
Precision Killing of M2 Macrophages with Phage-Displayed Peptide-Photosensitizer Conjugates



Abstract
:Simple Summary
Abstract
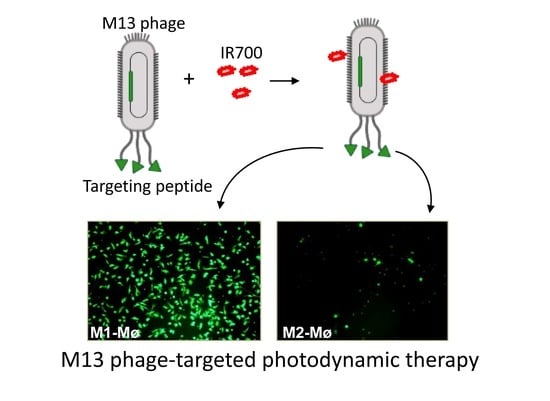
1. Introduction
2. Materials and Methods
2.1. Monocyte Purification from PBMCs
2.2. Generation of M1 and M2 Macrophages
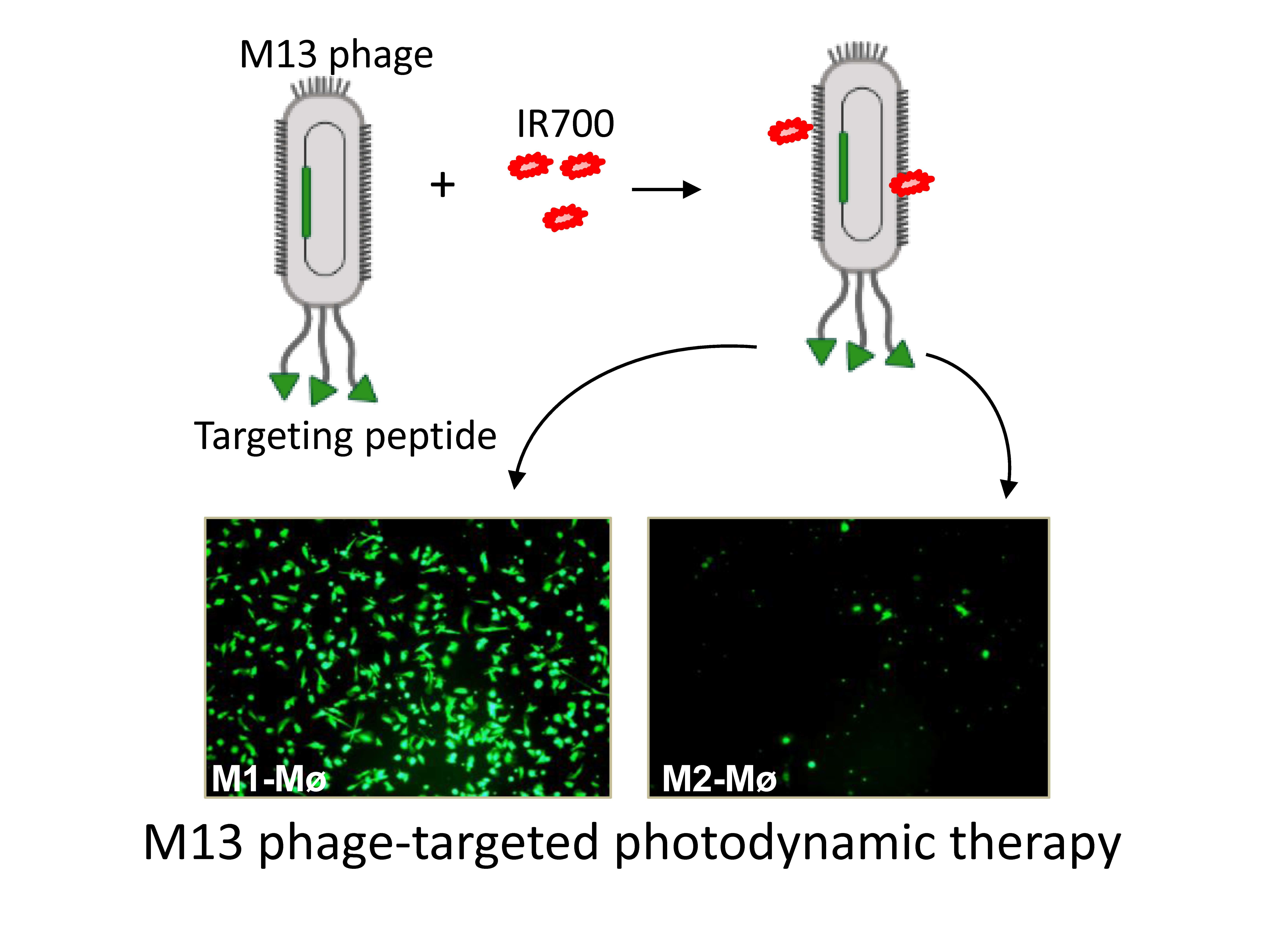
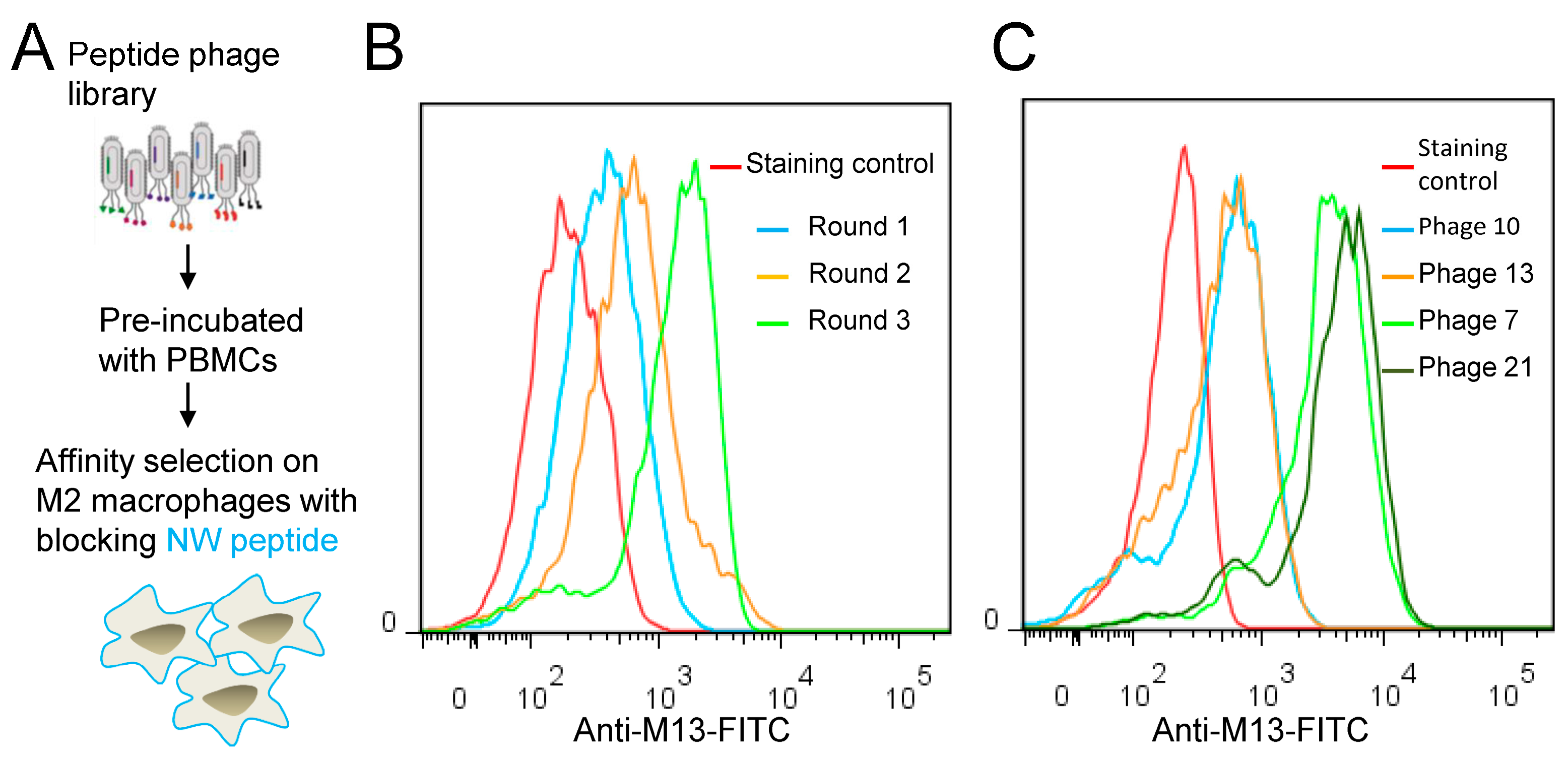
2.3. Biopanning of Phage Peptide Library on M2 Macrophages
2.4. Analysis of Phage Binding to Macrophages by Flow Cytometry
2.5. Analysis of the Phage-IR700 Binding to Macrophages
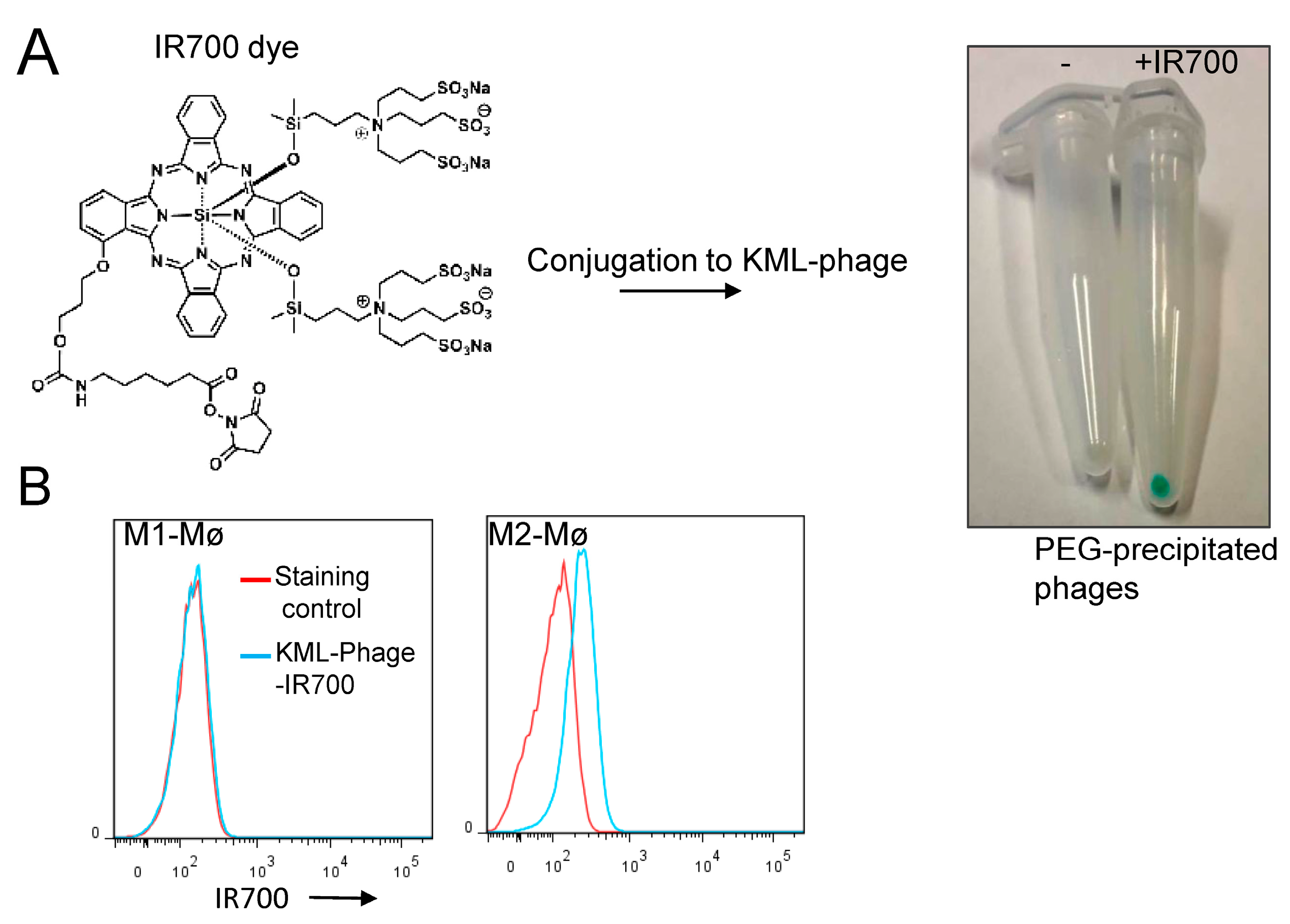
2.6. Conjugation of IR700 to Phage-Displayed Peptides
2.7. DNA Sequencing
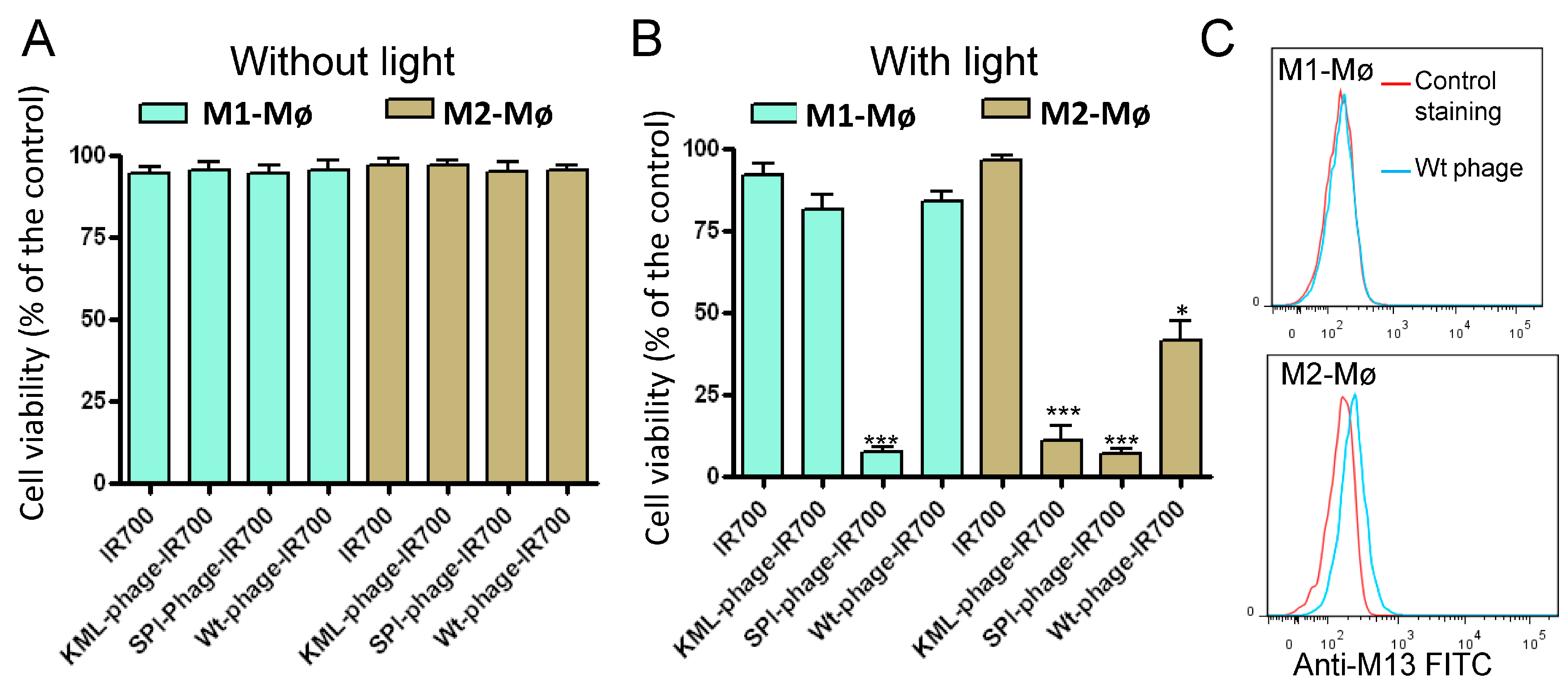
2.8. Photocytotoxicity
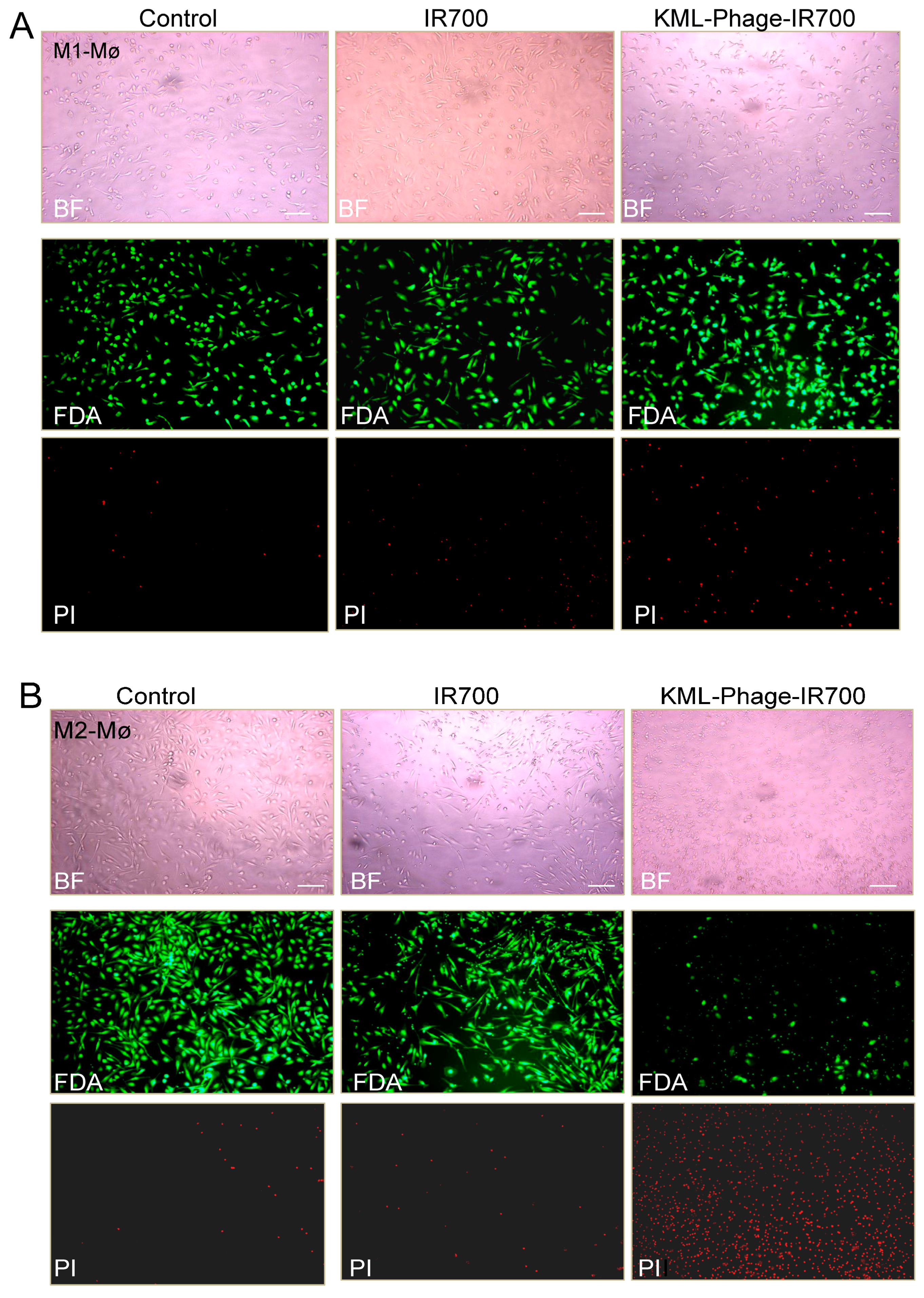
2.9. Bright-Field and Fluorescence Imaging
2.10. Docking
2.11. Statistical Analysis
3. Results
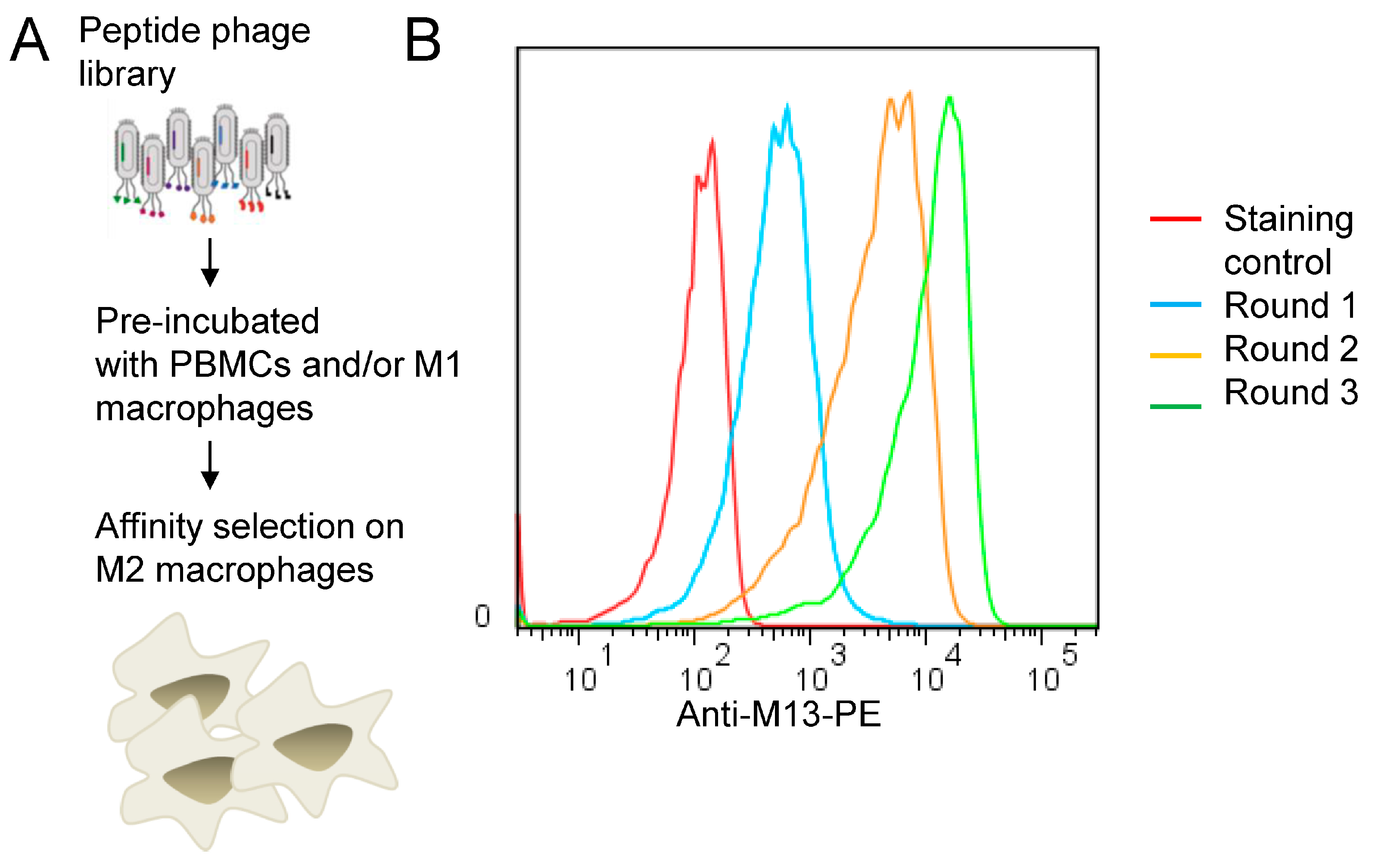
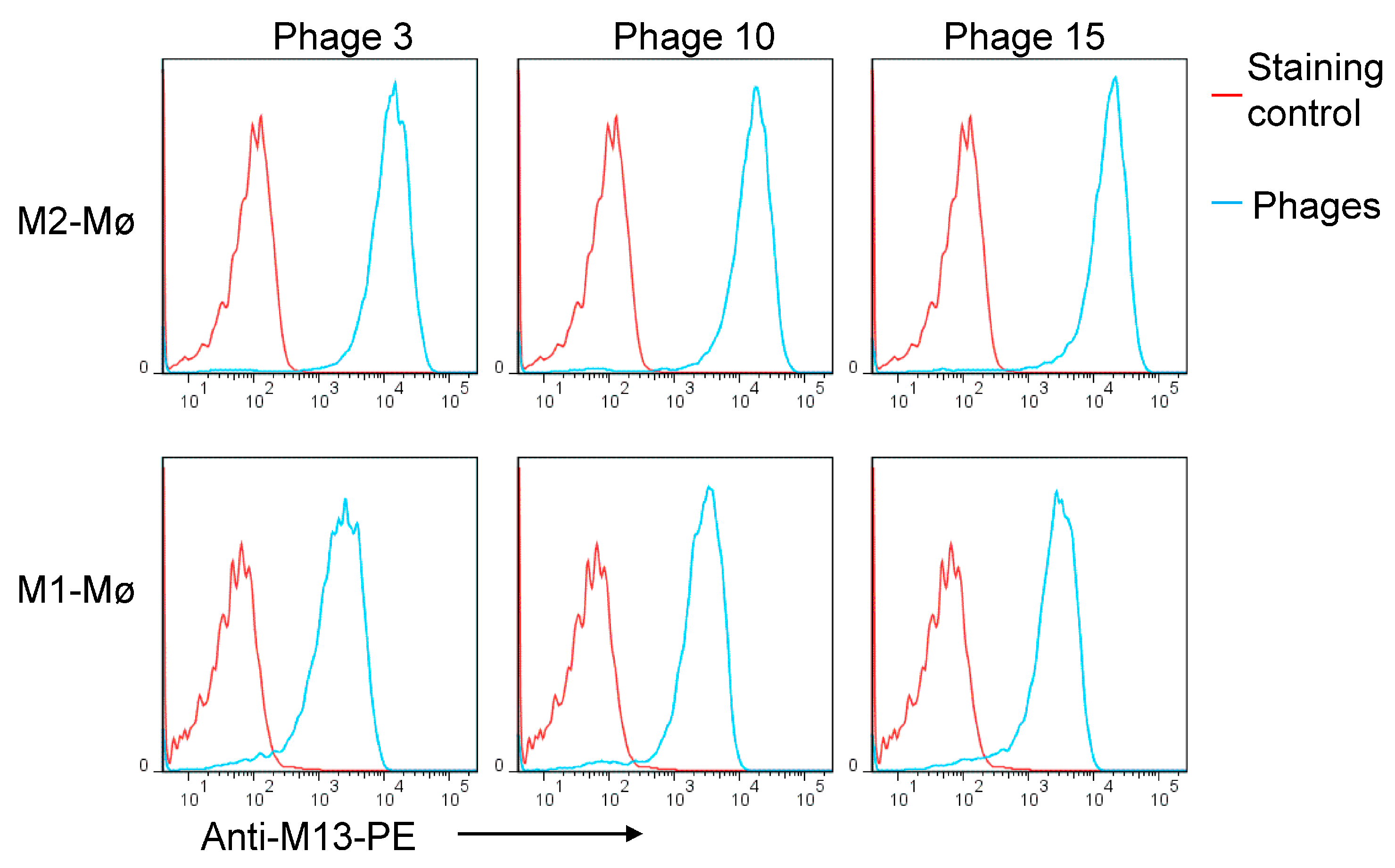
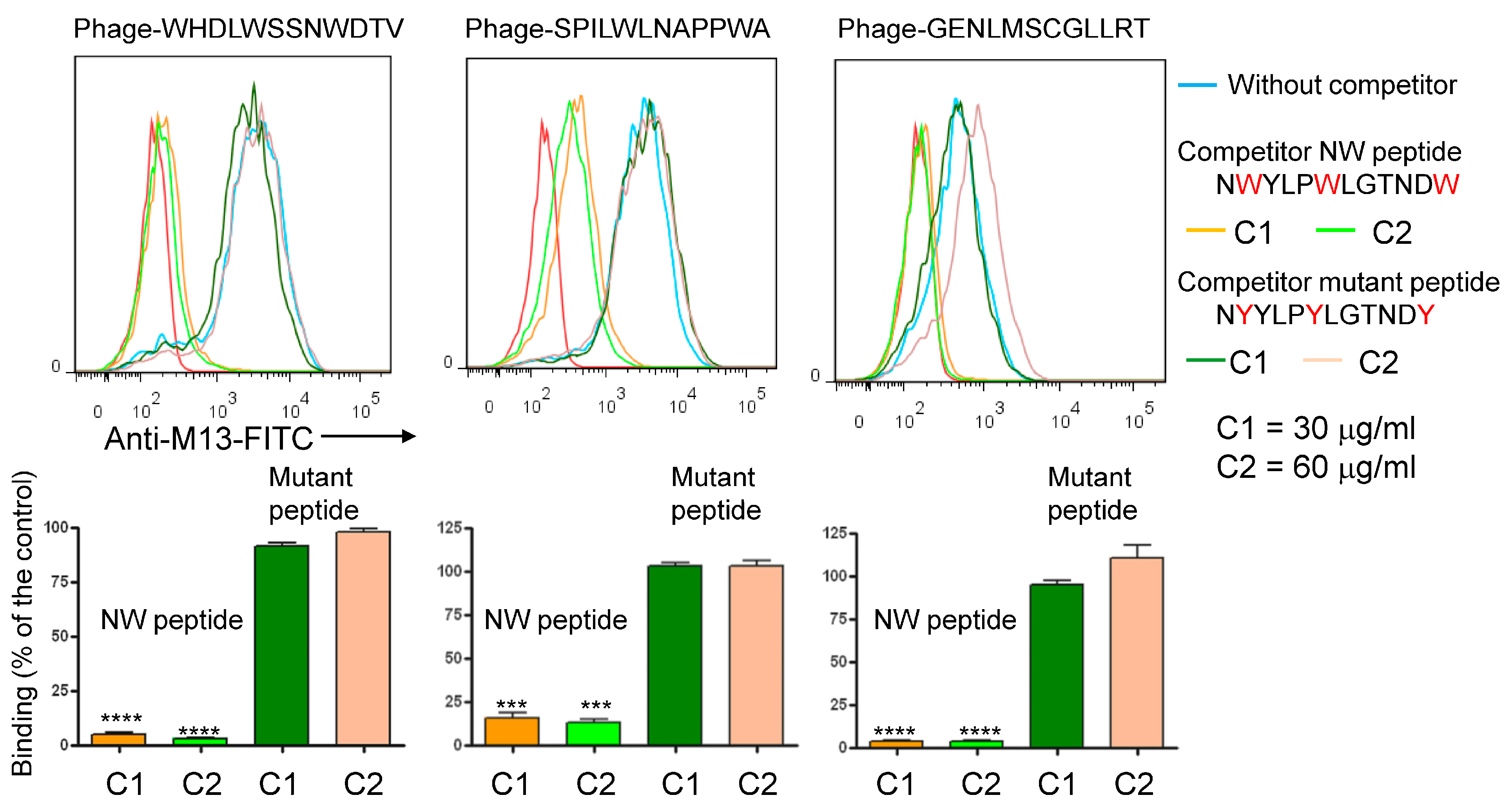
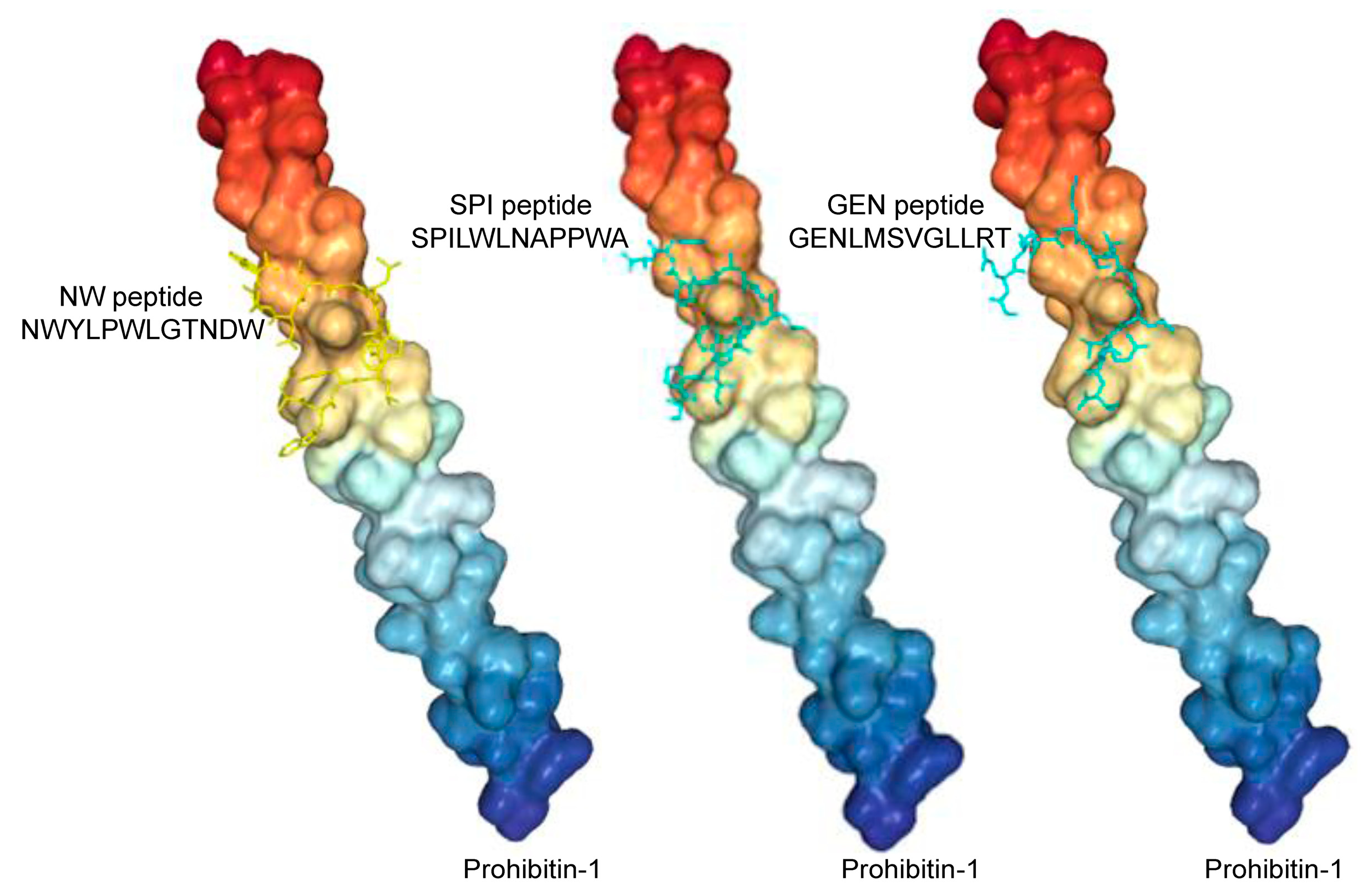
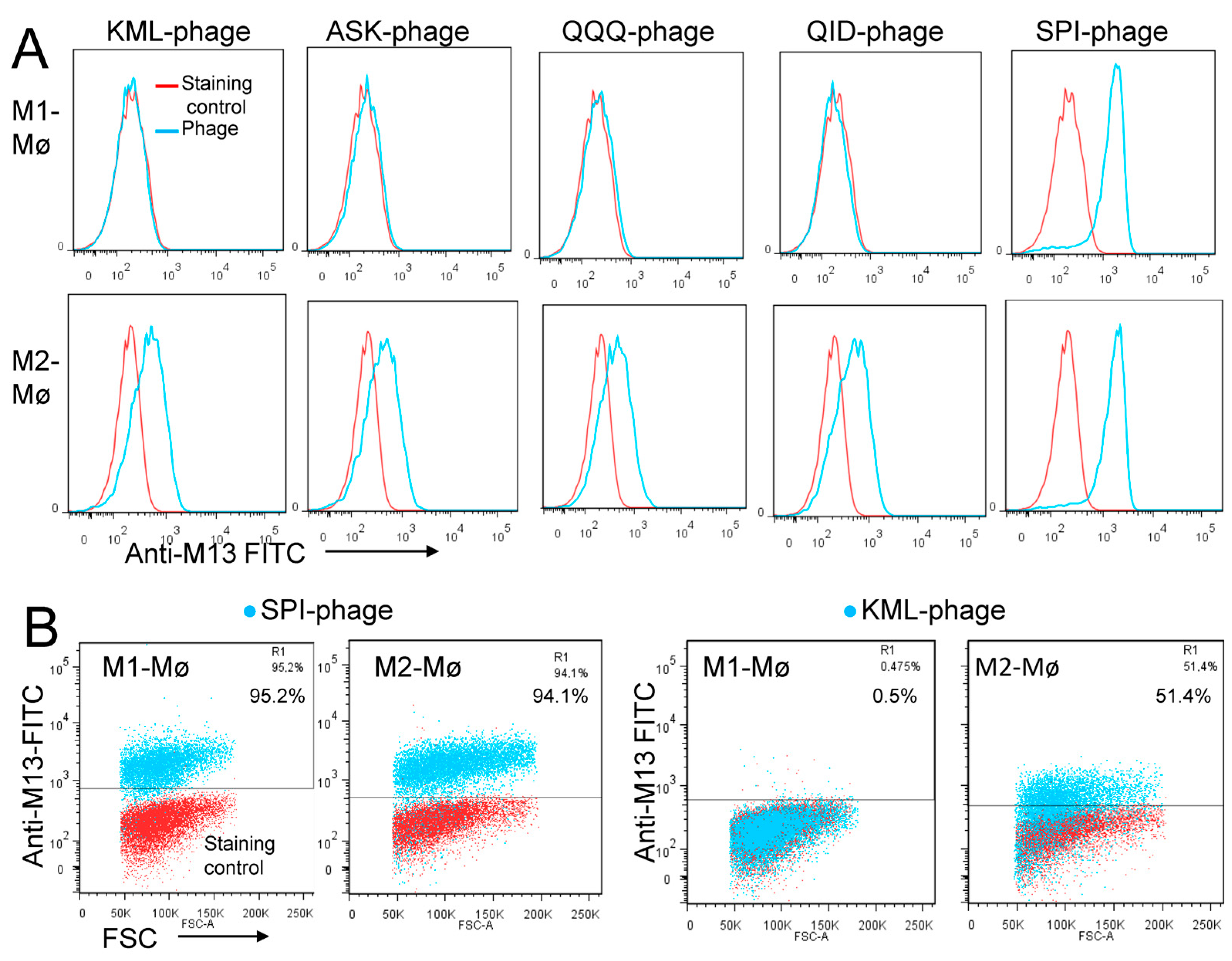
3.1. Biopanning on M2 Macrophages Reveals the Importance of Prohibitin-1 in Phage Selection
3.2. Blocking with NW Peptide Results in the Enrichment of M2-Binding Phages
3.3. Photocytotoxicity of the Phage Displayed Peptide-Photosensitizer Conjugates
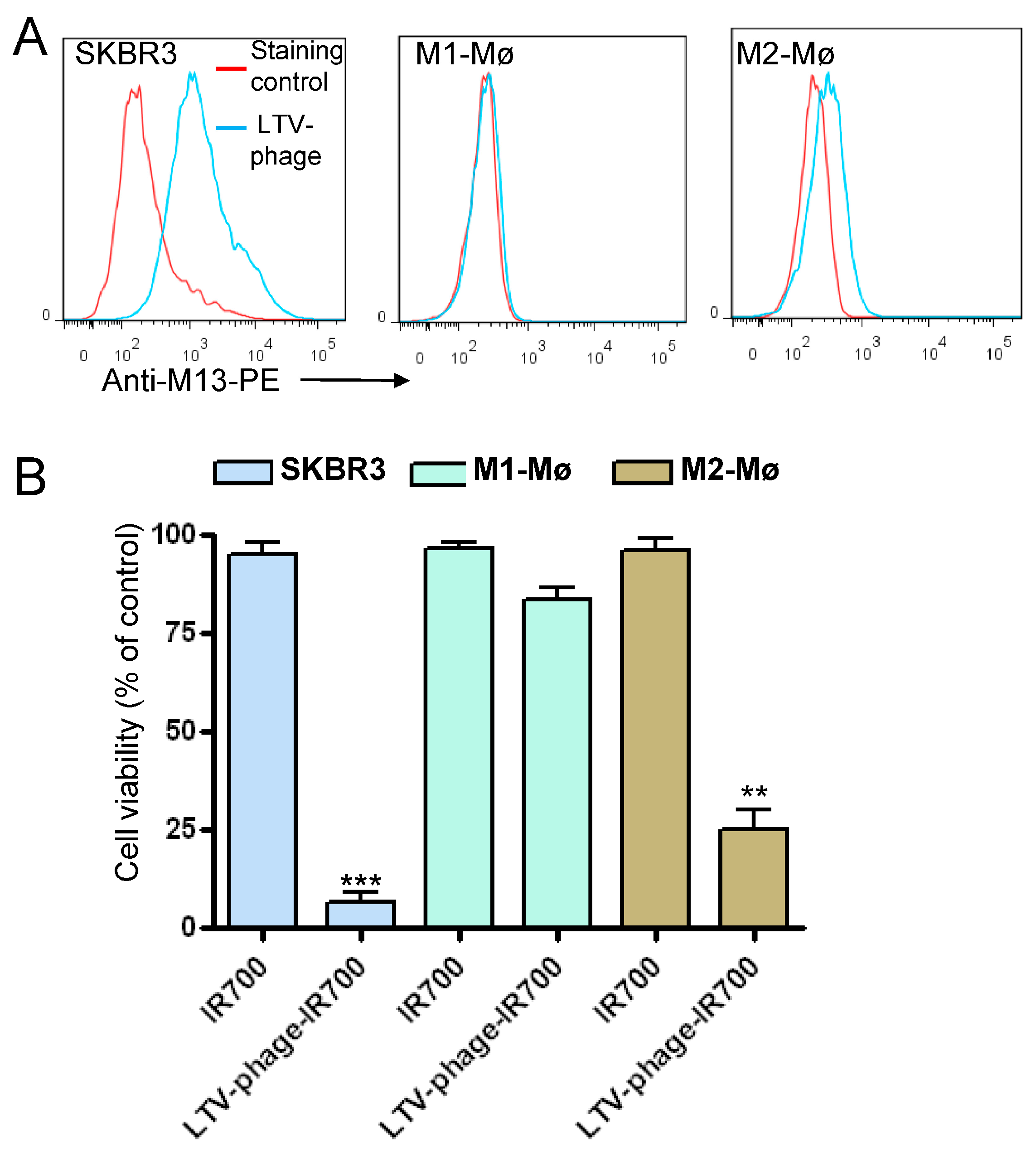
3.4. Co-Targeting Cancer Cells and M2 Macrophages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CSF-1 | Colony-stimulating factor 1 |
| CSF-1R | Colony-stimulating factor 1 receptor |
| FDA | Fluorescein diacetate |
| IL | Interleukin |
| LED | Light-emitting diode |
| NIR | Near infrared |
| PBMC | Peripheral blood mononuclear cell |
| PE | Phycoerythrin |
| PEG | Polyethylene glycol |
| PI | Propidium iodide |
| TGF-β | Transforming growth factor β |
| Wt | Wild type |
References
- Abbott, M.; Ustoyev, Y. Cancer and the Immune System: The History and Background of Immunotherapy. Semin. Oncol. Nurs. 2019, 35, 150923. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B.; Massa, C. Immune Therapy Resistance and Immune Escape of Tumors. Cancers 2021, 13, 511. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Teng, M.W.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Vesely, M.D.; Zhang, T.; Chen, L. Resistance Mechanisms to Anti-PD Cancer Immunotherapy. Annu. Rev. Immunol. 2022, 40, 45–74. [Google Scholar] [CrossRef]
- Jarosz-Biej, M.; Smolarczyk, R.; Cichoń, T.; Kułach, N. Tumor Microenvironment as A “Game Changer” in Cancer Radiotherapy. Int. J. Mol. Sci. 2019, 20, 3212. [Google Scholar] [CrossRef] [Green Version]
- Petty, A.J.; Yang, Y. Tumor-associated macrophages: Implications in cancer immunotherapy. Immunotherapy 2017, 9, 289–302. [Google Scholar] [CrossRef] [Green Version]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Hamidzadeh, K.; Goncalves, R. Macrophages and the maintenance of homeostasis. Cell. Mol. Immunol. 2021, 18, 579–587. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.V.; Ricardo, S.D. Macrophages and CSF-1: Implications for development and beyond. Organogenesis 2013, 9, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Aharinejad, S.; Paulus, P.; Sioud, M.; Hofmann, M.; Zins, K.; Schafer, R.; Stanley, E.R.; Abraham, D. Colony-stimulating factor-1 blockade by antisense oligonucleotides and small interfering RNAs suppresses growth of human mammary tumor xenografts in mice. Cancer Res. 2004, 64, 5378–5384. [Google Scholar] [CrossRef] [Green Version]
- Zins, K.; Abraham, D.; Sioud, M.; Aharinejad, S. Colon cancer cell-derived tumor necrosis factor-alpha mediates the tumor growth-promoting response in macrophages by up-regulating the colony-stimulating factor-1 pathway. Cancer Res. 2007, 67, 1038–1045. [Google Scholar] [CrossRef] [Green Version]
- Casagrande, N.; Borghese, C.; Favero, A.; Vicenzetto, C.; Aldinucci, D. Trabectedin overcomes doxorubicin-resistance, counteracts tumor-immunosuppressive reprogramming of monocytes and decreases xenograft growth in Hodgkin lymphoma. Cancer Lett. 2021, 500, 182–193. [Google Scholar] [CrossRef]
- Van Acker, H.H.; Anguille, S.; Willemen, Y.; Smits, E.L.; Van Tendeloo, V.F. Bisphosphonates for cancer treatment: Mechanisms of action and lessons from clinical trials. Pharmacol. Ther. 2016, 158, 24–40. [Google Scholar] [CrossRef]
- Zhang, Q.; Le, K.; Xu, M.; Zhou, J.; Xiao, Y.; Xiao, Y.; Jiang, Y.; Xi, Z.; Huang, T. Combined MEK inhibition and tumor-associated macrophages depletion suppresses tumor growth in a triple-negative breast cancer mouse model. Int. Immunopharmacol. 2019, 76, 105864. [Google Scholar] [CrossRef]
- Sioud, M. Phage Display Libraries: From Binders to Targeted Drug Delivery and Human Therapeutics. Mol. Biotechnol. 2019, 61, 286–303. [Google Scholar] [CrossRef]
- Sioud, M.; Skorstad, G.; Mobergslien, A.; Sæbøe-Larssen, S. A novel peptide carrier for efficient targeting of antigens and nucleic acids to dendritic cells. FASEB J. 2013, 27, 3272–3283. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M.; Pettersen, S.; Ailte, I.; Fløisand, Y. Targeted Killing of Monocytes/Macrophages and Myeloid Leukemia Cells with Pro-Apoptotic Peptides. Cancers 2019, 11, 1088. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Olberg, A.; Sioud, M. Structural Requirements for the Binding of a Peptide to Prohibitins on the Cell Surface of Monocytes/Macrophages. Int. J. Mol. Sci. 2022, 23, 4282. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.Y. HPEPDOCK: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M.; Oslo University Hospital, Oslo, Norway. Unpublished data. 2022.
- Sioud, M.; Juzenas, P.; Zhang, Q.; Kleinauskas, A.; Peng, Q. Evaluation of In Vitro Phototoxicity of a Minibody-IR700 Conjugate Using Cell Monolayer and Multicellular Tumor Spheroid Models. Cancers 2021, 13, 3356. [Google Scholar] [CrossRef]
- Kato, T.; Wakiyama, H.; Furusawa, A.; Choyke, P.L.; Kobayashi, H. Near Infrared Photoimmunotherapy: A Review of Targets for Cancer Therapy. Cancer 2021, 13, 2535. [Google Scholar] [CrossRef]
- Sato, K.; Ando, K.; Okuyama, S.; Moriguchi, S.; Ogura, T.; Totoki, S.; Hanaoka, H.; Nagaya, T.; Kokawa, R.; Takakura, H.; et al. Photoinduced Ligand Release from a Silicon Phthalocyanine Dye Conjugated with Monoclonal Antibodies: A Mechanism of Cancer Cell Cytotoxicity after Near-Infrared Photoimmunotherapy. ACS Cent. Sci. 2018, 4, 1559–1569. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.R.; Alberts, B.M.; Benzinger, R.; Lawhorne, L.; Treiber, G. Rapid bacteriophage sedimentation in the presence of polyethylene glycol and its application to large-scale virus purification. Virology 1970, 40, 734–744. [Google Scholar] [CrossRef]
- Ogawa, M.; Takakura, H. Photoimmunotherapy: A new cancer treatment using photochemical reactions. Bioorg. Med. Chem. 2021, 43, 116274. [Google Scholar] [CrossRef]
- Shadidi, M.; Sioud, M. Identification of novel carrier peptides for the specific delivery of therapeutics into cancer cells. FASEB J. 2003, 17, 256–258. [Google Scholar] [CrossRef]
- Neo, S.H.; Lew, Q.J.; Koh, S.M.; Zheng, L.; Bi, X.; Chao, S.H. Use of a novel cytotoxic HEXIM1 peptide in the directed breast cancer therapy. Oncotarget 2016, 7, 5483–5494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.F.; Birringer, M.; Dong, L.F.; Veprek, P.; Low, P.; Swettenham, E.; Stantic, M.; Yuan, L.H.; Zobalova, R.; Wu, K.; et al. A peptide conjugate of vitamin E succinate targets breast cancer cells with high ErbB2 expression. Cancer Res. 2007, 67, 3337–3344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Yang, J.; Jin, H.; Huang, C.; Fu, J.; Yang, F.; Gong, H.; Zeng, S.; Luo, Q.; Zhang, Z. Tetrameric far-red fluorescent protein as a scaffold to assemble an octavalent peptide nanoprobe for enhanced tumor targeting and intracellular uptake in vivo. FASEB J. 2011, 25, 1865–1873. [Google Scholar] [CrossRef]
- Moreno, M.; Zurita, E.; Giralt, E. Delivering wasp venom for cancer therapy. J. Control Release 2014, 182, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Pasqualini, R. Vascular targeting with phage peptide libraries. Q. J. Nucl. Med. 1999, 43, 159–162. [Google Scholar] [PubMed]
- Mishra, S.; Murphy, L.C.; Nyomba, B.G.; Murphy, L.J. Prohibitin: A potential target for new therapeutics. Trends Mol. Med. 2005, 11, 192–197. [Google Scholar] [CrossRef]
- Theiss, A.L.; Sitaraman, S.V. The role and therapeutic potential of prohibitin in disease. Biochim. Biophys. Acta 2011, 1813, 1137–1143. [Google Scholar] [CrossRef] [Green Version]
- Bichet, M.C.; Chin, W.H.; Richards, W.; Lin, Y.W.; Avellaneda-Franco, L.; Hernandez, C.A.; Oddo, A.; Chernyavskiy, O.; Hilsenstein, V.; Neild, A.; et al. Bacteriophage uptake by mammalian cell layers represents a potential sink that may impact phage therapy. iScience 2021, 24, 102287. [Google Scholar] [CrossRef]
- Petrov, G.; Dymova, M.; Richter, V. Bacteriophage-Mediated Cancer Gene Therapy. Int. J. Mol. Sci. 2022, 23, 14245. [Google Scholar] [CrossRef]
- Gustafson, H.H.; Olshefsky, A.; Sylvestre, M.; Sellers, D.L.; Pun, S.H. Current state of in vivo panning technologies: Designing specificity and affinity into the future of drug targeting. Adv. Drug Deliv. Rev. 2018, 130, 39–49. [Google Scholar] [CrossRef]
- Stephanopoulos, N.; Tong, G.J.; Hsiao, S.C.; Francis, M.B. Dual-surface modified virus capsids for targeted delivery of photodynamic agents to cancer cells. ACS Nano 2010, 4, 6014–6020. [Google Scholar] [CrossRef] [PubMed]
- Ulfo, L.; Cantelli, A.; Petrosino, A.; Costantini, P.E.; Nigro, M.; Starinieri, F.; Turrini, E.; Zadran, S.K.; Zuccheri, G.; Saporetti, R.; et al. Orthogonal nanoarchitectonics of M13 phage for receptor targeted anticancer photodynamic therapy. Nanoscale 2022, 14, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Bortot, B.; Apollonio, M.; Baj, G.; Andolfi, L.; Zupin, L.; Crovella, S.; Giosia, M.; Cantelli, A.; Saporetti, R.; Ulfo, L.; et al. Advanced photodynamic therapy with an engineered M13 phage targeting EGFR: Mitochondrial localization and autophagy induction in ovarian cancer cell lines. Free Radic. Biol. Med. 2022, 179, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Gandra, N.; Abbineni, G.; Qu, X.; Huai, Y.; Wang, L.; Mao, C. Bacteriophage bionanowire as a carrier for both cancer-targeting peptides and photosensitizers and its use in selective cancer cell killing by photodynamic therapy. Small 2013, 28, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suthiwangcharoen, N.; Li, T.; Li, K.; Thompson, P.; You, S.; Wang, Q. M13 Bacteriophage-Polymer Nanoassemblies as Drug Delivery Vehicles. Nano Res. 2011, 4, 483–493. [Google Scholar] [CrossRef]
- Ghosh, D.; Kohli, A.G.; Moser, F.; Endy, D.; Belcher, A.M. Refactored M13 bacteriophage as a platform for tumor cell imaging and drug delivery. ACS Synth. Biol. 2012, 1, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Quilbe, A.; Moralès, O.; Baydoun, M.; Kumar, A.; Mustapha, R.; Murakami, T.; Leroux, B.; Schutter, C.; Thecua, E.; Ziane, L.; et al. An Efficient Photodynamic Therapy Treatment for Human Pancreatic Adenocarcinoma. J. Clin. Med. 2020, 9, 192. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Wei, Y.; Jiang, X.; Wang, C.; Liu, M.; Yan, J.; Zhang, L.; Zhou, Y. Insight into the Prospects for Tumor Therapy Based on Photodynamic Immunotherapy. Pharmaceuticals 2022, 15, 1359. [Google Scholar] [CrossRef]
- Aniogo, E.C.; George, B.P.; Abrahamse, H. Molecular Effectors of Photodynamic Therapy-Mediated Resistance to Cancer Cells. Int. J. Mol. Sci. 2021, 22, 13182. [Google Scholar] [CrossRef]
- Biteghe, F.A.N.; Chalomie, N.E.T.; Mungra, N.; Vignaux, G.; Gao, N.; Vergeade, A.; Okem, A.; Naran, K.; Ndong, J.D.L.C.; Barth, S. Antibody-Based Immunotherapy: Alternative Approaches for the Treatment of Metastatic Melanoma. Biomedicines 2020, 8, 327. [Google Scholar] [CrossRef]
- Ulfo, L.; Costantini, P.E.; Di Giosia, M.; Danielli, A.; Calvaresi, M. EGFR-Targeted Photodynamic Therapy. Pharmaceutics 2022, 14, 241. [Google Scholar] [CrossRef]
- Wang, X.; Luo, D.; Basilion, J.P. Photodynamic Therapy: Targeting Cancer Biomarkers for the Treatment of Cancers. Cancers 2021, 13, 2992. [Google Scholar] [CrossRef]
- Beltrán Hernández, I.; Angelier, M.L.; Del Buono D’Ondes, T.; Di Maggio, A.; Yu, Y.; Oliveira, S. The Potential of Nanobody-Targeted Photodynamic Therapy to Trigger Immune Responses. Cancers 2020, 12, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, C.X.; Zhang, X.Z. Bacteriophage-mediated modulation of microbiota for diseases treatment. Adv. Drug. Deliv. Rev. 2021, 176, 113856. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selected Peptides | Binding | |
|---|---|---|
| M1-Mø | M2-Mø | |
| NWYLPWLGTNDW * | ++++ | ++++ |
| QWELPWLMQPPL | ++++ | ++++ |
| TWALPWLLEKPF | ++++ | ++++ |
| SPILWLNAPPWA | ++++ | ++++ |
| WHDLWSSNWDTV | ++++ | ++++ |
| GENLMSVGLLRT | ++ | ++ |
| Selected Peptides | Binding | |
|---|---|---|
| M1-Mø | M2-Mø | |
| NWYLPWLGTNDW * | ++++ | ++++ |
| TWDLPWLLEKPF | ++++ | ++++ |
| KMLPTMPRVLAG | - | ++ |
| DAAPTLPKGGVG | - | ++ |
| QIDTGYGLVSVS | - | ++ |
| GSKTGYLSETVR | - | ++ |
| ASKNAHLFLSSL | - | + |
| QQQYGTYVPTFG | - | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sioud, M.; Zhang, Q. Precision Killing of M2 Macrophages with Phage-Displayed Peptide-Photosensitizer Conjugates. Cancers 2023, 15, 2009. https://doi.org/10.3390/cancers15072009
Sioud M, Zhang Q. Precision Killing of M2 Macrophages with Phage-Displayed Peptide-Photosensitizer Conjugates. Cancers. 2023; 15(7):2009. https://doi.org/10.3390/cancers15072009
Chicago/Turabian StyleSioud, Mouldy, and Qindong Zhang. 2023. "Precision Killing of M2 Macrophages with Phage-Displayed Peptide-Photosensitizer Conjugates" Cancers 15, no. 7: 2009. https://doi.org/10.3390/cancers15072009
APA StyleSioud, M., & Zhang, Q. (2023). Precision Killing of M2 Macrophages with Phage-Displayed Peptide-Photosensitizer Conjugates. Cancers, 15(7), 2009. https://doi.org/10.3390/cancers15072009