sci. Rep. 2017, 7, 2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estaquier, J.; Vallette, F.; Vayssiere, J.-L.; Mignotte, B. The Mitochondrial Pathways of Apoptosis. Adv. Exp. Med. Biol. 2012, 942, 157–183. [Google Scholar] [CrossRef]
- Ramirez, M.L.G.; Salvesen, G.S. A Primer on Caspase Mechanisms. Semin. Cell Dev. Biol. 2018, 82, 79–85. [Google Scholar] [CrossRef]
- Julien, O.; Wells, J.A. Caspases and Their Substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg Effect: Essential Part of Metabolic Reprogramming and Central Contributor to Cancer Progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Hsu, C.-C.; Tseng, L.-M.; Lee, H.-C. Role of Mitochondrial Dysfunction in Cancer Progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Ma, J.; Lu, W. The Significance of Mitochondrial Dysfunction in Cancer. Int. J. Mol. Sci. 2020, 21, 5598. [Google Scholar] [CrossRef]
- Alberghina, L.; Gaglio, D. Redox Control of Glutamine Utilization in Cancer. Cell Death Dis. 2014, 5, e1561. [Google Scholar] [CrossRef] [Green Version]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. P53 Mutations in Human Cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotta, A.P.; Chipuk, J.E. Mitochondrial Dynamics as Regulators of Cancer Biology. Cell. Mol. Life Sci. 2017, 74, 1999–2017. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial Dynamics Regulates Migration and Invasion of Breast Cancer Cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef] [PubMed]
- Cleland, M.M.; Youle, R.J. Mitochondrial Dynamics and Apoptosis. In Mitochondrial Dynamics and Neurodegeneration; Springer: Berlin/Heidelberg, Germany, 2011; pp. 109–138. [Google Scholar]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcárcel-Ares, M.N. Mitochondrial Dysfunction and the Inflammatory Response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Maneiro, E.; López-Armada, M.J.; de Andres, M.C.; Caramés, B.; Martín, M.A.; Bonilla, A.; Del Hoyo, P.; Galdo, F.; Arenas, J.; Blanco, F.J. Effect of Nitric Oxide on Mitochondrial Respiratory Activity of Human Articular Chondrocytes. Ann. Rheum. Dis. 2005, 64, 388–395. [Google Scholar] [CrossRef]
- Guidarelli, A.; Cerioni, L.; Cantoni, O. Inhibition of Complex III Promotes Loss of Ca2+ Dependence for Mitochondrial Superoxide Formation and Permeability Transition Evoked by Peroxynitrite. J. Cell Sci. 2007, 120, 1908–1914. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Xu, M.; Xo, R.; Mates, A.; Wilson, G.L.; Pearsall, A.W., 4th; Grishko, V. Mitochondrial DNA Damage Is Involved in Apoptosis Caused by Pro-Inflammatory Cytokines in Human OA Chondrocytes. Osteoarthr. Cartil. 2010, 18, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-H.; Zhao, Y.-P.; Xue, M.; Ji, C.-B.; Gao, C.-L.; Zhu, J.-G.; Qin, D.-N.; Kou, C.-Z.; Qin, X.-H.; Tong, M.-L.; et al. TNF-Alpha Induces Mitochondrial Dysfunction in 3T3-L1 Adipocytes. Mol. Cell. Endocrinol. 2010, 328, 63–69. [Google Scholar] [CrossRef]
- Fermor, B.; Gurumurthy, A.; Diekman, B.O. Hypoxia, RONS and Energy Metabolism in Articular Cartilage. Osteoarthr. Cartil. 2010, 18, 1167–1173. [Google Scholar] [CrossRef] [Green Version]
- Tomita, M.; Sato, E.F.; Nishikawa, M.; Yamano, Y.; Inoue, M. Nitric Oxide Regulates Mitochondrial Respiration and Functions of Articular Chondrocytes. Arthritis Rheum. 2001, 44, 96–104. [Google Scholar] [CrossRef]
- Minelli, A.; Bellezza, I.; Conte, C.; Culig, Z. Oxidative Stress-Related Aging: A Role for Prostate Cancer? Biochim. Biophys. Acta 2009, 1795, 83–91. [Google Scholar] [CrossRef]
- Poyton, R.O.; Ball, K.A.; Castello, P.R. Mitochondrial Generation of Free Radicals and Hypoxic Signaling. Trends Endocrinol. Metab. 2009, 20, 332–340. [Google Scholar] [CrossRef]
- Walens, A.; DiMarco, A.V.; Lupo, R.; Kroger, B.R.; Damrauer, J.S.; Alvarez, J.V. CCL5 Promotes Breast Cancer Recurrence through Macrophage Recruitment in Residual Tumors. Elife 2019, 8, e43653. [Google Scholar] [CrossRef]
- Chao, T.; Furth, E.E.; Vonderheide, R.H. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-Cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2016, 4, 968–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keklikoglou, I.; Cianciaruso, C.; Güç, E.; Squadrito, M.L.; Spring, L.M.; Tazzyman, S.; Lambein, L.; Poissonnier, A.; Ferraro, G.B.; Baer, C.; et al. Chemotherapy Elicits Pro-Metastatic Extracellular Vesicles in Breast Cancer Models. Nat. Cell Biol. 2019, 21, 190–202. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Karin, M. NF-ΚB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Vurusaner, B.; Poli, G.; Basaga, H. Tumor Suppressor Genes and ROS: Complex Networks of Interactions. Free Radic. Biol. Med. 2012, 52, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Huang, X.; Xue, Z.; Cao, D.; Huang, K.; Chen, J.; Pan, Y.; Gao, Y. The Role of P21 in Apoptosis, Proliferation, Cell Cycle Arrest, and Antioxidant Activity in UVB-Irradiated Human HaCaT Keratinocytes. Med. Sci. Monit. Basic Res. 2015, 21, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.L.; Emerling, B.M.; Ricoult, S.J.H.; Guarente, L. SirT3 Suppresses Hypoxia Inducible Factor 1α and Tumor Growth by Inhibiting Mitochondrial ROS Production. Oncogene 2011, 30, 2986–2996. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Kepp, O.; Galluzzi, L.; Kroemer, G. Inflammasomes in Carcinogenesis and Anticancer Immune Responses. Nat. Immunol. 2012, 13, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, N.; Sasaki, S.; Baba, T. Chemokines in Cancer Development and Progression and Their Potential as Targeting Molecules for Cancer Treatment. Mediat. Inflamm. 2014, 2014, 170381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Ozga, A.J.; Chow, M.T.; Luster, A.D. Chemokines and the Immune Response to Cancer. Immunity 2021, 54, 859–874. [Google Scholar] [CrossRef]
- Dangaj, D.; Bruand, M.; Grimm, A.J.; Ronet, C.; Barras, D.; Duttagupta, P.A.; Lanitis, E.; Duraiswamy, J.; Tanyi, J.L.; Benencia, F. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019, 35, 885–900. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [Green Version]
- McTiernan, A.; Friedenreich, C.M.; Katzmarzyk, P.T.; Powell, K.E.; Macko, R.; Buchner, D.; Pescatello, L.S.; Bloodgood, B.; Tennant, B.; Vaux-Bjerke, A.; et al. Physical Activity in Cancer Prevention and Survival: A Systematic Review. Med. Sci. Sports Exerc. 2019, 51, 1252–1261. [Google Scholar] [CrossRef]
- Kruijsen-Jaarsma, M.; Révész, D.; Bierings, M.B.; Buffart, L.M.; Takken, T. Effects of Exercise on Immune Function in Patients with Cancer: A Systematic Review. Exerc. Immunol. Rev. 2013, 19, 120–143. [Google Scholar]
- Bianchini, F.; Kaaks, R.; Vainio, H. Weight Control and Physical Activity in Cancer Prevention. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2002, 3, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, M. The Second World Cancer Research Fund/American Institute for Cancer Research Expert Report. Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective. Proc. Nutr. Soc. 2008, 67, 253–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrens, G.; Jochem, C.; Keimling, M.; Ricci, C.; Schmid, D.; Leitzmann, M.F. The Association between Physical Activity and Gastroesophageal Cancer: Systematic Review and Meta-Analysis. Eur. J. Epidemiol. 2014, 29, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, A.K.; Dantas Florencio, G.L.; Maisonnette de Atayde Silva, M.J.; Cobucci, R.N.; Giraldo, P.C.; Cote, N.M. Effects of Physical Activity on Breast Cancer Prevention: A Systematic Review. J. Phys. Act. Health 2014, 11, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Keimling, M.; Behrens, G.; Schmid, D.; Jochem, C.; Leitzmann, M.F. The Association between Physical Activity and Bladder Cancer: Systematic Review and Meta-Analysis. Br. J. Cancer 2014, 110, 1862–1870. [Google Scholar] [CrossRef]
- Kenfield, S.A.; Van Blarigan, E.L.; DuPre, N.; Stampfer, M.J.; Giovannucci, E.L.; Chan, J.M. Selenium Supplementation and Prostate Cancer Mortality. JNCI J. Natl. Cancer Inst. 2015, 107, 360. [Google Scholar] [CrossRef] [Green Version]
- Meyerhardt, J.A.; Heseltine, D.; Niedzwiecki, D.; Hollis, D.; Saltz, L.B.; Mayer, R.J.; Thomas, J.; Nelson, H.; Whittom, R.; Hantel, A.; et al. Impact of Physical Activity on Cancer Recurrence and Survival in Patients with Stage III Colon Cancer: Findings from CALGB 89803. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 3535–3541. [Google Scholar] [CrossRef]
- Raglan, O.; Kalliala, I.; Markozannes, G.; Cividini, S.; Gunter, M.J.; Nautiyal, J.; Gabra, H.; Paraskevaidis, E.; Martin-Hirsch, P.; Tsilidis, K.K.; et al. Risk Factors for Endometrial Cancer: An Umbrella Review of the Literature. Int. J. Cancer 2019, 145, 1719–1730. [Google Scholar] [CrossRef] [Green Version]
- Speck, R.M.; Courneya, K.S.; Mâsse, L.C.; Duval, S.; Schmitz, K.H. An Update of Controlled Physical Activity Trials in Cancer Survivors: A Systematic Review and Meta-Analysis. J. Cancer Surviv. 2010, 4, 87–100. [Google Scholar] [CrossRef]
- Psaltopoulou, T.; Ntanasis-Stathopoulos, I.; Tzanninis, I.-G.; Kantzanou, M.; Georgiadou, D.; Sergentanis, T.N. Physical Activity and Gastric Cancer Risk: A Systematic Review and Meta-Analysis. Clin. J. Sport Med. Off. J. Can. Acad. Sport Med. 2016, 26, 445–464. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, W. Roles and Molecular Mechanisms of Physical Exercise in Cancer Prevention and Treatment. J. Sport Health Sci. 2021, 10, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Smith, M.; Lutgendorf, S.K.; Sood, A.K. Impact of Stress on Cancer Metastasis. Future Oncol. 2010, 6, 1863–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigley, A.B.; Spielmann, G.; LaVoy, E.C.P.; Simpson, R.J. Can Exercise-Related Improvements in Immunity Influence Cancer Prevention and Prognosis in the Elderly? Maturitas 2013, 76, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Timmons, B.W.; Cieslak, T. Human Natural Killer Cell Subsets and Acute Exercise: A Brief Review. Exerc. Immunol. Rev. 2008, 14, 8–23. [Google Scholar]
- Idorn, M.; Hojman, P. Exercise-Dependent Regulation of NK Cells in Cancer Protection. Trends Mol. Med. 2016, 22, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Walsh, N.P.; Gleeson, M.; Shephard, R.J.; Gleeson, M.; Woods, J.A.; Bishop, N.C.; Fleshner, M.; Green, C.; Pedersen, B.K.; Hoffman-Goetz, L.; et al. Position Statement. Part One: Immune Function and Exercise. Exerc. Immunol. Rev. 2011, 17, 6–63. [Google Scholar]
- Koelwyn, G.J.; Quail, D.F.; Zhang, X.; White, R.M.; Jones, L.W. Exercise-Dependent Regulation of the Tumour Microenvironment. Nat. Rev. Cancer 2017, 17, 620–632. [Google Scholar] [CrossRef]
- Dennett, A.M.; Peiris, C.L.; Shields, N.; Prendergast, L.A.; Taylor, N.F. Moderate-Intensity Exercise Reduces Fatigue and Improves Mobility in Cancer Survivors: A Systematic Review and Meta-Regression. J. Physiother. 2016, 6, 68–82. [Google Scholar] [CrossRef] [Green Version]
- Petersen, A.M.W.; Pedersen, B.K. The Anti-Inflammatory Effect of Exercise. J. Appl. Physiol. 2005, 98, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and Cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Pedersen, B.K. The Diseasome of Physical Inactivity-and the Role of Myokines in Muscle-Fat Cross Talk. J. Physiol. 2009, 587, 5559–5568. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marmot, M.; Atinmo, T.; Byers, T.; Chen, J.; Hirohata, T.; Jackson, A.; James, W.; Kolonel, L.; Kumanyika, S.; Leitzmann, C. Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective; World Cancer Research Fund/American Institute for Cancer Research: Washington, DC, USA, 2007. [Google Scholar]
- Lynch, B.M.; Neilson, H.K.; Friedenreich, C.M. Physical Activity and Breast Cancer Prevention. Recent Results Cancer Res. Fortschr. Krebsforsch Prog. Dans Rech. Cancer 2011, 186, 13–42. [Google Scholar] [CrossRef]
- Schmitz, K.H.; Courneya, K.S.; Matthews, C.; Demark-Wahnefried, W.; Galvão, D.A.; Pinto, B.M.; Irwin, M.L.; Wolin, K.Y.; Segal, R.J.; Lucia, A.; et al. American College of Sports Medicine Roundtable on Exercise Guidelines for Cancer Survivors. Med. Sci. Sports Exerc. 2010, 42, 1409–1426. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.; Findlay, M.; Von Dincklage, J.; Davidson, W.; Hill, J.; Isenring, E.; Talwar, B.; Bell, K.; Kiss, N.; Kurmis, R.; et al. Using a Wiki Platform to Promote Guidelines Internationally and Maintain Their Currency: Evidence-Based Guidelines for the Nutritional Management of Adult Patients with Head and Neck Cancer. J. Hum. Nutr. Diet. 2013, 26, 182–190. [Google Scholar] [CrossRef]
- Friedenreich, C.M.; Gregory, J.; Kopciuk, K.A.; Mackey, J.R.; Courneya, K.S. Prospective Cohort Study of Lifetime Physical Activity and Breast Cancer Survival. Int. J. Cancer 2009, 124, 1954–1962. [Google Scholar] [CrossRef]
- Meyerhardt, J.A.; Giovannucci, E.L.; Holmes, M.D.; Chan, A.T.; Chan, J.A.; Colditz, G.A.; Fuchs, C.S. Physical Activity and Survival after Colorectal Cancer Diagnosis. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 3527–3534. [Google Scholar] [CrossRef]
- Rogers, C.J.; Colbert, L.H.; Greiner, J.W.; Perkins, S.N.; Hursting, S.D. Physical Activity and Cancer Prevention: Pathways and Targets for Intervention. Sports Med. 2008, 38, 271–296. [Google Scholar] [CrossRef]
- Cramp, F.; Daniel, J. Exercise for the Management of Cancer-Related Fatigue in Adults. Cochrane Database Syst. Rev. 2008, 11, CD006145. [Google Scholar] [CrossRef]
- Segerstrom, S.C.; Miller, G.E. Psychological Stress and the Human Immune System: A Meta-Analytic Study of 30 Years of Inquiry. Psychol. Bull. 2004, 130, 601–630. [Google Scholar] [CrossRef] [Green Version]
- Melhem-Bertrandt, A.; Chavez-Macgregor, M.; Lei, X.; Brown, E.N.; Lee, R.T.; Meric-Bernstam, F.; Sood, A.K.; Conzen, S.D.; Hortobagyi, G.N.; Gonzalez-Angulo, A.-M. Beta-Blocker Use Is Associated with Improved Relapse-Free Survival in Patients with Triple-Negative Breast Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 2645–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Ma, Y.; Tanimoto, T.; Ozaki, A.; Chen, W.-L.; Wang, J.-Y.; Zhang, Y.-X.; Chen, L.-L.; Wang, J.-W.; Yu, J.-M. Effects of Physical Activity and Stress on the Relationship between Social Capital and Quality of Life among Breast Cancer Survivors. Sci. Rep. 2020, 10, 17746. [Google Scholar] [CrossRef] [PubMed]
- Nocon, M.; Hiemann, T.; Müller-Riemenschneider, F.; Thalau, F.; Roll, S.; Willich, S.N. Association of Physical Activity with All-Cause and Cardiovascular Mortality: A Systematic Review and Meta-Analysis. Eur. J. Cardiovasc. Prev. Rehabil. Off. J. Eur. Soc. Cardiol. Work. Groups Epidemiol. Prev. Card. Rehabil. Exerc. Physiol. 2008, 15, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Monninkhof, E.M.; Elias, S.G.; Vlems, F.A.; van der Tweel, I.; Schuit, A.J.; Voskuil, D.W.; van Leeuwen, F.E. Physical Activity and Breast Cancer: A Systematic Review. Epidemiology 2007, 18, 137–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovio, S.; Kåreholt, I.; Helkala, E.-L.; Viitanen, M.; Winblad, B.; Tuomilehto, J.; Soininen, H.; Nissinen, A.; Kivipelto, M. Leisure-Time Physical Activity at Midlife and the Risk of Dementia and Alzheimer’s Disease. Lancet Neurol. 2005, 4, 705–711. [Google Scholar] [CrossRef]
- Thompson, P.D.; Arena, R.; Riebe, D.; Pescatello, L.S. ACSM’s New Preparticipation Health Screening Recommendations from ACSM’s Guidelines for Exercise Testing and Prescription. Curr. Sports Med. Rep. 2013, 12, 215–217. [Google Scholar] [CrossRef]
- Christensen, J.F.; Simonsen, C.; Hojman, P. Exercise Training in Cancer Control and Treatment. Compr. Physiol. 2018, 9, 165–205. [Google Scholar] [CrossRef]
- Schmitz, K.H.; Campbell, A.M.; Stuiver, M.M.; Pinto, B.M.; Schwartz, A.L.; Morris, G.S.; Ligibel, J.A.; Cheville, A.; Galvão, D.A.; Alfano, C.M.; et al. Exercise Is Medicine in Oncology: Engaging Clinicians to Help Patients Move through Cancer. CA Cancer J. Clin. 2019, 69, 468–484. [Google Scholar] [CrossRef] [Green Version]
- The “war on Cancer” Isn’t yet Won. Nature 2022, 601, 297. [CrossRef]
- Liu, L.; Shi, Y.; Li, T.; Qin, Q.; Yin, J.; Pang, S.; Nie, S.; Wei, S. Leisure Time Physical Activity and Cancer Risk: Evaluation of the WHO’s Recommendation Based on 126 High-Quality Epidemiological Studies. Br. J. Sports Med. 2016, 50, 372–378. [Google Scholar] [CrossRef]
- Hojman, P.; Gehl, J.; Christensen, J.F.; Pedersen, B.K. Molecular Mechanisms Linking Exercise to Cancer Prevention and Treatment. Cell Metab. 2018, 27, 10–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashcraft, K.A.; Peace, R.M.; Betof, A.S.; Dewhirst, M.W.; Jones, L.W. Efficacy and Mechanisms of Aerobic Exercise on Cancer Initiation, Progression, and Metastasis: A Critical Systematic Review of In Vivo Preclinical Data. Cancer Res. 2016, 76, 4032–4050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollán, M.; Casla-Barrio, S.; Alfaro, J.; Esteban, C.; Segui-Palmer, M.A.; Lucia, A.; Martín, M. Exercise and Cancer: A Position Statement from the Spanish Society of Medical Oncology. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2020, 22, 1710–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinton, S.K.; Giovannucci, E.L.; Hursting, S.D. The World Cancer Research Fund/American Institute for Cancer Research Third Expert Report on Diet, Nutrition, Physical Activity, and Cancer: Impact and Future Directions. J. Nutr. 2020, 150, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.C.; Newton, R.U.; Spence, R.R.; Galvão, D.A. The Exercise and Sports Science Australia Position Statement: Exercise Medicine in Cancer Management. J. Sci. Med. Sport 2019, 22, 1175–1199. [Google Scholar] [CrossRef] [Green Version]
- Segal, R.; Zwaal, C.; Green, E.; Tomasone, J.R.; Loblaw, A.; Petrella, T. Exercise for People with Cancer: A Clinical Practice Guideline. Curr. Oncol. 2017, 24, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Cormie, P.; Atkinson, M.; Bucci, L.; Cust, A.; Eakin, E.; Hayes, S.; McCarthy, A.L.; Murnane, A.; Patchell, S.; Adams, D. Clinical Oncology Society of Australia Position Statement on Exercise in Cancer Care. Med. J. Aust. 2018, 209, 184–187. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.V.; Friedenreich, C.M.; Moore, S.C.; Hayes, S.C.; Silver, J.K.; Campbell, K.L.; Winters-Stone, K.; Gerber, L.H.; George, S.M.; Fulton, J.E.; et al. American College of Sports Medicine Roundtable Report on Physical Activity, Sedentary Behavior, and Cancer Prevention and Control. Med. Sci. Sports Exerc. 2019, 51, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.K.; Saltin, B. Evidence for Prescribing Exercise as Therapy in Chronic Disease. Scand. J. Med. Sci. Sports 2006, 16 (Suppl. 1), 3–63. [Google Scholar] [CrossRef]
- Singh, F.; Newton, R.U.; Galvão, D.A.; Spry, N.; Baker, M.K. A Systematic Review of Pre-Surgical Exercise Intervention Studies with Cancer Patients. Surg. Oncol. 2013, 22, 92–104. [Google Scholar] [CrossRef]
- Jones, L.W.; Eves, N.D.; Peddle, C.J.; Courneya, K.S.; Haykowsky, M.; Kumar, V.; Winton, T.W.; Reiman, T. Effects of Presurgical Exercise Training on Systemic Inflammatory Markers among Patients with Malignant Lung Lesions. Appl. Physiol. Nutr. Metab. Physiol. Appl. Nutr. Metab. 2009, 34, 197–202. [Google Scholar] [CrossRef]
- Ligibel, J.A.; Irwin, M.; Dillon, D.; Barry, W.; Giobbie-Hurder, A.; Frank, E.; Winer, E.P.; McTiernan, A.; Cornwell, M.; Pun, M. Abstract S5-05: Impact of Pre-Operative Exercise on Breast Cancer Gene Expression. Cancer Res. 2017, 77 (Suppl. 4), S5-05. [Google Scholar] [CrossRef]
- Gil-Rey, E.; Quevedo-Jerez, K.; Maldonado-Martin, S.; Herrero-Román, F. Exercise Intensity Guidelines for Cancer Survivors: A Comparison with Reference Values. Int. J. Sports Med. 2014, 35, e1–e9. [Google Scholar] [CrossRef]
- Campbell, K.L.; Winters-Stone, K.M.; Wiskemann, J.; May, A.M.; Schwartz, A.L.; Courneya, K.S.; Zucker, D.S.; Matthews, C.E.; Ligibel, J.A.; Gerber, L.H.; et al. Exercise Guidelines for Cancer Survivors: Consensus Statement from International Multidisciplinary Roundtable. Med. Sci. Sports Exerc. 2019, 51, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Schmid, D.; Leitzmann, M.F. Cardiorespiratory Fitness as Predictor of Cancer Mortality: A Systematic Review and Meta-Analysis. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Fuller, J.T.; Hartland, M.C.; Maloney, L.T.; Davison, K. Therapeutic Effects of Aerobic and Resistance Exercises for Cancer Survivors: A Systematic Review of Meta-Analyses of Clinical Trials. Br. J. Sports Med. 2018, 52, 1311. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.-L.; Zhang, F.-M.; Li, W.; Wang, K.-H.; Xu, H.-X.; Song, C.-H.; Guo, Z.-Q.; Shi, H.-P. Associations of Low Handgrip Strength with Cancer Mortality: A Multicentre Observational Study. J. Cachexia Sarcopenia Muscle 2020, 11, 1476–1486. [Google Scholar] [CrossRef]
- Ruiz, J.R.; Sui, X.; Lobelo, F.; Lee, D.-C.; Morrow, J.R.J.; Jackson, A.W.; Hébert, J.R.; Matthews, C.E.; Sjöström, M.; Blair, S.N. Muscular Strength and Adiposity as Predictors of Adulthood Cancer Mortality in Men. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2009, 18, 1468–1476. [Google Scholar] [CrossRef] [Green Version]
- Strasser, B.; Steindorf, K.; Wiskemann, J.; Ulrich, C.M. Impact of Resistance Training in Cancer Survivors: A Meta-Analysis. Med. Sci. Sports Exerc. 2013, 45, 2080–2090. [Google Scholar] [CrossRef]
- Padilha, C.S.; Marinello, P.C.; Galvão, D.A.; Newton, R.U.; Borges, F.H.; Frajacomo, F.; Deminice, R. Evaluation of Resistance Training to Improve Muscular Strength and Body Composition in Cancer Patients Undergoing Neoadjuvant and Adjuvant Therapy: A Meta-Analysis. J. Cancer Surviv. 2017, 11, 339–349. [Google Scholar] [CrossRef]
- Lee, J. The Effects of Resistance Training on Muscular Strength and Hypertrophy in Elderly Cancer Patients: A Systematic Review and Meta-Analysis. J. Sport Health Sci. 2022, 11, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Aagaard, P.; Simonsen, E.B.; Andersen, J.L.; Magnusson, P.; Dyhre-Poulsen, P. Neural Adaptation to Resistance Training: Changes in Evoked V-Wave and H-Reflex Responses. J. Appl. Physiol. 2002, 92, 2309–2318. [Google Scholar] [CrossRef] [PubMed]
- Al-Majid, S.; Waters, H. The Biological Mechanisms of Cancer-Related Skeletal Muscle Wasting: The Role of Progressive Resistance Exercise. Biol. Res. Nurs. 2008, 10, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Kangas, M.; Bovbjerg, D.H.; Montgomery, G.H. Cancer-Related Fatigue: A Systematic and Meta-Analytic Review of Non-Pharmacological Therapies for Cancer Patients. Psychol. Bull. 2008, 134, 700–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeely, M.L.; Courneya, K.S. Exercise Programs for Cancer-Related Fatigue: Evidence and Clinical Guidelines. J. Natl. Compr. Cancer Netw. 2010, 8, 945–953. [Google Scholar] [CrossRef]
- Cramp, F.; Byron-Daniel, J. Exercise for the Management of Cancer-Related Fatigue in Adults. Cochrane Database Syst. Rev. 2012, 11, CD006145. [Google Scholar] [CrossRef]
- Takemura, N.; Cheung, D.S.T.; Smith, R.; Deng, W.; Ho, K.Y.; Lin, J.; Kwok, J.Y.Y.; Lam, T.-C.; Lin, C.-C. Effectiveness of Aerobic Exercise and Mind-Body Exercise in Cancer Patients with Poor Sleep Quality: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Sleep Med. Rev. 2020, 53, 101334. [Google Scholar] [CrossRef]
- Berger, R.J.; Phillips, N.H. Comparative Aspects of Energy Metabolism, Body Temperature and Sleep. Acta Physiol. Scand. Suppl. 1988, 574, 21–27. [Google Scholar]
- Driver, H.S.; Taylor, S.R. Exercise and Sleep. Sleep Med. Rev. 2000, 4, 387–402. [Google Scholar] [CrossRef]
- Santos, R.V.T.; Tufik, S.; De Mello, M.T. Exercise, Sleep and Cytokines: Is There a Relation? Sleep Med. Rev. 2007, 11, 231–239. [Google Scholar] [CrossRef]
- Kovacevic, A.; Mavros, Y.; Heisz, J.J.; Fiatarone Singh, M.A. The Effect of Resistance Exercise on Sleep: A Systematic Review of Randomized Controlled Trials. Sleep Med. Rev. 2018, 39, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.L.; Jeong, Y. Quality of Life in Patients with Non-Small Cell Lung Cancer: Structural Equation Modeling. Cancer Nurs. 2019, 42, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Movsas, B.; Moughan, J.; Sarna, L.; Langer, C.; Werner-Wasik, M.; Nicolaou, N.; Komaki, R.; Machtay, M.; Wasserman, T.; Bruner, D.W. Quality of Life Supersedes the Classic Prognosticators for Long-Term Survival in Locally Advanced Non-Small-Cell Lung Cancer: An Analysis of RTOG 9801. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 5816–5822. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Li, X.-X.; Ma, H.-K.; Zhang, X.; Wang, B.-W.; Guo, T.-T.; Xiao, Y.; Bing, Z.-T.; Ge, L.; Yang, K.-H.; et al. Exercise Training for Improving Patient-Reported Outcomes in Patients with Advanced-Stage Cancer: A Systematic Review and Meta-Analysis. J. Pain Symptom Manag. 2020, 59, 734–749.e10. [Google Scholar] [CrossRef]
- Fukushima, T.; Nakano, J.; Hashizume, K.; Ueno, K.; Matsuura, E.; Ikio, Y.; Ishii, S.; Morishita, S.; Tanaka, K.; Kusuba, Y. Effects of Aerobic, Resistance, and Mixed Exercises on Quality of Life in Patients with Cancer: A Systematic Review and Meta-Analysis. Complement. Ther. Clin. Pract. 2021, 42, 101290. [Google Scholar] [CrossRef] [PubMed]
- Craft, L.L.; Vaniterson, E.H.; Helenowski, I.B.; Rademaker, A.W.; Courneya, K.S. Exercise Effects on Depressive Symptoms in Cancer Survivors: A Systematic Review and Meta-Analysis. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2012, 21, 3–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massie, M.J. Prevalence of Depression in Patients with Cancer. JNCI Monogr. 2004, 2004, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Pirl, W.F. Evidence Report on the Occurrence, Assessment, and Treatment of Depression in Cancer Patients. J. Natl. Cancer Inst. Monogr. 2004, 2004, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Fleishman, S.B. Treatment of Symptom Clusters: Pain, Depression, and Fatigue. J. Natl. Cancer Inst. Monogr. 2004, 2004, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Wipfli, B.M.; Rethorst, C.D.; Landers, D.M. The Anxiolytic Effects of Exercise: A Meta-Analysis of Randomized Trials and Dose–Response Analysis. J. Sport Exerc. Psychol. 2008, 30, 392–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and Classification of Cancer Cachexia: An International Consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, M.; Murton, A.J.; Wilcock, A. Therapeutic Exercise in Cancer Cachexia. Crit. Rev. Oncogenes. 2012, 17, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Grande, A.J.; Silva, V.; Riera, R.; Medeiros, A.; Vitoriano, S.G.P.; Peccin, M.S.; Maddocks, M. Exercise for Cancer Cachexia in Adults. Cochrane Database Syst. Rev. 2014, 11, CD010804. [Google Scholar] [CrossRef] [Green Version]
- Grande, A.J.; Silva, V.; Sawaris Neto, L.; Teixeira Basmage, J.P.; Peccin, M.S.; Maddocks, M. Exercise for Cancer Cachexia in Adults. Cochrane Database Syst. Rev. 2021, 3, CD010804. [Google Scholar] [CrossRef] [PubMed]
- Heywood, R.; McCarthy, A.L.; Skinner, T.L. Safety and Feasibility of Exercise Interventions in Patients with Advanced Cancer: A Systematic Review. Support. Care Cancer Off. J. Multinatl. Assoc. Support. Care Cancer 2017, 25, 3031–3050. [Google Scholar] [CrossRef] [PubMed]
- Batalik, L.; Winnige, P.; Dosbaba, F.; Vlazna, D.; Janikova, A. Home-Based Aerobic and Resistance Exercise Interventions in Cancer Patients and Survivors: A Systematic Review. Cancers 2021, 13, 1915. [Google Scholar] [CrossRef]
- Kulhánová, I.; Znaor, A.; Shield, K.D.; Arnold, M.; Vignat, J.; Charafeddine, M.; Fadhil, I.; Fouad, H.; Al-Omari, A.; Al-Zahrani, A.S.; et al. Proportion of Cancers Attributable to Major Lifestyle and Environmental Risk Factors in the Eastern Mediterranean Region. Int. J. Cancer 2020, 146, 646–656. [Google Scholar] [CrossRef]
- Poirier, A.E.; Ruan, Y.; Volesky, K.D.; King, W.D.; O’Sullivan, D.E.; Gogna, P.; Walter, S.D.; Villeneuve, P.J.; Friedenreich, C.M.; Brenner, D.R. The Current and Future Burden of Cancer Attributable to Modifiable Risk Factors in Canada: Summary of Results. Prev. Med. 2019, 122, 140–147. [Google Scholar] [CrossRef]
- Guthold, R.; Stevens, G.A.; Riley, L.M.; Bull, F.C. Worldwide Trends in Insufficient Physical Activity from 2001 to 2016: A Pooled Analysis of 358 Population-Based Surveys with 1.9 Million Participants. Lancet Glob. Health 2018, 6, e1077–e1086. [Google Scholar] [CrossRef] [Green Version]
- NCD Risk Factor Collaboration. Trends in Adult Body-Mass Index in 200 Countries from 1975 to 2014: A Pooled Analysis of 1698 Population-Based Measurement Studies with 19·2 Million Participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef] [Green Version]
- Di Cesare, M.; Obreja, G. Worldwide Trends in Body-Mass Index, Underweight, Overweight, and Obesity from 1975 to 2016: A Pooled Analysis of 2416 Population-Based Measurement Studies in 128·9 Million Children, Adolescents, and Adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef] [Green Version]
- Bandera, E.V.; Alfano, C.M.; Qin, B.; Kang, D.-W.; Friel, C.P.; Dieli-Conwright, C.M. Harnessing Nutrition and Physical Activity for Breast Cancer Prevention and Control to Reduce Racial/Ethnic Cancer Health Disparities. Am. Soc. Clin. Oncol. Educ. Book 2021, 41, e62–e78. [Google Scholar] [CrossRef] [PubMed]
- Wesselink, E.; van Baar, H.; van Zutphen, M.; Tibosch, M.; Kouwenhoven, E.A.; Keulen, E.T.P.; Kok, D.E.; van Halteren, H.K.; Breukink, S.O.; de Wilt, J.H.W. Inflammation Is a Mediating Factor in the Association between Lifestyle and Fatigue in Colorectal Cancer Patients. Cancers 2020, 12, 3701. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [Green Version]
- Mahamat-Saleh, Y.; Al-Rahmoun, M.; Severi, G.; Ghiasvand, R.; Veierod, M.B.; Caini, S.; Palli, D.; Botteri, E.; Sacerdote, C.; Ricceri, F.; et al. Baseline and Lifetime Alcohol Consumption and Risk of Skin Cancer in the European Prospective Investigation into Cancer and Nutrition Cohort (EPIC). Int. J. Cancer 2022, 152, 348–362. [Google Scholar] [CrossRef]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer Treatment and Survivorship Statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Huang, X.; Zhou, S.; Ding, Y.; Wang, H.; Jiang, W.; Xu, M. IL1RN and PRRX1 as a Prognostic Biomarker Correlated with Immune Infiltrates in Colorectal Cancer: Evidence from Bioinformatic Analysis. Int. J. Genom. 2022, 2022, 2723264. [Google Scholar] [CrossRef]
- Wolin, K.Y.; Yan, Y.; Colditz, G.A.; Lee, I.-M. Physical Activity and Colon Cancer Prevention: A Meta-Analysis. Br. J. Cancer 2009, 100, 611–616. [Google Scholar] [CrossRef]
- Li, X.; Marcus, D.; Russell, J.; Aboagye, E.O.; Ellis, L.B.; Sheeka, A.; Park, W.-H.E.; Bharwani, N.; Ghaem-Maghami, S.; Rockall, A.G. An Integrated Clinical-MR Radiomics Model to Estimate Survival Time in Patients with Endometrial Cancer. J. Magn. Reson. Imaging 2022. [Google Scholar] [CrossRef]
- Schmid, D.; Behrens, G.; Keimling, M.; Jochem, C.; Ricci, C.; Leitzmann, M. A Systematic Review and Meta-Analysis of Physical Activity and Endometrial Cancer Risk. Eur. J. Epidemiol. 2015, 30, 397–412. [Google Scholar] [CrossRef]
- Cust, A.E.; Armstrong, B.K.; Friedenreich, C.M.; Slimani, N.; Bauman, A. Physical Activity and Endometrial Cancer Risk: A Review of the Current Evidence, Biologic Mechanisms and the Quality of Physical Activity Assessment Methods. Cancer Causes Control 2007, 18, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.; Ju, W.; Lee, D.H.; Ding, E.L.; Hsieh, C.C.; Goodman, J.E.; Giovannucci, E.L. Leisure-Time Physical Activity and Endometrial Cancer Risk: Dose-Response Meta-Analysis of Epidemiological Studies. Int. J. Cancer 2014, 135, 682–694. [Google Scholar] [CrossRef]
- Tavani, A.; Bravi, F.; Dal Maso, L.; Zucchetto, A.; Bosetti, C.; Pelucchi, C.; Montella, M.; Franceschi, S.; La Vecchia, C. Physical Activity and Risk of Endometrial Cancer: An Italian Case-Control Study. Eur. J. Cancer Prev. Off. J. Eur. Cancer Prev. Organ. 2009, 18, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Friberg, E.; Mantzoros, C.S.; Wolk, A. Physical Activity and Risk of Endometrial Cancer: A Population-Based Prospective Cohort Study. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2006, 15, 2136–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitani, S.; Kawakami, H.; Shiraishi, O.; Kanemura, H.; Suzuki, S.; Haratani, K.; Hayashi, H.; Yonesaka, K.; Chiba, Y.; Yasuda, T.; et al. Implication of Changes in PD-L1 Expression during Neoadjuvant Chemotherapy with Docetaxel, Cisplatin, and 5-Fluorouracil (DCF) Regimen in Esophageal Squamous Cell Carcinoma. Esophagus 2022. [Google Scholar] [CrossRef]
- Cook, M.B.; Matthews, C.E.; Gunja, M.Z.; Abid, Z.; Freedman, N.D.; Abnet, C.C. Physical Activity and Sedentary Behavior in Relation to Esophageal and Gastric Cancers in the NIH-AARP Cohort. PLoS ONE 2013, 8, e84805. [Google Scholar] [CrossRef]
- Teras, L.R.; Gapstur, S.M.; Diver, W.R.; Birmann, B.M.; Patel, A.V. Recreational Physical Activity, Leisure Sitting Time and Risk of Non-Hodgkin Lymphoid Neoplasms in the American Cancer Society Cancer Prevention Study II Cohort. Int. J. Cancer 2012, 131, 1912–1920. [Google Scholar] [CrossRef]
- Liss, M.; Natarajan, L.; Hasan, A.; Noguchi, J.L.; White, M.; Parsons, J.K. Physical Activity Decreases Kidney Cancer Mortality. Curr. Urol. 2017, 10, 193–198. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Bai, Y.-P.; Fan, X.-X. Association Between Physical Activity and Lower Risk of Lung Cancer: A Meta-Analysis of Cohort Studies. Front. Oncol. 2019, 9, 5. [Google Scholar] [CrossRef] [Green Version]
- Walter, R.B.; Buckley, S.A.; White, E. Regular Recreational Physical Activity and Risk of Hematologic Malignancies: Results from the Prospective VITamins and Lifestyle (VITAL) Study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, 1370–1377. [Google Scholar] [CrossRef]
- Van Veldhoven, C.M.; Khan, A.E.; Teucher, B.; Rohrmann, S.; Raaschou-Nielsen, O.; Tjønneland, A.; Overvad, K.; Vigl, M.; Boeing, H.; Benetou, V.; et al. Physical Activity and Lymphoid Neoplasms in the European Prospective Investigation into Cancer and Nutrition (EPIC). Eur. J. Cancer 2011, 47, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Maes, M. Gut Dysbiosis Dysregulates Central and Systemic Homeostasis via Suboptimal Mitochondrial Function: Assessment, Treatment and Classification Implications. Curr. Top. Med. Chem. 2020, 20, 524–539. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Tumour Microenvironment: Roles of the Aryl Hydrocarbon Receptor, O-GlcNAcylation, Acetyl-CoA and Melatonergic Pathway in Regulating Dynamic Metabolic Interactions across Cell Types-Tumour Microenvironment and Metabolism. Int. J. Mol. Sci. 2020, 22, 141. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Suárez, V.J.; Bustamante-Sanchez, Á.; Tornero-Aguilera, J.F.; Ruisoto, P.; Mielgo-Ayuso, J. Inflammation in COVID-19 and the Effects of Non-Pharmacological Interventions during the Pandemic: A Review. Int. J. Mol. Sci. 2022, 23, 15584. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Powell, C.; Phillips, C.M.; Millar, S.R.; Carson, B.P.; Dowd, K.P.; Perry, I.J.; Kearney, P.M.; Harrington, J.M.; O′Toole, P.W.; et al. The Influence of Different Physical Activity Behaviours on the Gut Microbiota of Older Irish Adults. J. Nutr. Health Aging 2021, 25, 854–861. [Google Scholar] [CrossRef]
- Cucielo, M.S.; Cesário, R.C.; Silveira, H.S.; Gaiotte, L.B.; Dos Santos, S.A.A.; de Campos Zuccari, D.A.P.; Seiva, F.R.F.; Reiter, R.J.; de Almeida Chuffa, L.G. Melatonin Reverses the Warburg-Type Metabolism and Reduces Mitochondrial Membrane Potential of Ovarian Cancer Cells Independent of MT1 Receptor Activation. Molecules 2022, 27, 4350. [Google Scholar] [CrossRef]
- Escames, G.; Ozturk, G.; Baño-Otálora, B.; Pozo, M.J.; Madrid, J.A.; Reiter, R.J.; Serrano, E.; Concepción, M.; Acuña-Castroviejo, D. Exercise and melatonin in humans: Reciprocal benefits. J. Pineal Res. 2012, 52, 1–11. [Google Scholar] [CrossRef]
- Schmudde, M.; Braun, A.; Pende, D.; Sonnemann, J.; Klier, U.; Beck, J.F.; Moretta, L.; Bröker, B.M. Histone deacetylase inhibitors sensitize tumour cells for cytotoxic effects of natural killer cells. Cancer Lett. 2008, 272, 110–121. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clemente-Suárez, V.J.; Martín-Rodríguez, A.; Redondo-Flórez, L.; Ruisoto, P.; Navarro-Jiménez, E.; Ramos-Campo, D.J.; Tornero-Aguilera, J.F. Metabolic Health, Mitochondrial Fitness, Physical Activity, and Cancer. Cancers 2023, 15, 814. https://doi.org/10.3390/cancers15030814
Clemente-Suárez VJ, Martín-Rodríguez A, Redondo-Flórez L, Ruisoto P, Navarro-Jiménez E, Ramos-Campo DJ, Tornero-Aguilera JF. Metabolic Health, Mitochondrial Fitness, Physical Activity, and Cancer. Cancers. 2023; 15(3):814. https://doi.org/10.3390/cancers15030814
Chicago/Turabian StyleClemente-Suárez, Vicente Javier, Alexandra Martín-Rodríguez, Laura Redondo-Flórez, Pablo Ruisoto, Eduardo Navarro-Jiménez, Domingo Jesús Ramos-Campo, and José Francisco Tornero-Aguilera. 2023. "Metabolic Health, Mitochondrial Fitness, Physical Activity, and Cancer" Cancers 15, no. 3: 814. https://doi.org/10.3390/cancers15030814
APA StyleClemente-Suárez, V. J., Martín-Rodríguez, A., Redondo-Flórez, L., Ruisoto, P., Navarro-Jiménez, E., Ramos-Campo, D. J., & Tornero-Aguilera, J. F. (2023). Metabolic Health, Mitochondrial Fitness, Physical Activity, and Cancer. Cancers, 15(3), 814. https://doi.org/10.3390/cancers15030814