
Treatment Pathways and Prognosis in Advanced Sarcoma with Peritoneal Sarcomatosis
 , , ,
, , ,  , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Patient Characteristics
3.2. State of Disease
3.3. Treatment
3.4. Follow-Up
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ressing, M.; Wardelmann, E.; Hohenberger, P.; Jakob, J.; Kasper, B.; Emrich, K.; Eberle, A.; Blettner, M.; Zeissig, S.R. Strengthening health data on a rare and heterogeneous disease: Sarcoma incidence and histological subtypes in Germany. BMC Public Health 2018, 18, 235. [Google Scholar] [CrossRef] [Green Version]
- Gatta, G.; van der Zwan, J.M.; Casali, P.G.; Siesling, S.; Dei Tos, A.P.; Kunkler, I.; Otter, R.; Licitra, L.; Mallone, S.; Tavilla, A.; et al. Rare cancers are not so rare: The rare cancer burden in Europe. Eur. J. Cancer 2011, 47, 2493–2511. [Google Scholar] [CrossRef]
- Lam, S.W.; Silva, T.M.; Bovée, J.V.M.G. New molecular entities of soft tissue and bone tumors. Curr. Opin. Oncol. 2022, 34, 354–361. [Google Scholar] [CrossRef]
- Vay, C.; Schlünder, P.M.; Dizdar, L.; Esposito, I.; Ghadimi, M.P.H.; Knoefel, W.T.; Krieg, A. Targeting abundant survivin expression in liposarcoma: Subtype dependent therapy responses to YM155 treatment. J. Cancer Res. Clin. Oncol. 2022, 148, 633–645. [Google Scholar] [CrossRef]
- Mersch, S.; Riemer, J.C.; Schlünder, P.M.; Ghadimi, M.P.; Ashmawy, H.; Möhlendick, B.; Topp, S.A.; Arent, T.; Kröpil, P.; Stoecklein, N.H.; et al. Peritoneal sarcomatosis: Site of origin for the establishment of an in vitro and in vivo cell line model to study therapeutic resistance in dedifferentiated liposarcoma. Tumour Biol. 2016, 37, 2341–2351. [Google Scholar] [CrossRef]
- Pilati, P.; Rossi, C.R.; Mocellin, S.; Foletto, M.; Scagnet, B.; Pasetto, L.; Lise, M. Multimodal treatment of peritoneal carcinomatosis and sarcomatosis. Eur. J. Surg. Oncol. 2001, 27, 125–134. [Google Scholar] [CrossRef]
- Sugarbaker, P.H. Intraperitoneal chemotherapy and cytoreductive surgery for the prevention and treatment of peritoneal carcinomatosis and sarcomatosis. Semin. Surg. Oncol. 1998, 14, 254–261. [Google Scholar] [CrossRef]
- Rossi, C.R.; Casali, P.; Kusamura, S.; Baratti, D.; Deraco, M. The consensus statement on the locoregional treatment of abdominal sarcomatosis. J. Surg. Oncol. 2008, 98, 291–294. [Google Scholar] [CrossRef]
- Wickham, H.; Bryan, J. Readxl: Read Excel Files R Package Version 1.3.1. Available online: https://cran.r-project.org/web/packages/readxl/index.html (accessed on 13 March 2019).
- Kassambara, A.; Kosinski, M.; Biecek, P.; Scheipl, F. Survminer: Drawing Survival Curves using ‘ggplot2’. Available online: https://cran.r-project.org/web/packages/survminer/index.html (accessed on 9 March 2021).
- Therneau, T.M. A Package for Survival Analysis in R. R package version 3.4-0. Available online: https://cran.r-project.org/web/packages/survival/index.html (accessed on 9 August 2022).
- Gronchi, A. Surgery in soft tissue sarcoma: The thin line between a surgical or more conservative approach. Future Oncol. 2021, 17, 3–6. [Google Scholar] [CrossRef]
- Wong, L.C.K.; Li, Z.; Fan, Q.; Tan, J.W.; Tan, Q.X.; Wong, J.S.M.; Ong, C.J.; Chia, C.S. Cytoreductive surgery (CRS) with hyperthermic intraperitoneal chemotherapy (HIPEC) in peritoneal sarcomatosis—A systematic review and meta-analysis. Eur. J. Surg. Oncol. 2022, 48, 640–648. [Google Scholar] [CrossRef]
- Zerhouni, S.; Van Coevorden, F.; Swallow, C.J. The role and outcomes of palliative surgery for retroperitoneal sarcoma. J. Surg. Oncol. 2018, 117, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Ratan, R.; Patel, S.R. Chemotherapy for soft tissue sarcoma. Cancer 2016, 122, 2952–2960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, E.; Nessim, C. Retroperitoneal Sarcoma Care in 2021. Cancers 2022, 14, 1293. [Google Scholar] [CrossRef] [PubMed]
- Gough, N.; Koffman, J.; Ross, J.R.; Riley, J.; Judson, I. Does palliative chemotherapy really palliate and are we measuring it correctly? A mixed methods longitudinal study of health related quality of life in advanced soft tissue sarcoma. PLoS ONE 2019, 14, e0210731. [Google Scholar] [CrossRef] [Green Version]
- Grad, I.; Hanes, R.; Ayuda-Durán, P.; Kuijjer, M.L.; Enserink, J.M.; Meza-Zepeda, L.A.; Myklebost, O. Discovery of novel candidates for anti-liposarcoma therapies by medium-scale high-throughput drug screening. PLoS ONE 2021, 16, e0248140. [Google Scholar] [CrossRef]
- Ghadimi, M.P.; Young, E.D.; Belousov, R.; Zhang, Y.; Lopez, G.; Lusby, K.; Kivlin, C.; Demicco, E.G.; Creighton, C.J.; Lazar, A.J.; et al. Survivin is a viable target for the treatment of malignant peripheral nerve sheath tumors. Clin. Cancer Res. 2012, 18, 2545–2557. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Li, X.; Yu, S. Molecular targeted therapy for advanced or metastatic soft tissue sarcoma. Cancer Control 2021, 28, 10732748211038424. [Google Scholar] [CrossRef]
- Loosen, S.H.; Krieg, S.; Eschrich, J.; Luedde, M.; Krieg, A.; Schallenburger, M.; Schwartz, J.; Neukirchen, M.; Luedde, T.; Kostev, K.; et al. The Landscape of Outpatient Palliative Care in Germany: Results from a Retrospective Analysis of 14,792 Patients. Int. J. Environ. Res. Public Health 2022, 19, 14885. [Google Scholar] [CrossRef]
- Brandes, F.; Striefler, J.K.; Dörr, A.; Schmiester, M.; Märdian, S.; Koulaxouzidis, G.; Kaul, D.; Behzadi, A.; Thuss-Patience, P.; Ahn, J.; et al. Impact of a specialised palliative care intervention in patients with advanced soft tissue sarcoma—A single-centre retrospective analysis. BMC Palliat. Care 2021, 20, 16. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Serre, D.; Reichardt, P.; Martín-Broto, J.; Bauer, S. Options for treating different soft tissue sarcoma subtypes. Future Oncol. 2018, 14, 25–49. [Google Scholar] [CrossRef]
- Katz, D.; Palmerini, E.; Pollack, S.M. More Than 50 Subtypes of Soft Tissue Sarcoma: Paving the Path for Histology-Driven Treatments. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, F.C.; Viterbo, A. Histologic and genetic advances in refining the diagnosis of “undifferentiated pleomorphic sarcoma”. Cancers 2013, 5, 218–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes-Jordan, A.; LaQuaglia, M.P.; Modak, S. Management of desmoplastic small round cell tumor. Semin. Pediatr. Surg. 2016, 25, 299–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerald, W.L.; Ladanyi, M.; de Alava, E.; Cuatrecasas, M.; Kushner, B.H.; LaQuaglia, M.P.; Rosai, J. Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): Desmoplastic small round-cell tumor and its variants. J. Clin. Oncol. 1998, 16, 3028–3036. [Google Scholar] [CrossRef]
- Lal, D.R.; Su, W.T.; Wolden, S.L.; Loh, K.C.; Modak, S.; La Quaglia, M.P. Results of multimodal treatment for desmoplastic small round cell tumors. J. Pediatr. Surg. 2005, 40, 251–255. [Google Scholar] [CrossRef]
- Hayes-Jordan, A.A.; Coakley, B.A.; Green, H.L.; Xiao, L.; Fournier, K.F.; Herzog, C.E.; Ludwig, J.A.; McAleer, M.F.; Anderson, P.M.; Huh, W.W. Desmoplastic Small Round Cell Tumor Treated with Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy: Results of a Phase 2 Trial. Ann. Surg. Oncol. 2018, 25, 872–877. [Google Scholar] [CrossRef]
- Wessalowski, R.; Schneider, D.T.; Mils, O.; Hannen, M.; Calaminus, G.; Engelbrecht, V.; Pape, H.; Willers, R.; Engert, J.; Harms, D.; et al. An approach for cure: PEI-chemotherapy and regional deep hyperthermia in children and adolescents with unresectable malignant tumors. Klin. Padiatr. 2003, 215, 303–309. [Google Scholar] [CrossRef]
- Harmon, R.L.; Sugarbaker, P.H. Prognostic indicators in peritoneal carcinomatosis from gastrointestinal cancer. Int. Semin. Surg. Oncol. 2005, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Jacquet, P.; Sugarbaker, P.H. Clinical research methodologies in diagnosis and staging of patients with peritoneal carcinomatosis. Cancer Treat. Res. 1996, 82, 359–374. [Google Scholar] [CrossRef]
- Salti, G.I.; Ailabouni, L.; Undevia, S. Cytoreductive surgery and hyperthermic intraperitoneal chemotherapy for the treatment of peritoneal sarcomatosis. Ann. Surg. Oncol. 2012, 19, 1410–1415. [Google Scholar] [CrossRef]
- Berthet, B.; Sugarbaker, T.A.; Chang, D.; Sugarbaker, P.H. Quantitative methodologies for selection of patients with recurrent abdominopelvic sarcoma for treatment. Eur. J. Cancer 1999, 35, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Baratti, D.; Pennacchioli, E.; Kusamura, S.; Fiore, M.; Balestra, M.R.; Colombo, C.; Mingrone, E.; Gronchi, A.; Deraco, M. Peritoneal sarcomatosis: Is there a subset of patients who may benefit from cytoreductive surgery and hyperthermic intraperitoneal chemotherapy? Ann. Surg. Oncol. 2010, 17, 3220–3228. [Google Scholar] [CrossRef]
- Haddox, C.L.; Riedel, R.F. Recent advances in the understanding and management of liposarcoma. Fac. Rev. 2021, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Martin-Broto, J.; Mondaza-Hernandez, J.L.; Moura, D.S.; Hindi, N. A Comprehensive Review on Solitary Fibrous Tumor: New Insights for New Horizons. Cancers 2021, 13, 2913. [Google Scholar] [CrossRef] [PubMed]
- Serpico, R.; Brown, J.; Blank, A.; Jones, K.; Randall, R.L.; Groundland, J. Metastasis of Osteosarcoma to the Abdomen: A Report of Two Cases and a Review of the Literature. Case Rep. Oncol. 2021, 14, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Toulmonde, M.; Bonvalot, S.; Méeus, P.; Stoeckle, E.; Riou, O.; Isambert, N.; Bompas, E.; Jafari, M.; Delcambre-Lair, C.; Saada, E.; et al. Retroperitoneal sarcomas: Patterns of care at diagnosis, prognostic factors and focus on main histological subtypes: A multicenter analysis of the French Sarcoma Group. Ann. Oncol. 2014, 25, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, M.; Gonzalez, R.J.; Strauss, D.; Raut, C.P. Current principles of surgery for retroperitoneal sarcomas. J. Surg. Oncol. 2018, 117, 33–41. [Google Scholar] [CrossRef]
- Bredbeck, B.C.; Delaney, L.D.; Kathawate, V.G.; Harter, C.A.; Wilkowski, J.; Chugh, R.; Cuneo, K.C.; Dossett, L.A.; Sabel, M.S.; Angeles, C.V. Factors associated with disease-free and abdominal recurrence-free survival in abdominopelvic and retroperitoneal sarcomas. J. Surg. Oncol. 2022, 125, 1292–1300. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Montesco, M.C.; Coindre, J.M.; Dei Tos, A.P.; Lurkin, A.; Ranchère-Vince, D.; Vecchiato, A.; Decouvelaere, A.V.; Mathoulin-Pélissier, S.; Albert, S.; et al. Sarcoma: Concordance between initial diagnosis and centralized expert review in a population-based study within three European regions. Ann. Oncol. 2012, 23, 2442–2449. [Google Scholar] [CrossRef]
- Meyer, M.; Seetharam, M. First-Line Therapy for Metastatic Soft Tissue Sarcoma. Curr. Treat. Options Oncol. 2019, 20, 6. [Google Scholar] [CrossRef]
- Blay, J.Y.; Soibinet, P.; Penel, N.; Bompas, E.; Duffaud, F.; Stoeckle, E.; Mir, O.; Adam, J.; Chevreau, C.; Bonvalot, S.; et al. Improved survival using specialized multidisciplinary board in sarcoma patients. Ann. Oncol. 2017, 28, 2852–2859. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.J.; Singer, S.; Brennan, M.F.; Jaques, D.P. Effectiveness of palliative procedures for intra-abdominal sarcomas. Ann. Surg. Oncol. 2005, 12, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Thalji, S.Z.; Tsai, S.; Gamblin, T.C.; Clarke, C.; Christians, K.; Charlson, J.; Ethun, C.G.; Poultsides, G.; Grignol, V.P.; Roggin, K.K.; et al. Outcomes of palliative-intent surgery in retroperitoneal sarcoma-Results from the US Sarcoma Collaborative. J. Surg. Oncol. 2020, 121, 1140–1147. [Google Scholar] [CrossRef] [PubMed]




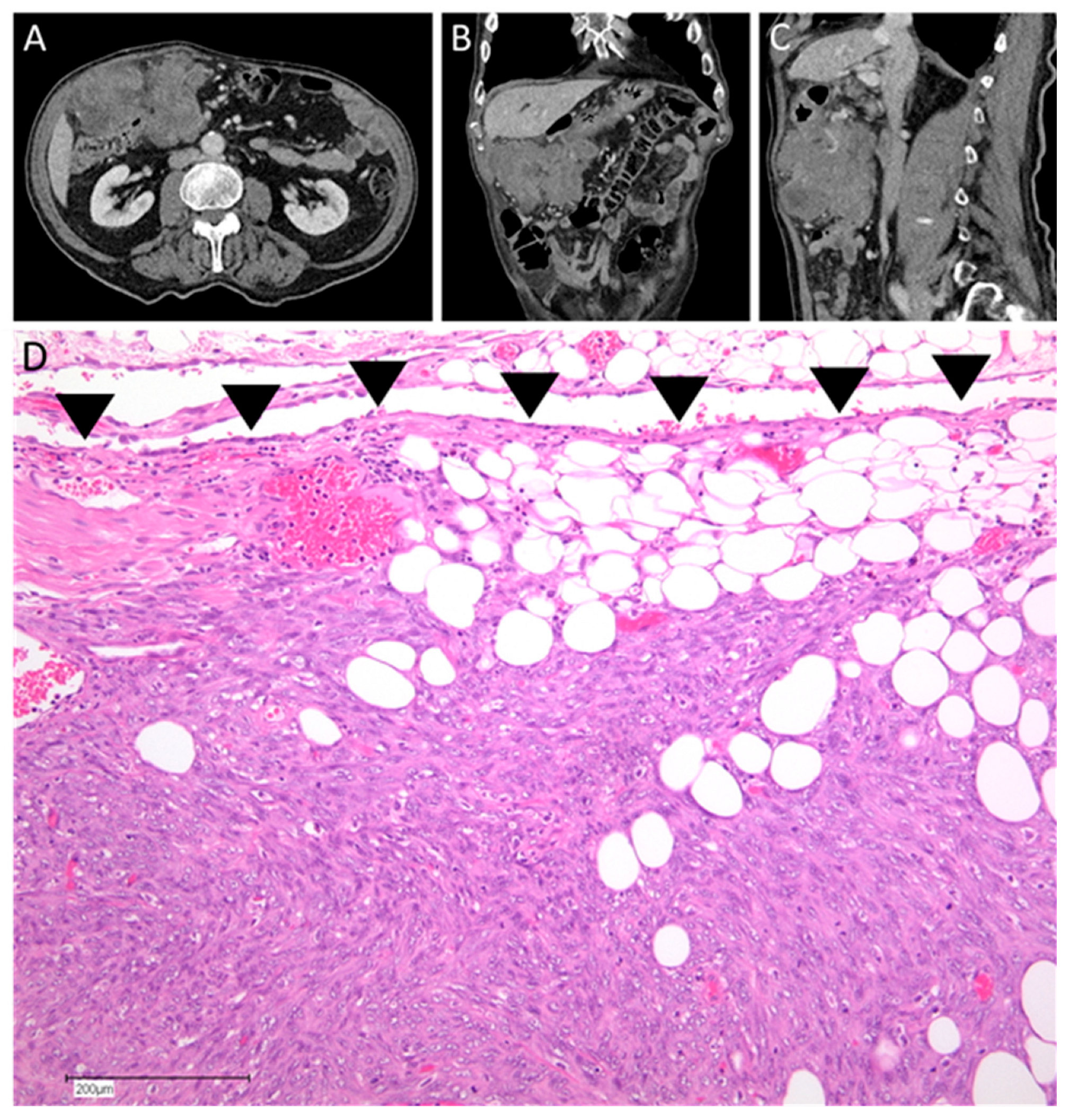{kind=link}
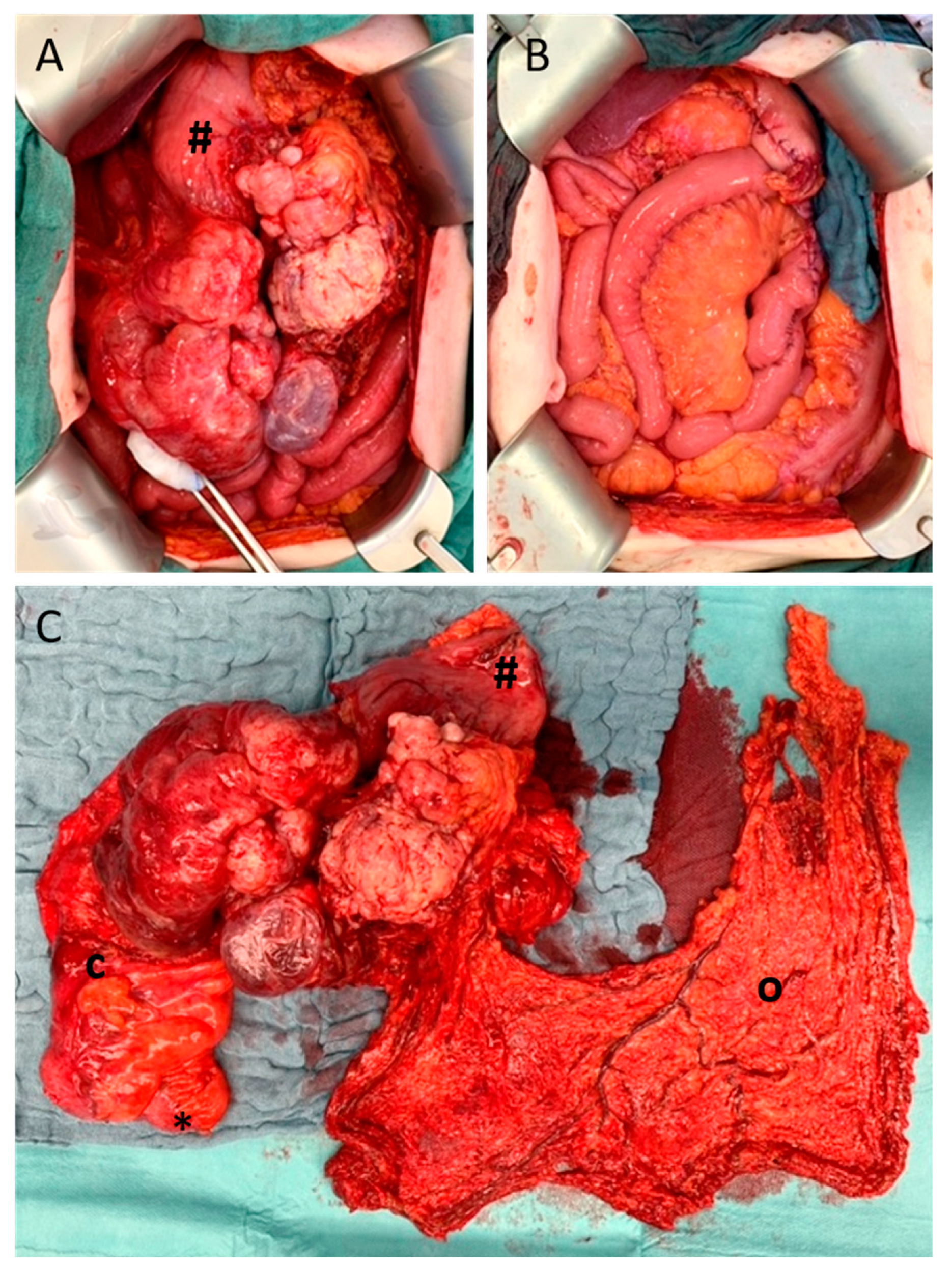{kind=link}
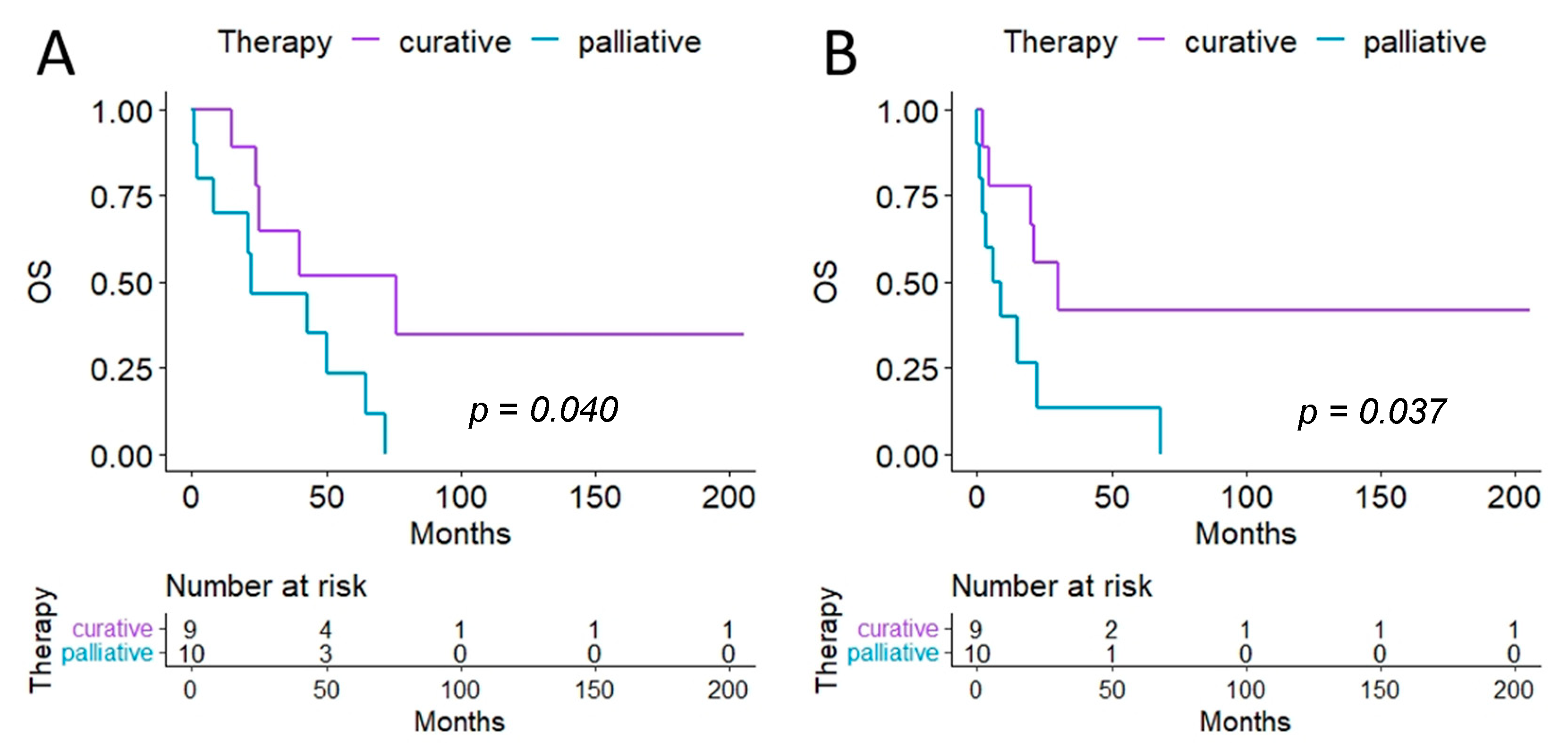{kind=link}
| Tumor Subtype | Number of Patients | Median Age (Range) | Sex Male/Female |
|---|---|---|---|
| NOS | 5 | 67 (46–88) | 3/2 |
| DSRCT | 4 | 18 (18–24) | 2/2 |
| Dedifferentiated liposarcoma | 3 | 65 (36–84) | 1/2 |
| Myxoid liposarcoma | 2 | 61 (44–64) | 2/0 |
| Leiomyosarcoma | 2 | 51 (44–58) | 0/2 |
| MPNST | 1 | 24 | 0/1 |
| SFT | 1 | 68 | 1/0 |
| Osteosarcoma | 1 | 26 | 1/0 |
| Total | 19 | 45.9 (18–88) | 10/9 |
| Tumor Subtype | Number of Patients | Mean Time Since Primary Diagnosis (SD) | Prior External Treatment | Mean Prior Resections (Range) | Prior R1/R2 | Distant Metastasis |
|---|---|---|---|---|---|---|
| NOS | 5 | 18 (26.1) | 40% | 3.4 (0–10) | 60% | 20% |
| DSRCT | 4 | 6 (9.6) | 75% | 0.3 (0–1) | 25% | 50% |
| Dedifferentiated liposarcoma | 3 | 7 (10) | 33% | 1.7 (0–4) | 66% | 100% |
| Myxoid liposarcoma | 2 | 40 (7.4) | 100% | 1.5 (1–2) | 0% | 100% |
| Leiomyosarcoma | 2 | 9 (11.8) | 100% | 1.5 (1–2) | ND | 50% |
| MPNST | 1 | 13 | 100% | 3 | 100% | 100% |
| SFT | 1 | 27 | 0% | 1 | 0% | 0% |
| Osteosarcoma | 1 | 34 | 0% | 2 | 0% | 100% |
| Total | 19 | 16.2 (17.94) | 57.9% | 1.8 | 42.1% | 52.6% |
| Tumor Subtype | Number of Patients | Total Number of Resections per Patient | Surgical Treatment Spectrum | |||
|---|---|---|---|---|---|---|
| Exploration and Biopsy | Palliative Procedure | Limited Resection | Multivisceral Resection | |||
| NOS | 5 | 5 | 15 | 11 | ||
| DSRCT | 4 | 2.75 | 2 | 1 | 7 | |
| Dedifferentiated liposarcoma | 3 | 2 | 2 | 1 | 2 | 3 |
| Myxoid liposarcoma | 2 | 5 | 1 | 3 | 6 | |
| Leiomyosarcoma | 2 | 6 | 2 | 5 | 6 | |
| MPNST | 1 | 4 | 2 | 2 | ||
| SFT | 1 | 2 | 1 | 1 | 1 | |
| Osteosarcoma | 1 | 3 | 2 | 1 | ||
| Total | 19 | 3.8 | 7 | 2 | 31 | 37 |
| Tumor Subtype | Number of Patients | Mean Survival in Months Since Primary Diagnosis (SD) | Mean Survival in Months after PS | Major Complications (Clavien–Dindo 3 or 4) | Mean Follow-Up in Months (SD) |
|---|---|---|---|---|---|
| NOS | 5 | 38 (31.2) | 20 (27.8) | 60% | 17 (25.3) |
| DSRCT | 4 | 24 (1.0) | 17 (9.2) | 25% | 13 (7.4) |
| Dedifferentiated liposarcoma | 3 | 10 (10.3) | 3 (2.5) | 33% | 3 (2.3) |
| Myxoid liposarcoma | 2 | 63 (18.3) | 22 (10.9) | 0% | 20 (9.6) |
| Leiomyosarcoma | 2 | 151 (76.5) | 143 (88.3) | 0% | 115 (80.4) |
| MPNST | 1 | 15 | 2 | 100% | 2 |
| SFT | 1 | 62 | 36 | 100% | 3 |
| Osteosarcoma | 1 | 43 | 9 | 0% | 8 |
| Total | 19 | 46 (47.4) | 30 (47.9) | 31.6% | 22 (40.2) |
| Number of Patients | Age in Years | Major Complications (Clavien–Dindo 3 or 4) | Hospital Stay in Days (SD) | |
|---|---|---|---|---|
| Curative Intention | 9 | 40 | 22% | 29 (16) |
| Palliative Intention | 10 | 55 | 40% | 27 (21) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klingler, F.; Ashmawy, H.; Häberle, L.; Esposito, I.; Schimmöller, L.; Knoefel, W.T.; Krieg, A. Treatment Pathways and Prognosis in Advanced Sarcoma with Peritoneal Sarcomatosis. Cancers 2023, 15, 1340. https://doi.org/10.3390/cancers15041340
Klingler F, Ashmawy H, Häberle L, Esposito I, Schimmöller L, Knoefel WT, Krieg A. Treatment Pathways and Prognosis in Advanced Sarcoma with Peritoneal Sarcomatosis. Cancers. 2023; 15(4):1340. https://doi.org/10.3390/cancers15041340
Chicago/Turabian StyleKlingler, Fabian, Hany Ashmawy, Lena Häberle, Irene Esposito, Lars Schimmöller, Wolfram Trudo Knoefel, and Andreas Krieg. 2023. "Treatment Pathways and Prognosis in Advanced Sarcoma with Peritoneal Sarcomatosis" Cancers 15, no. 4: 1340. https://doi.org/10.3390/cancers15041340
APA StyleKlingler, F., Ashmawy, H., Häberle, L., Esposito, I., Schimmöller, L., Knoefel, W. T., & Krieg, A. (2023). Treatment Pathways and Prognosis in Advanced Sarcoma with Peritoneal Sarcomatosis. Cancers, 15(4), 1340. https://doi.org/10.3390/cancers15041340