
c-MYC-Induced AP4 Attenuates DREAM-Mediated Repression by p53



Abstract
:Simple Summary
Abstract
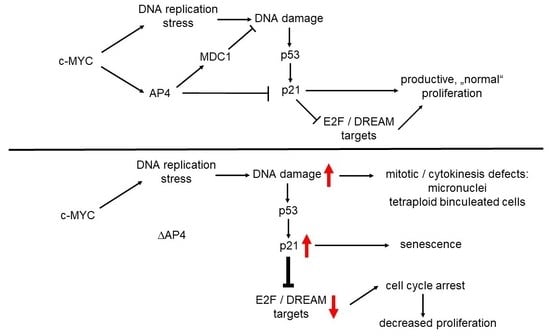
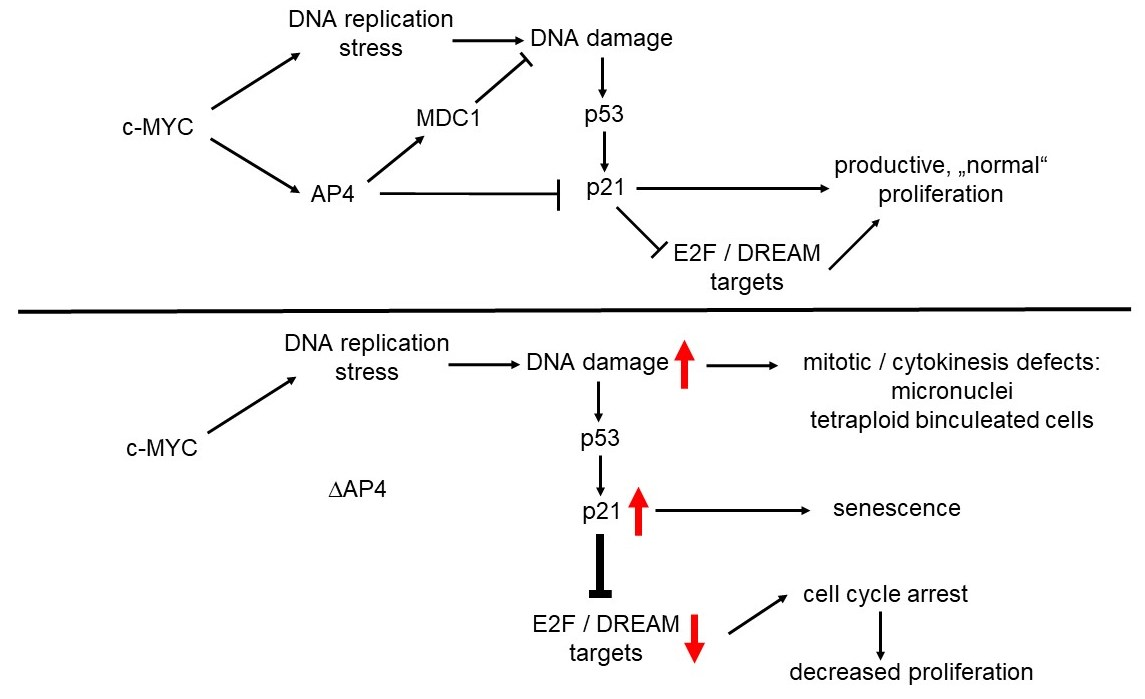{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Generation of AP4-Deficient MCF-7 Cells
2.3. Generation of p53-Deficient Cell Pools
2.4. Western Blot Analysis
2.5. Beta-Galactosidase (β-Gal) Staining
2.6. Immunofluorescence Analysis
2.7. Comet Assay
2.8. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qPCR) Analysis
2.9. Assessment of Proliferation by Real-Time Impedance Measurement
2.10. Colony Formation Assay
2.11. Transcriptomic Analysis
2.12. Analysis of ChIP-Seq, RNA Expression, and Clinical Data from Public Databases
2.13. Statistical Analysis
3. Results
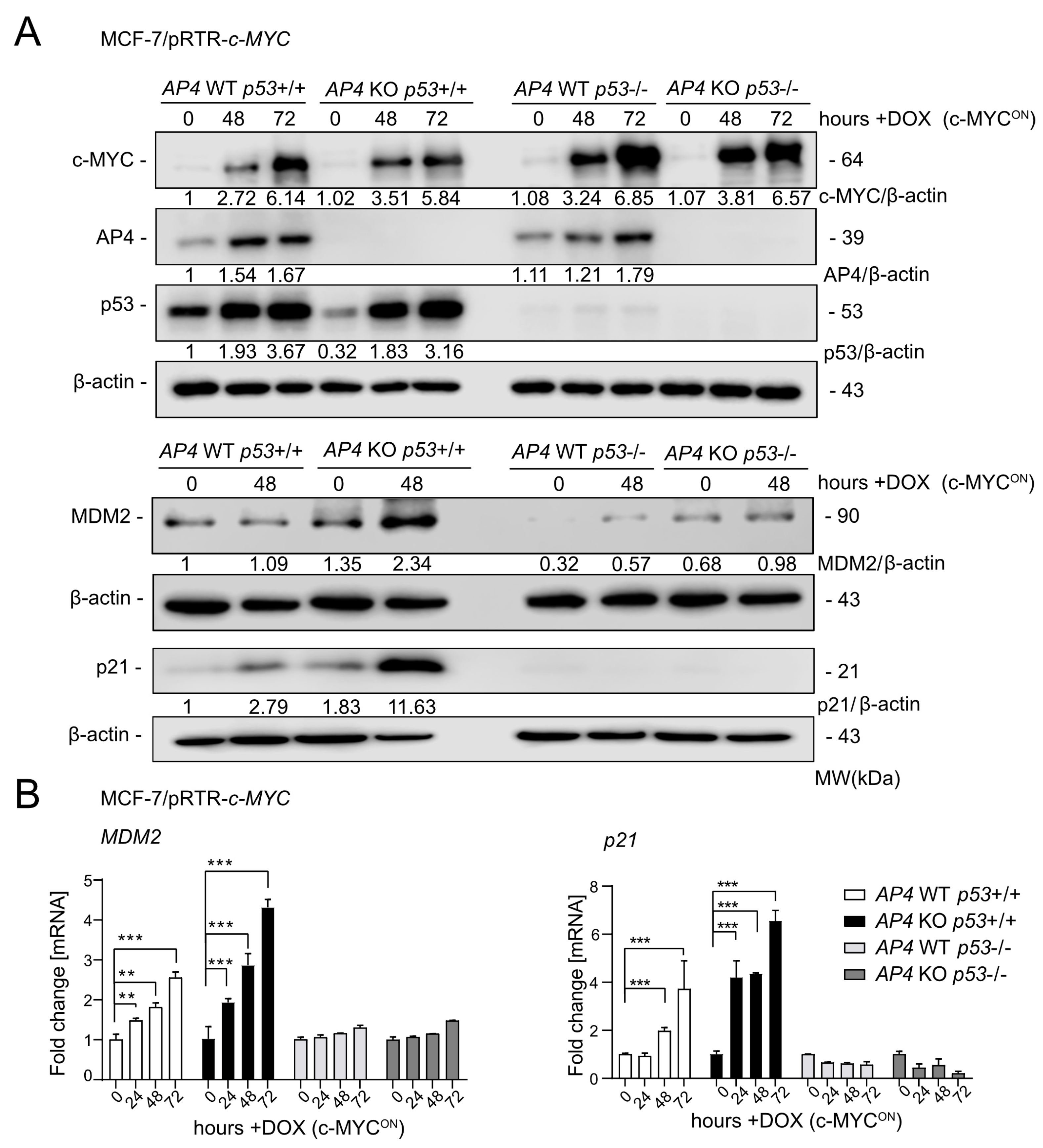
3.1. Generation and Characterization of AP4-and/or p53-Deficient MCF-7/pRTR-c-MYC Cell Lines
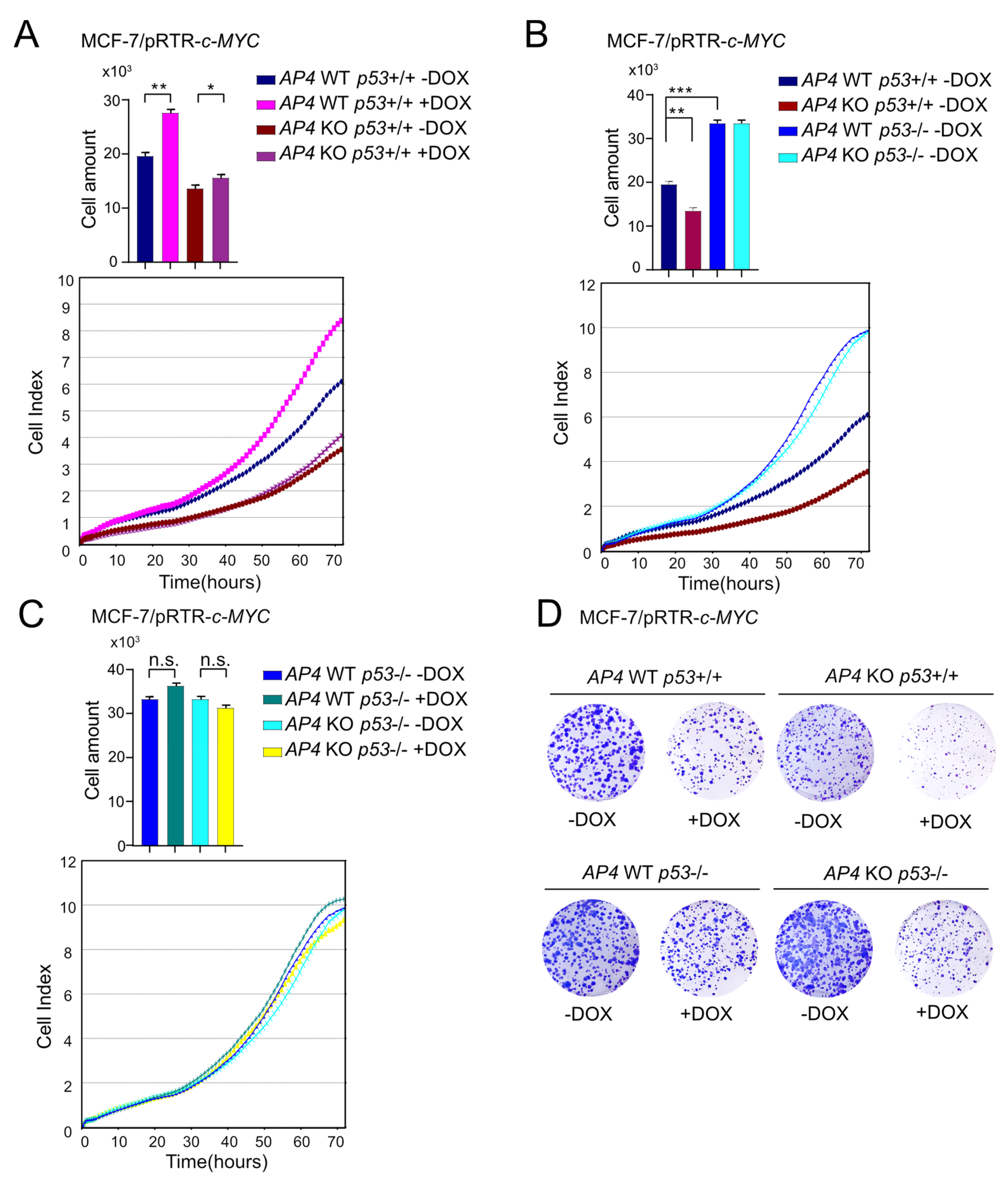
3.2. Loss of AP4 Suppresses Induction of Cell Proliferation by Ectopic c-MYC
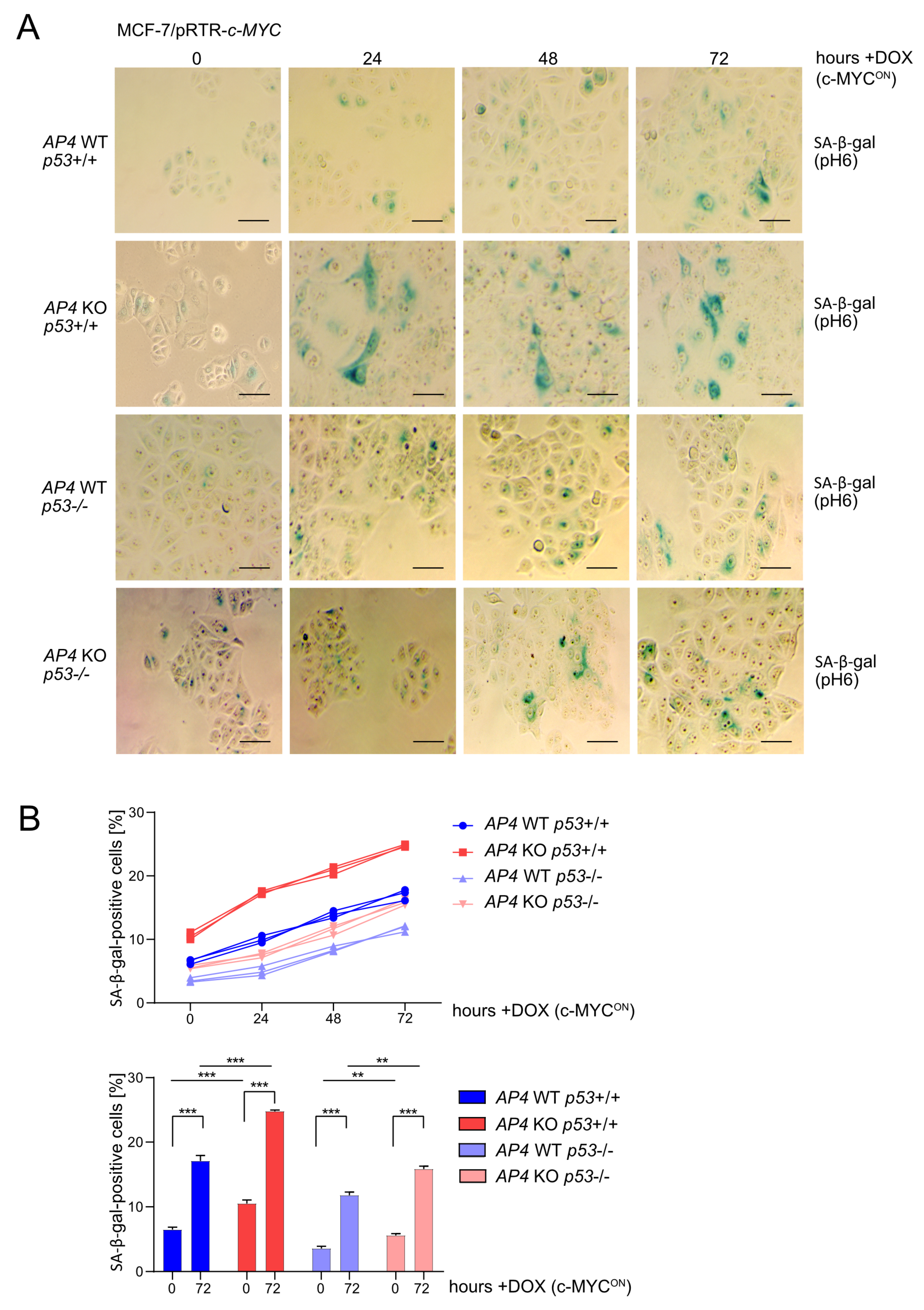
3.3. Loss of AP4 Causes Senescence in Breast Cancer Cells, Which Is Dependent on Wild-Type p53
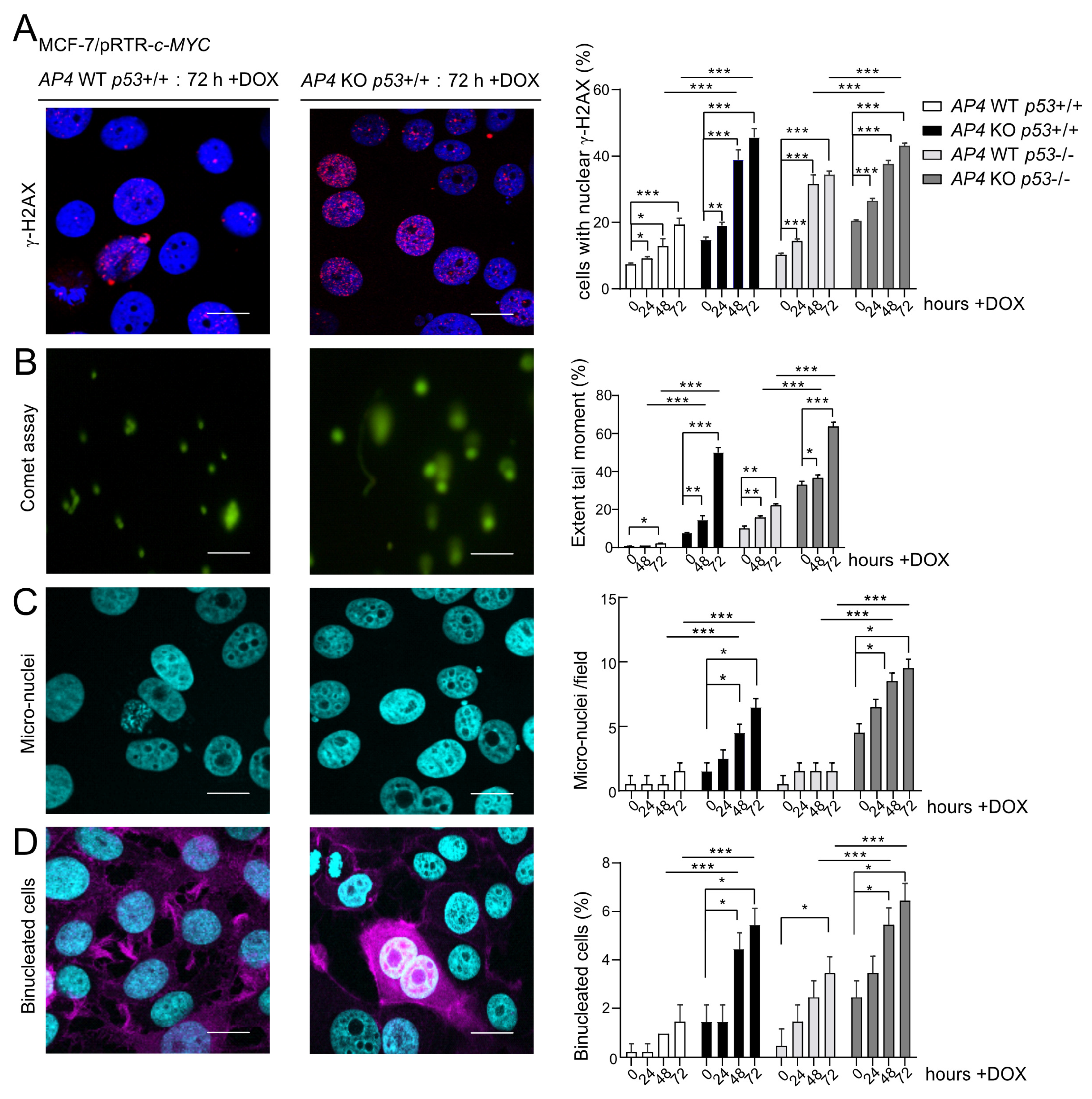
3.4. Deletion of AP4 or p53 Increases Spontaneous and c-MYC-Induced DNA Damage in Breast Cancer Cells
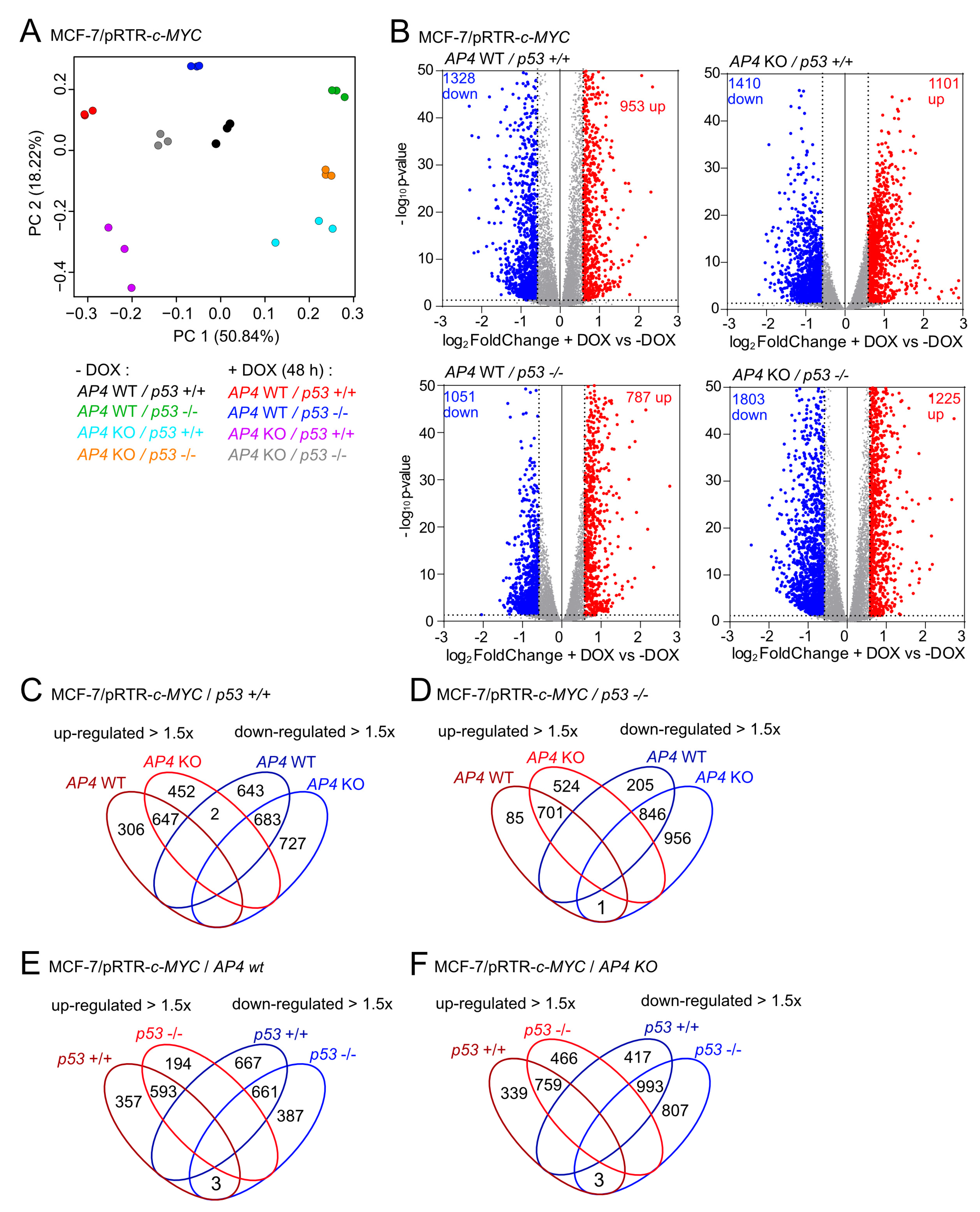
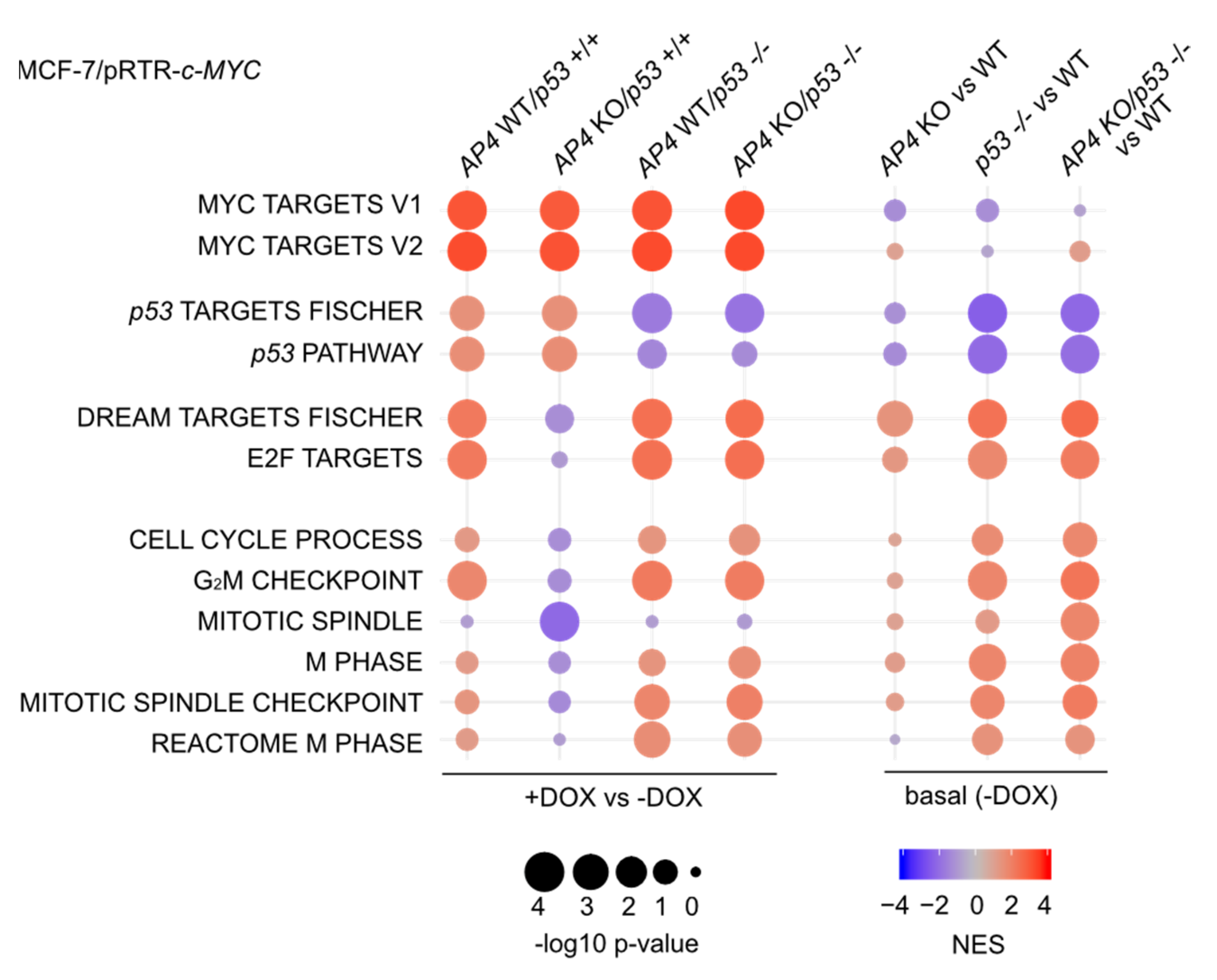
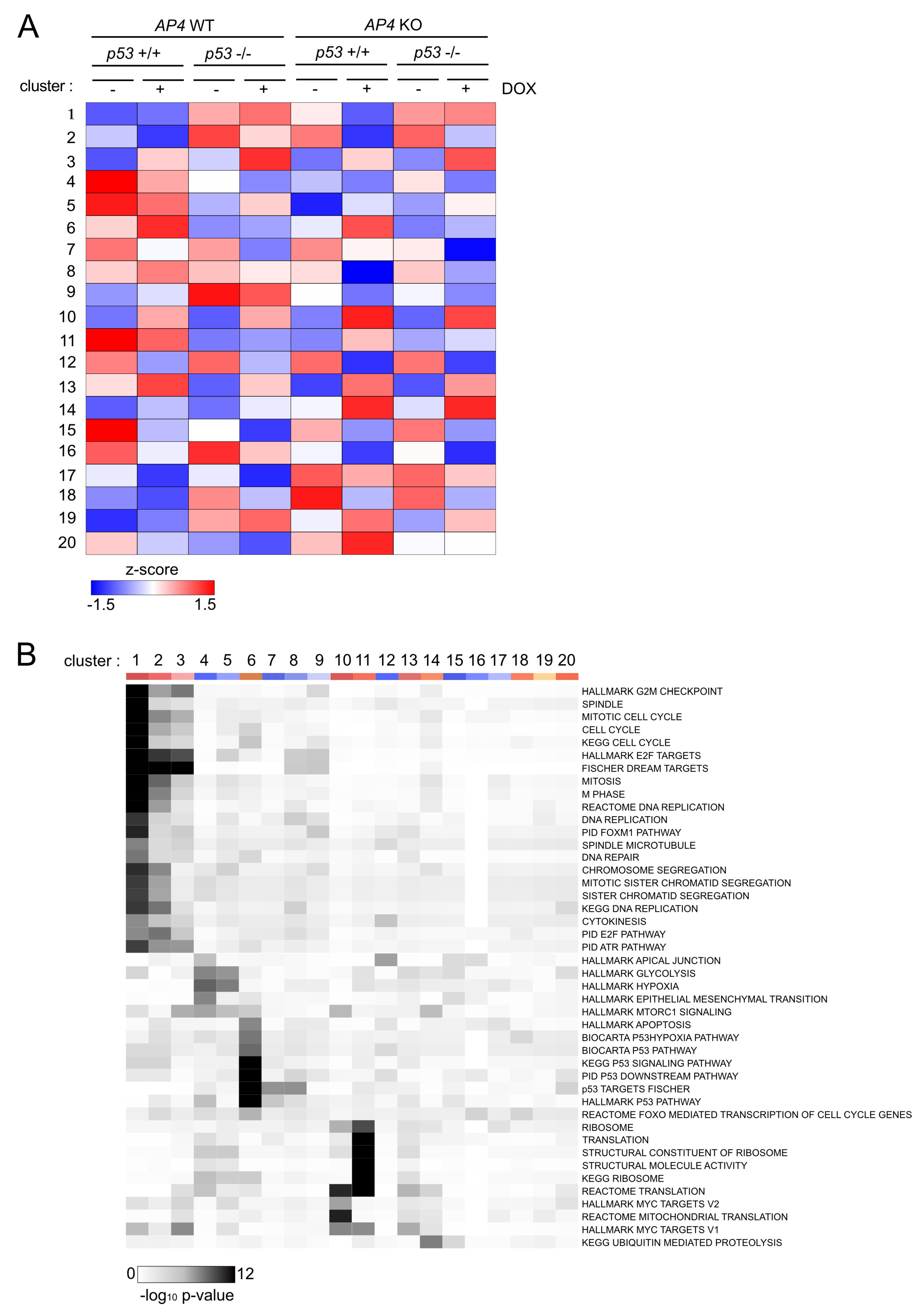
3.5. Characterization of AP4- and p53-Dependent Effects in the c-MYC-Regulated Transcriptome
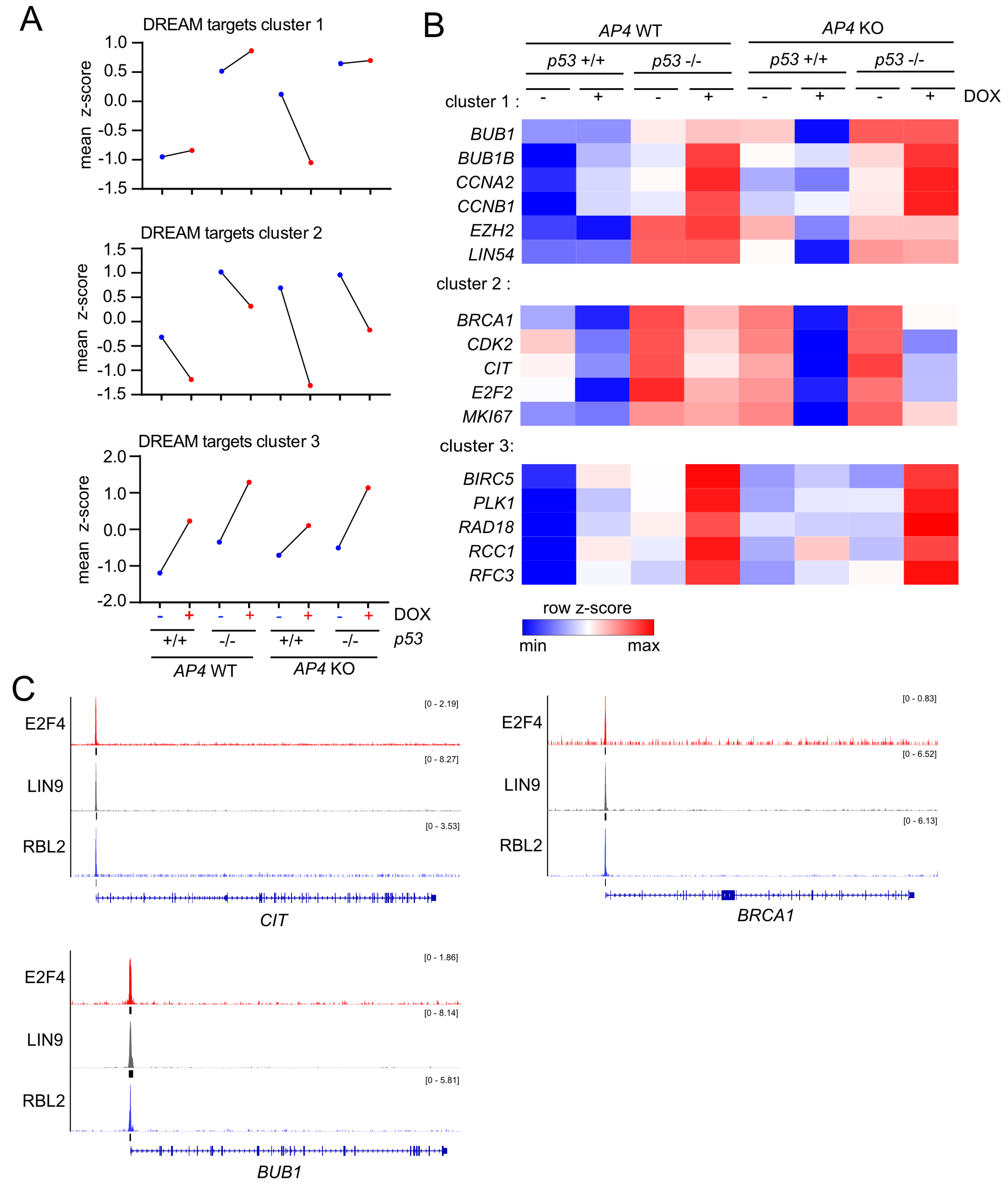
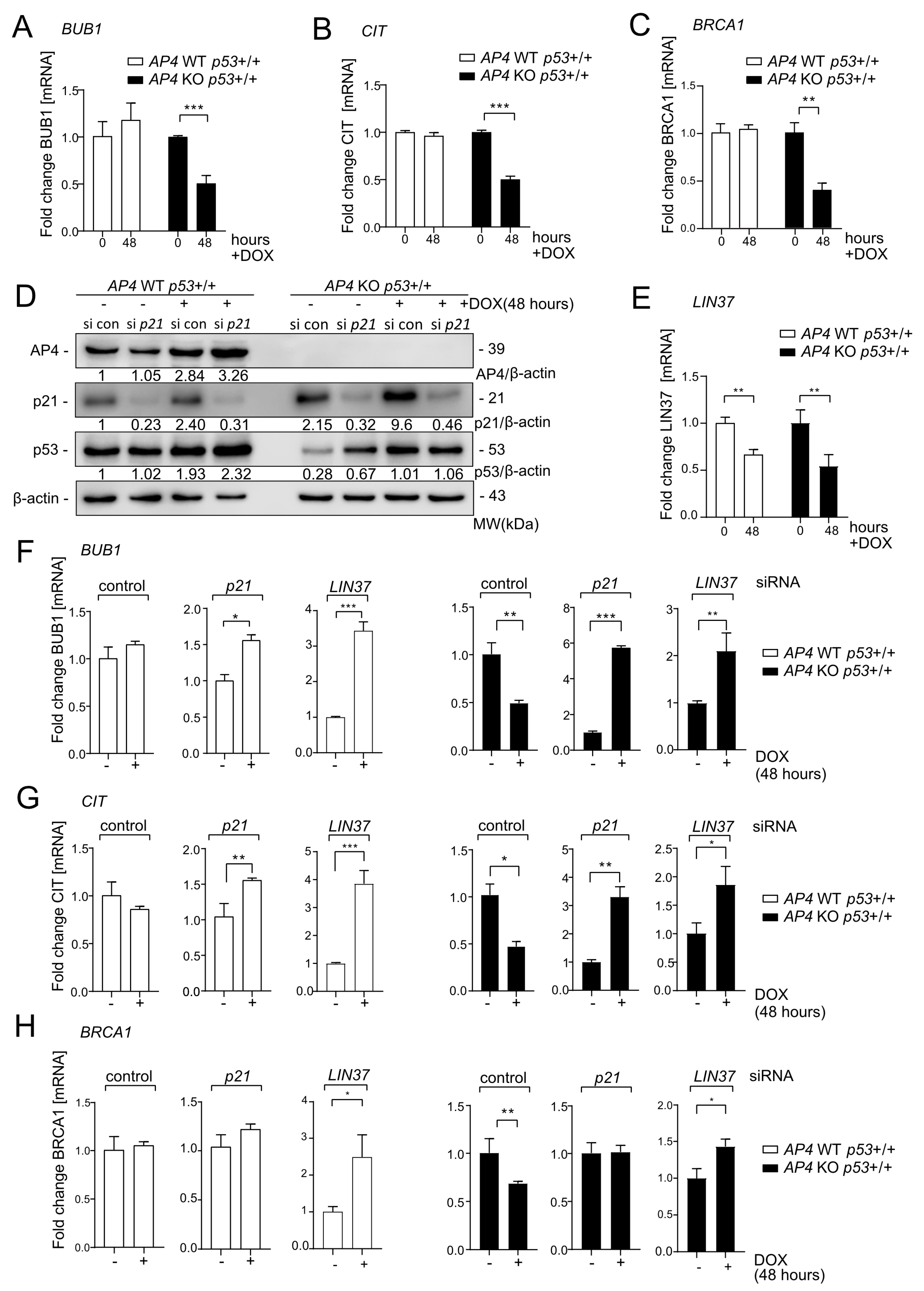
3.6. Repression of DREAM Targets after c-MYC Activation Is Mediated by p21 and LIN37
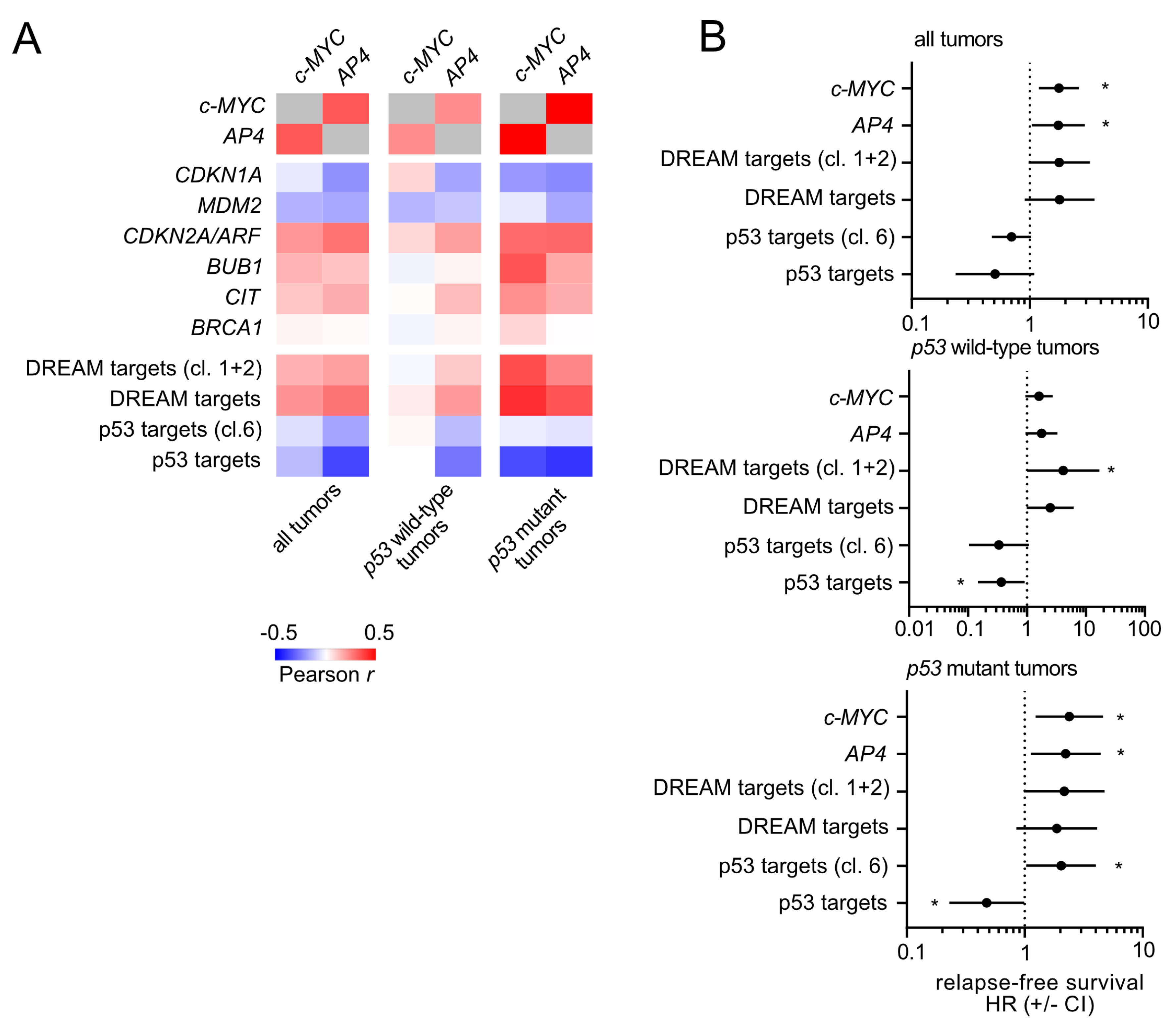
3.7. Association of c-MYC and AP4 Expression with p21, DREAM Targets and Patient Survival Is Dependent on p53 Status
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef] [PubMed]
- Green, A.R.; Aleskandarany, M.A.; Agarwal, D.; Elsheikh, S.; Nolan, C.C.; Diez-Rodriguez, M.; Macmillan, R.D.; Ball, G.R.; Caldas, C.; Madhusudan, S.; et al. MYC functions are specific in biological subtypes of breast cancer and confers resistance to endocrine therapy in luminal tumours. Br. J. Cancer 2016, 114, 917–928. [Google Scholar] [CrossRef]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Kokontis, J.M.; Hay, N. Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21waf1/cip1. Genes Dev. 1994, 8, 2817–2830. [Google Scholar] [CrossRef]
- Hermeking, H.; Eick, D. Mediation of c-Myc-induced apoptosis by p53. Science 1994, 265, 2091–2093. [Google Scholar] [CrossRef] [PubMed]
- Eischen, C.M.; Weber, J.D.; Roussel, M.F.; Sherr, C.J.; Cleveland, J.L. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999, 13, 2658–2669. [Google Scholar] [CrossRef]
- Kung, C.P.; Weber, J.D. It’s Getting Complicated-A Fresh Look at p53-MDM2-ARF Triangle in Tumorigenesis and Cancer Therapy. Front. Cell Dev. Biol. 2022, 10, 818744. [Google Scholar] [CrossRef] [PubMed]
- Farooq, U.; Notani, D. Transcriptional regulation of INK4/ARF locus by cis and trans mechanisms. Front. Cell Dev. Biol. 2022, 10, 948351. [Google Scholar] [CrossRef]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Menssen, A.; Epanchintsev, A.; Lodygin, D.; Rezaei, N.; Jung, P.; Verdoodt, B.; Diebold, J.; Hermeking, H. c-MYC delays prometaphase by direct transactivation of MAD2 and BubR1: Identification of mechanisms underlying c-MYC-induced DNA damage and chromosomal instability. Cell Cycle 2007, 6, 339–352. [Google Scholar] [CrossRef] [Green Version]
- Hermeking, H.; Funk, J.O.; Reichert, M.; Ellwart, J.W.; Eick, D. Abrogation of p53-induced cell cycle arrest by c-Myc: Evidence for an inhibitor of p21WAF1/CIP1/SDI1. Oncogene 1995, 11, 1409–1415. [Google Scholar]
- Jung, P.; Menssen, A.; Mayr, D.; Hermeking, H. AP4 encodes a c-MYC-inducible repressor of p21. Proc. Natl. Acad. Sci. USA 2008, 105, 15046–15051. [Google Scholar] [CrossRef]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- Jackstadt, R.; Roh, S.; Neumann, J.; Jung, P.; Hoffmann, R.; Horst, D.; Berens, C.; Bornkamm, G.W.; Kirchner, T.; Menssen, A.; et al. AP4 is a mediator of epithelial-mesenchymal transition and metastasis in colorectal cancer. J. Exp. Med. 2013, 210, 1331–1350. [Google Scholar] [CrossRef]
- Huang, Q.; Raya, A.; DeJesus, P.; Chao, S.H.; Quon, K.C.; Caldwell, J.S.; Chanda, S.K.; Izpisua-Belmonte, J.C.; Schultz, P.G. Identification of p53 regulators by genome-wide functional analysis. Proc. Natl. Acad. Sci. USA 2004, 101, 3456–3461. [Google Scholar] [CrossRef]
- Ku, W.C.; Chiu, S.K.; Chen, Y.J.; Huang, H.H.; Wu, W.G.; Chen, Y.J. Complementary quantitative proteomics reveals that transcription factor AP-4 mediates E-box-dependent complex formation for transcriptional repression of HDM2. Mol. Cell. Proteom. 2009, 8, 2034–2050. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef]
- Fischer, M.; Schwarz, R.; Riege, K.; DeCaprio, J.A.; Hoffmann, S. TargetGeneReg 2.0: A comprehensive web-atlas for p53, p63, and cell cycle-dependent gene regulation. NAR Cancer 2022, 4, zcac009. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef] [PubMed]
- Dubik, D.; Shiu, R.P. Transcriptional regulation of c-myc oncogene expression by estrogen in hormone-responsive human breast cancer cells. J. Biol. Chem. 1988, 263, 12705–12708. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; Swarbrick, A.; Musgrove, E.A.; Sutherland, R.L. Mechanisms of growth arrest by c-myc antisense oligonucleotides in MCF-7 breast cancer cells: Implications for the antiproliferative effects of antiestrogens. Cancer Res. 2002, 62, 3126–3131. [Google Scholar]
- Prall, O.W.; Rogan, E.M.; Musgrove, E.A.; Watts, C.K.; Sutherland, R.L. c-Myc or cyclin D1 mimics estrogen effects on cyclin E-Cdk2 activation and cell cycle reentry. Mol. Cell Biol. 1998, 18, 4499–4508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, J.F. ICI 182,780 (Fulvestrant)—The first oestrogen receptor down-regulator--current clinical data. Br. J. Cancer 2001, 85 (Suppl. S2), 11–14. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Kaller, M.; Jaeckel, S.; Rokavec, M.; Hermeking, H. AP4 suppresses DNA damage, chromosomal instability and senescence via inducing MDC1/Mediator of DNA damage Checkpoint 1 and repressing MIR22HG/miR-22-3p. Mol. Cancer 2022, 21, 120. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2019, 060012. [Google Scholar] [CrossRef]
- Stephens, M. False discovery rates: A new deal. Biostatistics 2016, 18, 275–294. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Oki, S.; Ohta, T.; Shioi, G.; Hatanaka, H.; Ogasawara, O.; Okuda, Y.; Kawaji, H.; Nakaki, R.; Sese, J.; Meno, C. ChIP-Atlas: A data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018, 19, e46255. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Jackstadt, R.; Jung, P.; Hermeking, H. AP4 directly downregulates p16 and p21 to suppress senescence and mediate transformation. Cell Death Dis. 2013, 4, e775. [Google Scholar] [CrossRef]
- Fischer, M.; Grossmann, P.; Padi, M.; DeCaprio, J.A. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016, 44, 6070–6086. [Google Scholar] [CrossRef]
- Fischer, M.; Quaas, M.; Steiner, L.; Engeland, K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 2016, 44, 164–174. [Google Scholar] [CrossRef]
- Uxa, S.; Bernhart, S.H.; Mages, C.F.S.; Fischer, M.; Kohler, R.; Hoffmann, S.; Stadler, P.F.; Engeland, K.; Muller, G.A. DREAM and RB cooperate to induce gene repression and cell-cycle arrest in response to p53 activation. Nucleic Acids Res. 2019, 47, 9087–9103. [Google Scholar] [CrossRef]
- Jaeckel, S.; Kaller, M.; Jackstadt, R.; Gotz, U.; Muller, S.; Boos, S.; Horst, D.; Jung, P.; Hermeking, H. Ap4 is rate limiting for intestinal tumor formation by controlling the homeostasis of intestinal stem cells. Nat. Commun. 2018, 9, 3573. [Google Scholar] [CrossRef]
- Jackstadt, R.; Hermeking, H. AP4 is required for mitogen- and c-MYC-induced cell cycle progression. Oncotarget 2014, 5, 7316–7327. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Tumor surveillance via the ARF-p53 pathway. Genes Dev. 1998, 12, 2984–2991. [Google Scholar] [CrossRef]
- Craig, C.; Kim, M.; Ohri, E.; Wersto, R.; Katayose, D.; Li, Z.; Choi, Y.H.; Mudahar, B.; Srivastava, S.; Seth, P.; et al. Effects of adenovirus-mediated p16INK4A expression on cell cycle arrest are determined by endogenous p16 and Rb status in human cancer cells. Oncogene 1998, 16, 265–272. [Google Scholar] [CrossRef]
- Hollestelle, A.; Nagel, J.H.; Smid, M.; Lam, S.; Elstrodt, F.; Wasielewski, M.; Ng, S.S.; French, P.J.; Peeters, J.K.; Rozendaal, M.J.; et al. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res. Treat. 2010, 121, 53–64. [Google Scholar] [CrossRef]
- Ray, S.; Atkuri, K.R.; Deb-Basu, D.; Adler, A.S.; Chang, H.Y.; Herzenberg, L.A.; Felsher, D.W. MYC can induce DNA breaks in vivo and in vitro independent of reactive oxygen species. Cancer Res. 2006, 66, 6598–6605. [Google Scholar] [CrossRef]
- Bretones, G.; Delgado, M.D.; Leon, J. Myc and cell cycle control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Hermeking, H.; Rago, C.; Schuhmacher, M.; Li, Q.; Barrett, J.F.; Obaya, A.J.; O’Connell, B.C.; Mateyak, M.K.; Tam, W.; Kohlhuber, F.; et al. Identification of CDK4 as a target of c-MYC. Proc. Natl. Acad. Sci. USA 2000, 97, 2229–2234. [Google Scholar] [CrossRef]
- Amati, B.; Alevizopoulos, K.; Vlach, J. Myc and the cell cycle. Front. Biosci 1998, 3, d250–d268. [Google Scholar] [CrossRef]
- Lutz, W.; Leon, J.; Eilers, M. Contributions of Myc to tumorigenesis. Biochim. Biophys. Acta 2002, 1602, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.M.; Joyson, S.M.; Hermeking, H.; Chiu, S.K. Transcription Factor AP4 Mediates Cell Fate Decisions: To Divide, Age, or Die. Cancers 2021, 13, 676. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaller, M.; Shi, W.; Hermeking, H. c-MYC-Induced AP4 Attenuates DREAM-Mediated Repression by p53. Cancers 2023, 15, 1162. https://doi.org/10.3390/cancers15041162
Kaller M, Shi W, Hermeking H. c-MYC-Induced AP4 Attenuates DREAM-Mediated Repression by p53. Cancers. 2023; 15(4):1162. https://doi.org/10.3390/cancers15041162
Chicago/Turabian StyleKaller, Markus, Wenjing Shi, and Heiko Hermeking. 2023. "c-MYC-Induced AP4 Attenuates DREAM-Mediated Repression by p53" Cancers 15, no. 4: 1162. https://doi.org/10.3390/cancers15041162
APA StyleKaller, M., Shi, W., & Hermeking, H. (2023). c-MYC-Induced AP4 Attenuates DREAM-Mediated Repression by p53. Cancers, 15(4), 1162. https://doi.org/10.3390/cancers15041162